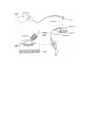
Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Ebola virus disease wikipedia , lookup
Social history of viruses wikipedia , lookup
Plant virus wikipedia , lookup
Introduction to viruses wikipedia , lookup
Human Endogenous Retrovirus-W wikipedia , lookup
History of virology wikipedia , lookup
Endogenous retrovirus wikipedia , lookup
Virus quantification wikipedia , lookup
Negative-sense single-stranded RNA virus wikipedia , lookup
DEVELOPMENT AND CHARACTERIZATION OF A TRANSGENIC MOUSE MODEL FOR POLIOMYELITIS Ruibao Ren Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 1992 ABSTRACT Development and characterization of a transgenic mouse model for poliomyelitis Ruibao Ren In this work, transgenic mice containing a human poliovirus receptor (PVR) gene in the germ line were established. inoculation of PVR transgenic mice with poliovirus of all three serotypes leads to the development of a fatal paralytic disease that clinically and histopathologically resembles human poliomyelitis. This study demonstrates that the absence of PVR is the determinant of poliovirus host range restriction in mice. The transgenic mice express PVR transcripts and poliovirus binding sites wide range of tissues. The expression of PVR RNA in transgenic mice generally mimics that in human. The study of PVR gene expression in human adult and embryonic tissues by in situ hybridization provides a basis for understanding the normal function of PVR, which is a novel member of the immunoglobulin superfamily of proteins. To characterize the tropism of poliovirus infection in PVR transgenic mice, poliovirus replication sites were examined by in situ hybridization. These studies demonstrate that poliovirus tissue tropism is not governed solely by expression of the PVR gene nor by accessibility of cells to virus infection. Another fundamental unsolved issue in poliovirus pathogenesis, the route by which virus spreads to the central nervous system (CNS), was studied in PVR transgenic mice. The results demonstrate that poliovirus enters into the CNS through peripheral nerves. Although they are susceptible to neurovirulent poliovirus strains, the PVR transgenic mice inoculated with attenuated poliovirus of all three serotypes do not develop signs of disease. To identify the determinants that attenuate a vaccine-related poliovirus type 2 strain, P2/P712, genomic recombinants between P2/P712 and a poliovirus type 2 neurovirulent strain, P2/Lansing, were constructed. Using transgenic and nontransgenic mice, the major determinants of P2/P712 were identified as nucleotide 481 in the viral noncoding region and amino acid residue 143 in the capsid polypeptide VP1. These results establish the transgenic mouse expressing human poliovirus receptor as a new model for studying poliovirus neurovirulence, attenuation, and pathogenesis. The transgenic mouse model for poliomyelitis could constitute an alternative host for safety testing of poliovirus vaccines, replacing the costly neurovirulence test in monkeys. TABLE OF CONTENTS List of tables vi List of figures vii List of abbreviations ix Acknowledgement xi Chapter 1. Introduction 1 1. Structure of poliovirus 2 2. Poliovirus replication 8 a) An overview of the poliovirus life cycle 9 b) Early stages of poliovirus infection 9 c) Poliovirus translation 11 3. Pathogenesis of poliomyelitis 13 a) Clinical features 14 b) Course of poliovirus infection 15 c) Infection of the CNS 16 d) Pathology 19 e) Tissue tropism 20 f) Host range 22 4. Poliovirus receptor 24 5. Poliovirus attenuation 26 a) isolation of attenuated virus strains 26 b) Determinants of attenuation 28 Chapter II. Materials and Methods 30 Cells, virus and antibody 30 Virus growth and assay 32 RNA and DNA isolation 33 Construction of Hela cell genomic cosmid library 33 DNA transformation 34 Microinjection and production of transgenic mice 34 Poliovirus receptor binding assay 35 Neurovirulence assay 36 ii Assay for viral replication in mouse brain and spinal cord 37 Animal inoculation and tissue sampling 37 Sciatic nerve transection 37 Neuropathology 38 Hybridization probe synthesis 39 In situ hybridization 39 PCR amplification of cDNA 40 Construction of viral recombinants 40 Mutagenesis of viral recombinants 41 Nucleotide sequencing 41 Chapter III. Transgenic mice expressing a human poliovirus receptor: a new model for poliomyelitis 42 Isolation of a human poliovirus receptor gene 43 Generation of transgenic mice carrying a human poliovirus receptor gene 45 Expression of poliovirus receptor RNA in transgenic mouse tissues 45 Poliovirus receptor binding activity in transgenic mouse tissues 49 Infection of PVR transgenic mice with poliovirus 52 Neuropathology of PVR transgenic mice infected with poliovirus 55 Chapter IV. Human poliovirus receptor gene expression in human and transgenic mouse 63 Localization of PVR RNA in transgenic mouse tissues Expression of the alternative spliced forms of PVR transcripts in iii 64 transgenic mouse tissues 67 Expression of PVR RNA in transgenic mouse embryo and placenta 67 Expression of PVR RNA in human adult and embryonic tissues 71 Expression of PVR RNA in human placenta 72 Chapter V. Poliovirus tissue tropism in transgenic mice 78 Poliovirus replication sites in the CNS of transgenic mice 79 Poliovirus susceptibility of transgenic mouse nonneural tissues 82 Susceptibility of cultured PVR transgenic mouse kidney cells to poliovirus 87 Poliovirus infection in newborn mice following peroral inoculation 89 Chapter VI. Poliovirus spreads from muscle to the central nervous system by neural pathways 95 Efficiency of induction of poliomyelitis by different inoculation routes 96 Paralysis following intramuscular inoculation of poliovirus 96 Spread of poliovirus to the CNS 99 Effect of nerve transection on poliovirus infection 101 Chapter VII. Attenuation determinants in a vaccine-related type 2 poliovirus P2/P712 103 Mapping an attenuation determinant in the coding region of P2/P712 104 Identification of the major attenuation determinant in capsid protein VP1 of P2/P712 104 Identification of the major attenuation determinant in the 5’ noncoding region of P2/P712 107 Neurovirulence of recombinant viruses in transgenic mice iv expressing human poliovirus receptors 108 Poliovirus replication in PVR transgenic mouse skeletal muscle 111 Temperature sensitivity of polioviruses in transgenic mouse primary muscle culture 111 Chapter VIII. Discussion 119 Determinant of poliovirus host range in mice 120 PVR gene expression in transgenic mice 121 PVR gene expression in human 123 Poliovirus tissue tropism 126 Histopathology of experimental poliomyelitis in PVR transgenic mice 129 Poliovirus pathogenesis 131 Attenuating determinants of a vaccine-related type 2 poliovirus 141 Molecular basis of poliovirus temperature sensitivity and attenuation 144 Conclusions and Perspectives 148 References 151 v LIST OF TABLES 1. Yields of poliovirus after infection of mouse cells transformed with poliovirus receptor cosmid clones. 44 2. Susceptibility of mice to poliovirus infection. 54 3. Summary of clinical and neuropathological findings of 21 day neurovirulence test. 4. 58 Susceptibility of PVR transgenic mouse kidney cells after in vitro cultivation. 90 5. Poliovirus binding to dispersed PVR transgenic mouse kidney cells. 90 6. Susceptibility of suckling mice to poliovirus infection following peroral inoculation. 7. 91 Effect of route of inoculation on LD50 Of Poliovirus Pl/Mahoney in PVR transgenic mice. 8. 97 Localization of initial paralysis in mice inoculated intramuscularly with poliovirus. 9. 98 Effect of sciatic nerve transection on poliovirus-induced lethality in TgPVR mice. 10. 102 The temperature sensitivity of poliovirus strains on MPMC monolayers. 117 vi LIST OF FIGURES 1. Map of the poliovirus genome. 4 2. Atomic structure of the pentamer of Pl/Mahoney. 6 3. The pathogenesis of poliomyelitis in primates. 17 4. Identification of transgenic mice containing PVR DNA. 46 5. Northern hybridization analysis of mouse tissue RNAs. 50 6. Poliovirus binding activity in mouse tissue homogenates. 53 7. Time course of paralysis and poliovirus replication in mice. 56 8. Neuropathology of poliovirus infected transgenic mice. 60 9. PVR mRNA expression in transgenic mouse tissues. 65 10. Detection of alternatively spliced PVR RNA. 68 11. PVR mRNA expression in the prenatal transgenic mouse. 69 12. PVR mRNA expression in human adult and fetal tissues. 73 13. PVR mRNA expression in human placenta. 75 14. In situ detection of poliovirus RNA in spinal cord of PVR transgenic mice infected intraperitoneally with poliovirus. 15. 80 In situ detection of poliovirus RNA in infected PVR transgenic mouse brain. 83 16. Poliovirus replication in PVR transgenic mouse skeletal muscle. 85 17. Poliovirus replication in PVR transgenic mouse kidney. 88 18. In situ detection of poliovirus RNA in suckling PVR transgenic 19. mouse orally infected with Pl/Mahoney. 92 Time course of poliovirus replication in the CNS. 100 vii 20. Constitution and mouse neurovirulence of P2/P712-P2/Lansing capsid protein VP1 coding sequences in recombinant and mutant viruses. 21. 105 Constitution and mouse neurovirulence of P2/P712-P2/Lansing 5'-ncr recombinant and mutant viruses. 109 22. Time course of poliovirus replication in skeletal muscle. 112 23. Cytopathic changes in MPMC cells infected with type 2 polioviruses. 114 24. Cytopathic changes in MPMC cells infected with type 1 polioviruses. 115 25. Possible route of poliovirus spread from muscle to the CNS in mice. 135 26. Possible scheme of poliovirus pathogenesis in humans. 139 viii LIST OF ABBREVIATIONS BBB: Blood-Brain Barrier CNS: Central Nervous system CPE: Cytopathic Effect cs: cold sensitive DMEM: Dulbecco’s Modified Eagle's Medium 5’-ncr: 5' noncoding region of poliovirus RNA H&E: Hematoxylin and Eosin hPL: human placental lactogen Ig: Immunoglobulin ile: isoleucine IRES: Internal Ribosome Entry Site isc: the inferior spinal cord Kb: Kilobase Kd: Kilodalton LD50: The amount of the virus which cause death in 50% of mice MOI: Multiplicity-Of-Infection MPMC: Mouse Primary Muscle Culture N-Agl: Neutralization antigenic site 1 ntg: nontransgenic mice ORF: Open Reading Frame PBS: Phosphate Buffered Saline PCR: Polymerase Chain Reaction ix PFU: Plaque-Forming Units phe: phenylalanine PRG: Poliovirus Receptor Genornic DNA PVR: Poliovirus Receptor RLP: Ribosome Landing Pad ser: serine ssc: the superior spinal cord TCID: Tissue Culture Infective Doses TgPVR: Transgenic mice expressing a human PVR thr: threonine ts: temperature sensitive x ACKNOWLEDGEMENTS Vincent Racaniello, my advisor, gave me the opportunity to work in his lab and developed the goals for this project. He has provided constant input, critical judgement, advice, and enthusiasm at every level of this research. I will be forever grateful for his guidance, patience, and friendship. Frank Costantini and Edward Gorgacz, our generous collaborators, made crucial contributions to this project. To them, I give many sincere thanks. I would like to thank Saul Silverstein for his enthusiasm, judgment, personal help, and guidance in the preparation of this dissertation. My thanks also go to Harnish Young for his critical judgment and support. Eric Moss, Liz Colston, and Mary Morrison have helped the progress of the work presented here and contributed enormously to my development as a scientist. I am grateful for their encouragement, personal help, and friendship. I am also grateful for advice, technical assistance, and friendship to Cathy Mendelsohn, Robert O'Neill, Michael Shepley, Michael Bouchard, Gerardo Kaplan, Marion Freistadt, Xiuxuan Zhu, Jason Chen, Roy Bohenzky, Michael Reach, Tom DeChiara, Christos Panagiotidis, Suhua Zhang, Yanzhang Dong, Sa Liao, Alan Dove, Chu-Hui Peng, Yuang-Jing He, David Peters, William Dundon, Du Lam, Tony Wild, and Marie Waddell. I gratefully acknowledge Bernard Fields and Lynda Morrison for assistance with sciatic nerve transection and peroral inoculation of suckling mice; James Lee, Leah Jaffe for assistance with in situ hybridization, and Vivette D'Agati, Kathleen O'Toole, Laura Hair and John Pintar for histological consultations. xi And last but by no means least I would like to thank my wife, Youzhen Wang, for her love, faith, and support. xii Chapter I. Introduction 1 The importance of poliovirus as a serious human pathogen was initially responsible for extensive investigation into its biology. In the past decade, studies on the molecular biology, structure, and genetics of poliovirus have made this one of the best understood viruses of eukaryotic cells. However, because of the absence of a convenient animal model, studies on the pathogenesis of poliomyelitis have not kept up with the progress in understanding other aspects of poliovirus replication. Although killed and live attenuated virus vaccines have effectively controlled paralytic poliomyelitis since the 1950's, poliomyelitis remains a serious health risk in many countries. Countries using the live attenuated vaccine still experience a low level of poliomyelitis, at least some of which is caused by reversion to virulence of the vaccine strain. As a result of vaccine-associated disease, there is some impetus to construct, by genetic engineering, vaccine strains which do not revert to neurovirulence. A significant obstacle to the development of new poliovirus vaccines is the cost and availability of the large number of monkeys that would be required for neurovirulence testing of candidate vaccine strains. It is important to identify the determinants of poliovirus host range restriction and generate a more convenient and less expensive laboratory animal. The initial goal of this work was to study the role of human poliovirus receptor (PVR) in viral host range restriction. This was achieved by generating transgenic mice containing the PVR gene in the germ line. The second goal was to use the PVR transgenic mouse model to study the mechanism of poliovirus tissue tropism, to identify the route by which poliovirus reaches the central nervous system (CNS), and to identify the determinants of an attenuated poliovirus type 2 strain. 1. Structure of poliovirus. Poliovirus is a member of the picornaviridae, a large virus family that contains enterovirus (e.g. polioviruses, coxsackieviruses, echoviruses, and hepatitis virus A), rhinovirus, aphthovirus (e.g. foot-and-mouth 2 disease virus), cardiovirus (e.g. encephalomyocarditis and mengovirus), and some unclassified viruses (Rueckert, 1990). It is a small non-enveloped, icosahedral particle consisting of a single-stranded messagesense RNA genome that is surrounded by 60 copies each of capsid proteins VP1 and VP3, 58 to 59 copies of VP2 and VP4, and 1 to 2 copies of VP0, the precursor to VP2 and VP4 (Kitamura et al., 1981; Rueckert, 1990). Polioviruses are grouped into three serotypes based on antigenicity of the capsid. The 7.5 kilobase (Kb) RNA genome of poliovirus is linked to a small viral protein, VPg, at its 5' end and is polyadenylated. The RNA encodes a single long open reading frame (OFR) preceded by an approximately 0.75 kb 5' noncoding region (5'-ncr) that is involved in translation initiation and genome replication (reviewed in (Sarnow et al., 1990). The 5'-one-third of the open reading frame encodes the four capsid proteins (VP4, VP2, VP3, and VP1), and the remainder encodes seven nonstructural proteins (2A-C and 3A-D) that perform functions required for virus replication. Proteins 2A and 3C are proteinases; Proteins 3B and 3D are the genome linked protein VPg and the viral RNA polymerase respectively. Figure 1 shows a map of the poliovirus genome. Atomic structures of three poliovirus strains have been determined by X-ray crystallography to high resolution. They are P1/Mahoney (Hogle et al., 1985), the vaccine strain P3/Sabin (Filman et al., 1989), and a mouse-adapted type2/type1 poliovirus chimera (Yeates et al., 1991). The capsid proteins VP1, VP2, and VP3 are similar in size and share a common folding pattern. Each of the proteins contains a conserved core that is an eight-stranded antiparallel beta barrel. The three proteins differ in the size and conformation 3 of the loops that 0 1 2 3 4 5 6 7 kb 5'nc VP4 VP2 VP3 VP1 2A 2B 2C 3A 3B 3C RNA 3D 3'nc AAA Figure 1. Map of the poliovirus genome. The open box represents the open reading frame. The sequences encoding viral proteins are indicated above the open box. The circle at the 5' terminus represents the genome linked protein VPg. The 5' noncoding region and 3' noncoding region are indicated by 5'nc and 3'nc respectively. The size of each region of viral RNA is shown in scale above the viral RNA genome. 4 connect the strands of the beta barrels and in the extensions at their amino and carboxyl termini. One copy of each capsid protein forms a protomer. Five protomers assemble into a pentamer and 12 pentamers assemble into the viral capsid. Figure 2 shows the architecture of a pentamer. VP1 encircles the fivefold axis of symmetry, VP2 and VP3 alternate around the threefold axis. The small protein VP4, which functions in some respects as the detached amino terminus of VP2, is on the virion interior. In a virus particle, the outer surface of the virion is dominated by two sets of prominent radial extensions: a fivefold peak formed by residues from VP1, and a threefold plateau formed by residues from VP2 and VP3. Three loops (BC, DE, and HI) of the VP1 barrel are exposed at the summit of the fivefold peaks (Hogle et al., 1985). The fivefold peaks are surrounded by broad valleys, known as the canyon in rhinovirus 14 (Rossmann et al., 1985). The canyon was proposed to be the site on the viral surface which binds to cellular receptors (Hogle et al., 1985; Rossmann et al., 1985). In addition to the proteins, two other large molecules assemble into the capsid: myristic acid residues attach to the amino-terminus of each copy of VP4 (Chow et al., 1987), and a hydrocarbon, resembling sphingosine, is inserted into a hydrophobic pocket (hydrocarbon-binding pocket) in each VP1 (Filman et al., 1989). Two structures in the capsid are believed to be important for the strong association of the subunits and therefore must play significant roles in virion assembly and disassembly: a β-tube formed from the amino-termini of five copies of VP3, VP4 and a small part of VP1 which encircle the fivefold axis on the virion interior, and a sevenstranded beta sheet formed by residues from three proteins from pairs of threefold related pentamers (Filman et al., 1989; Hogle et al., 1985). Both of these structures depend for their formation on the association of capsid precursors. Formation of the β-tube depends both on the cleavage between VP0 and VP3 to free the amino terminus of VP3 and on the 5 Figure 2. Atomic structure of the pentamer of P1/Mahoney. All images are α-carbon tracings. VP1 is blue, VP2 is yellow, VP3 is red, and VP4 is green. Top) Five protomers that constitute a pentamer are shown from the outside of the particle looking down the fivefold axis of icosahedral symmetry. Highlighted is the interface between fivefold related protomers. Bottom) A pentamer is shown from the side with the outside of the particle at the top of the image and the interior at the bottom. 6 association of protomers to form pentamers. Formation of the seven-stranded beta sheet requires the association of pentamers and the cleavage of VP0 to free the amino terminus of VP2, which is the final step in virion maturation. Other important structural units involved in conformational transitions during viral assembly and disassembly are: the interface between fivefold related protomers (Figure 2) and the hydrocarbon-binding pocket of VP1. The involvement of the interface in conformational transitions of virus during viral assembly and disassembly is suggested by analogy with the structurally similar T=3 plant viruses (e.g. tomato bushy stunt virus). In these plant viruses, the corresponding interface is disrupted during the expansion of the particle which is induced by the depletion of divalent cations at basic p H (Robinson and Harrison, 1982). The hydrocarbon-binding pocket of VP1 in poliovirus is nearly identical to the site which binds a class of antiviral drugs in rhinovirus 14 (Badger et al., 1988). Once bound, these compounds prevent a variety of conformational rearrangements of the virus, including those required for productive cell entry, and those associated with thermal inactivation (McSharry et al., 1979). The hydrocarbon-binding pocket, therefore, may be normally used to modulate the stability of the viruses. The poliovirus structures share features of construction with several other small spherical RNA viruses, which include the rhinovirus (Rossmann et al., 1985), mengovirus (Luo et al., 1987), food-and-mouth disease virus (Acharya et al., 1989), a number of plant viruses, and an insect virus (Harrison, 1990). They have in common the general folding motif of the major capsid proteins, an eight-stranded β-barrel, and the arrangement of these proteins into subunits and the capsid (Harrison, 1990). The structural similarities among these icosahedral, positive-stranded, RNA viruses implies a common solution to the problem of how to design a vehicle for delivery of the RNA genome from one host to another, in which assembly and disassembly of the vehicle is required. 2. Poliovirus replication. 8 a) An overview of the poliovirus life cycle. Poliovirus infects a cell by interacting with a specific receptor at the cell surface, followed by entry through cell membranes, and release of viral RNA into the cytoplasm. In the cytoplasm, viral RNA is translated into a polyprotein. Virally encoded proteinases (2Apro and 3Cpro) cleave the polyprotein into functional polypeptides (reviewed in (Semler et al., 1988). Poliovirus then prevents cellular cap-dependent translation shortly after infection. Viral RNA is translated by an cap-independent mechanism (see part b, poliovirus translation). The viral genome is copied into a negative-strand intermediate by the viral RNA polymerase and associated viral and cellular proteins. The negative-strand RNA in turn serves a template for synthesis of positive-strand RNAs. As the concentration of viral proteins increases, an increasing fraction of the positive-strand RNAs in the replication complex is packaged into virions. Viral assembly begins when the capsid protein precursor P1 is cleaved to form a protomer composed of VP0,VP3, and VP1. Five protomers assemble into a pentamer, 12 of which are required to form a 60-subunit protein shell enveloping the RNA genome. New viral particles are released by infection-mediated disintegration of the host cell. Poliovirus replicates rapidly, requiring 6-8 hours from adsorption to cell lysis (reviewed in (Racaniello, 1988; Rueckert, 1990; Wimmer et al., 1987). b) Early stages of poliovirus infection. The life cycle of poliovirus involves several early stages before translation and replication of the viral RNA begins. These include binding of the virion to the cell, alteration of the capsid, and the entrance of viral RNA into the cytoplasm (Holland and Hoyer, 1962). Attachment of virions to the surface of a susceptible cell occurs at temperatures ranging from 0 to 37°C (LonbergHolm and Philipson, 1974). Below 25°C, the bound virions may be recovered as intact infectious virus by a number of treatments, including exposure to 6M LiCl, 8M urea, low pH, and detergents (Lonberg-Holm and Philipson, 1974). When binding is carried out at 37°C, a substantial fraction of the bound virus is eluted from the cell in altered form. The altered particles sediment at 135S (versus 160S for native virion), have lost the internal 9 capsid protein VP4, have changes in antigenicity and protease sensitivity, and are no longer able to attach to susceptible cells (Hogle et al., 1990; Lonberg-Holm et al., 1975). The conformational transition leading to the altered particle can also be induced by extracts of membranes from susceptible cells (Guttman and Baltimore, 1977). Recently it was shown that the alteration of poliovirus may result from interaction with the soluble PVR at neutral pH in the absence of membranes (Kaplan et al., 1990). The altered particles contain infectious viral RNA. Neither the soluble PVR nor the membrane bound PVR can induce release of RNA (Guttman and Baltimore, 1977; Holland and Hoyer, 1962; Kaplan et al., 1990). The alteration of the virus capsid induced by PVR is thought to be the first stage in release of the genome from its durable protein shell (Fricks and Hogle, 1990; Holland and Hoyer, 1962; Kaplan et al., 1990). Consistent with this idea, altered particles similar to those eluted from cells have been found to be the dominant form of the virus inside cells early in infection (Everaert et al., 1989). The antiviral compounds (including arildone and the WIN compounds) bind virus and appear to exert their antiviral activities by preventing the formation of both the intracellular and the extracellular altered particles, suggesting that altered particle is a necessary intermediate in the cell entry process (Caliguiri et al., 1980; McSharry et al., 1979). Altered particles are more hydrophobic than native virions as a result of a conformational alteration and exposure of the amino terminus of VP1 (Fricks and Hogle, 1990). The hydrophobicity of altered particles might serve to embed the virus in the plasma membrane, providing a mechanism for passage of the viral genome into the cytoplasm (Fricks and Hogle, 1990). Subsequent stages leading to uncoating are not well understood. Since the particle-to-PFU ratio of poliovirus is high (Lonberg-Holm and Philipson, 1974), it is difficult to assess the role of the intermediates in the early stages of infection. The actual route of entry that leads to productive infection has not been established. It has been suggested that the mechanism of poliovirus penetration and uncoating resembles that of enveloped viruses such as Semliki Forest virus and influenza virus (Madshus et 10 al., 1984a; Madshus et al., 1984b; Zeichhardt et al., 1985). These viruses penetrate the cell by adsorptive endocytosis. Acidification of the endosomes induces a conformational change in a spike glycoprotein that leads to release of the viral genome into the cytoplasm. It was shown that poliovirus particles can be seen in coated pits and endosomes shortly after adsorption, suggesting that entry occurs by receptor-mediated endocytosis (Willingmann et al., 1989; Zeichhardt et al., 1985). Raising the pH after adsorption with the carboxylic ionophore monensin or with weak bases inhibits uncoating (Madshus et al., 1984a; Madshus et al., 1984b; Zeichhardt et al., 1985). However, the results of experiments with inhibitors of endosome acidification are conflicting. It was reported recently that elevation of endosome pH does not affect virus uncoating. (Gromeier and Wetz, 1990). The pathway of poliovirus entry and the mechanism of uncoating remain unsolved. It is not clear if PVR on the cell surface is sufficient to mediate virus entry. It is possible that receptor-mediated alteration may result in anchoring of virus to the plasma membrane via exposed hydrophobic domains and uncoating of the viral RNA may then occur at the plasma membrane or within the cytoplasm. Alternatively, endocytosis of the altered particle and additional modification may be required for uncoating of the genome. c) Poliovirus translation. Initiation of translation of eukaryotic mRNA is accomplished by a cap- and 5' end-dependent mechanism. A scanning model proposed that the 40S ribosomal subunit and initiation factors first bind at the 5'-end of the mRNA in a process that is facilitated by the presence of a cap structure (m 7GpppN, where N is any nucleotide). The 40S ribosomal subunit then scans the mRNA in a 5' to 3' direction until it encounters an appropriate initiator AUG, where the 60S ribosomal subunit joins (Kozak, 1989). In contrast to most eukaryotic mRNAs, poliovirus RNA does not have cap structure at its 5' terminus (Nomoto et al., 1976). Instead, the viral RNA has a small polypeptide, VPg, covalently linked to its 5' end (Nomoto et al., 1977). Vpg is removed 11 by a cellular factor in the cytoplasm after entry, leaving pUpU as the 5'-terminal structure of polysomal viral RNA (Ambros et al., 1978). In addition, the unusually long 5'-ncr of poliovirus RNA contains multiple cryptic upstream AUGs. The translation products originating from these ORFs have not been observed in vivo or in vitro and are not necessary for viral replication (Pelletier et al., 1988a). These characteristics render poliovirus RNA incompatible with the scanning model of translation initiation. In fact, in poliovirus infected cells cap-dependent translation is shut-off (see review in (Sonenberg, 1990), and addition of a cap structure to the 5'-ncr of poliovirus RNA inhibits its translation in mammalian cells (Hambidge and Sarnow, 1991). Translation initiation of poliovirus RNA occurs by a cap-independent mechanism. It was shown that an internal sequence (nucleotides 140 to 630) in the 5'-ncr of poliovirus RNA is required for cap-independent translation (Pelletier et al., 1988b; Pelletier and Sonenberg, 1988; Trono et al., 1988), and this sequence can also confer cap-independent translation to heterologous mRNAs (Pelletier et al., 1988b). Moreover, it was shown that eukaryotic ribosomes can bind internally to the 5'-ncr of poliovirus RNA (Pelletier and Sonenberg, 1988; Pelletier and Sonenberg, 1989). The internal cis-acting element in the 5'ncr of poliovirus RNA required for ribosome binding is termed the ribosome landing pad (RLP) (Pelletier and Sonenberg, 1988). An element similar to the RLP of poliovirus has also been identified in encephalomyocarditis virus (Jang et al., 1988), and referred to as an internal ribosome entry site (IRES), and in foot-and-mouth disease virus (Kuhn et al., 1990). Interestingly, internal binding of ribosomes can also occur in cellular mRNA (Sarnow, 1989). It is not clear how ribosomes bind to RLP and reach the poliovirus initiator AUG at position 745. Recently, it was shown that efficient function of RLP involves two appropriately spaced elements: an oligopyrimidine tract UUUCC at position 559, which is considered to be an analog of the prokaryotic Shine-Dalgarno sequence because of its complementarity to a segment at the 3' end of the 18S rRNA, and an AUG at position 12 586 (Pilipenko et al., 1992). It was proposed that 40S ribosome interaction with these two elements and a secondary structure domain from nucleotides 542-556 is required for efficient ribosome-template interaction. It is thought that the secondary and tertiary structural motifs in the RLP region are recognized by proteins that facilitate internal binding (Sonenberg, 1990). It was shown that cellular proteins bind multiple sites within the 5'-ncr of poliovirus RNA (Del Angel et al., 1989). Eukaryotic initiation factor eIF-2 is part of the complex formed with sequences from nucleotides 97-182 and 510-629. The polypeptide that is complexed with the 559-624 RNA fragment was identified as a cellular protein termed p52 (Meerovitch et al., 1989). A cellular protein p57, which binds a stem-loop structure near the 5' border of the IRES element of encephalomyocarditis virus, has also been identified (Jang and Wimmer, 1990). These cellular proteins may be involved in cap-independent ribosome binding (Sonenberg, 1991). Many features of the RPL and IRES elements in different picornaviruses appear to be quite different (Jang and Wimmer, 1990; Skinner et al., 1989). The differences are suggested to play a role in determining host range or tissue tropism: the elements may require different sets of factors to function efficiently in different cells (Jang and Wimmer, 1990). It was shown that poliovirus RNA is translated inefficiently in reticulocyte lysates and wheat-germ extracts (Pelletier et al., 1988c). Purified p52 stimulated preferentially the translation of poliovirus 5'-ncr containing mRNA in a reticulocyte lysate (Sonenberg, 1990). Moreover, a correlation between neurovirulence and translation efficiency in a neuroblastoma cell line, but not in Hela cells was demonstrated (La Monica and Racaniello, 1989). However, it is not clear if differences in translation factors between tissues account for differential translation. Answers to this question require further characterization of the structural requirements for the RPL element and the transacting factors that promote the process of ribosome internal binding. 3. Pathogenesis of poliomyelitis. 13 a) Clinical features. Poliovirus is the causative agent of paralytic poliomyelitis. Until the 1900s poliomyelitis was a disease primarily of infants. But with improved sanitation in many countries epidemics increased, the age distribution advanced, and the disease showed increasing severity as it appeared in young adults (Ginsberg, 1988). This paradoxical response to improved sanitation can be explained by three later findings. First, in areas of poor sanitation, essentially everyone was infected at a very early stage; but the paralytic form of infection was, and still is very rare in infancy. Second, early infection resulted in life-time immunity via neutralization antibodies. Third, infected adults who had no previous exposure to the virus had a much higher incidence of developing the paralytic disease than infants. When an individual is infected with poliovirus, one of the following responses may occur: inapparent infection without symptoms, mild (minor) illness, aseptic meningitis, or paralytic poliomyelitis (Melnick, 1990). The vast majority of individuals infected with poliovirus experience no symptoms. Others (4-8%) experience the minor illness, which is characterized by fever, malaise, drowsiness, headache, nausea, vomiting, constipation, or sore throat in various combinations. The patient recovers in a few days. Some infected individuals experienced aseptic meningitis, which includes the above syndrome, along with stiffness and pain in the back and neck. The disease lasts 2-10 days, and recovery is rapid and complete. Only about 1-2% of infected individuals develop paralytic poliomyelitis during an epidemic condition (Bodian and Horstmann, 1965). The major illness, paralysis, may occur following the minor illness or occur without an antecedent first phase. Cases are classified anatomically as spinal poliomyelitis, if paralysis is limited to muscles supplied by motor neurons in the cord, and bulbar poliomyelitis if the cranial nerve nuclei or medullary centers are involved. A combination of the two forms, bulbospinal poliomyelitis, beginning with paralysis of the legs and ascending to involve abdominal and thoracic muscles of respiration, arms, and finally medullary centers and cranial nerve nuclei occurs in the most severe cases, 14 particularly in adults. The predominating form is flaccid paralysis resulting from lower motor neuron damage (spinal poliomyelitis). The legs are affected more frequently than the arms. Bulbar poliomyelitis is often fatal due to respiratory or cardiac failure. Survivors of spinal poliomyelitis often recover with varying degrees of physical deficit and deformity (Bodian and Horstmann, 1965). b) Course of poliovirus infection. Poliovirus is an enteric virus. In a typical infection, virus is ingested and initially multiplies in the oropharyngeal and the intestinal mucosa (Bodian and Horstmann, 1965; Sabin, 1956). Virus has first been observed in throat secretions and in feces. It is not known, however, whether virus multiplies in epithelial or lymphoid cells of the alimentary tract. Significant pathological lesions were not found in the alimentary tract (Bodian and Horstmann, 1965; Sabin, 1956). Examination of tissues in the presymptomatic period in chimpanzees has revealed the presence of virus primarily in tonsillopharyngeal tissue and in the Peyer's patches of the ileum (Bodian and Horstmann, 1965). In human necropsy material, virus has been isolated with relative ease from the central nervous system (CNS), tonsillopharyngeal tissue, wall of the ileum, and lymph nodes (Sabin and Ward, 1941; Wenner and Rabe, 1951). It is not clear whether virus replicates in these lymphoid tissues or virus is absorbed into the regional lymph nodes after replication in superficial epithelial cells. In addition, polioviruses are found in peripheral ganglia of the alimentary tract prior to invasion of the CNS in monkeys fed poliovirus (Faber, 1956; Sabin, 1956). The role of peripheral ganglia in poliovirus pathogenesis is not clear, but transmission of virus along nerve fibers from peripheral ganglia might provide a route for entry into the CNS. Studies on reoviruses showed that viruses selectively bind to specialized microfold cells (M cells) overlying ileal Peyer's patches and are transcytosed into lymphoid tissue (Bass et al., 1988). Virus undergoes primary replication in the mononuclear cells of ileal Peyer's patches and in neurons of the adjacent myenteric plexus 15 (Morrison et al., 1991; Wolf et al., 1981). Virus spreads directly from the intestinal lumen to the CNS through vagal autonomic nerve fibers (Morrison et al., 1991). From the primary sites of propagation the virus drains into deep cervical and mesenteric lymph nodes. From the nodes the virus drains into blood, resulting in a transient viremia which disseminates virus to other susceptible tissues (Bodian and Horstmann, 1965). Virus has been readily detected in the blood of monkeys, chimpanzees, and humans in the early stages of infection (Bodian, 1954a; Bodian, 1954b; Bodian, 1955; Horstmann, 1952; Horstmann et al., 1954). It is believed that viral replication in extraneural tissues results in maintenance of viremia beyond the first stage, but the sites at which this replication occurs in humans is not known. In the experimentally infected chimpanzee, virus is found in very high concentration in the brown fat of suprasternal, upper axillary, and paravertebral regions (Bodian, 1955). In monkeys that were infected intramuscularly, large amounts of virus are found in lymph nodes, axillary fat, adrenals, as well as the inoculated muscle (Wenner and Kamitsuka, 1956; Wenner and Kamitsuka, 1957). There is also evidence that replication may occur in cells of the reticuloendothelial system and in the vascular endothelium in monkeys (Blinzinger et al., 1969; Kanamitsu et al., 1967). Maintenance of a persisting viremia is believed to be required for viral invasion of the CNS (Bodian and Horstmann, 1965). In most natural infections only transient viremia occurs. In 1-2% of infected individuals, the virus enters the CNS by incompletely understood routes (see part c, infection of the CNS). In the CNS, poliovirus replicates primarily in motor neurons within the anterior horn of the spinal cord, the brain stem, and the motor cortex, destroying these cells and producing the characteristic paralysis. Figure 3 illustrates the scheme of poliomyelitis pathogenesis in chimpanzees and humans. c) Infection of the CNS. The route by which poliovirus enters the CNS is not completely understood. Two possibilities have been suggested which are not 16 ingested virus intestinal mucosa oropharyngeal mucosa virus in throat virus in feces Peyer's patches tonsils deep cervical lymph nodes mesenteric lymph nodes BLOOD other susceptible extraneural tissues central nervous system regional nerve ganglia Figure 3. The pathogenesis of poliomyelitis in primates. (adapted from Ginsberg, 1988; see Bodian, 1955; Sabin, 1956). 17 mutually exclusive: the virus enters the CNS from blood across the blood-brain barrier (BBB), or it enters a peripheral nerve and is transmitted to the CNS (Blinzinger and Anzil, 1974; Bodian, 1959; Hurst, 1936; Melnick, 1985; Morrison and Fields, 1991; Sabin, 1957; Wyatt, 1990). The general opinion currently favors blood-borne entry into the CNS. First, the viremia preceding paralytic infection and appears necessary for virus entry into the CNS. the virulence of different strains correlates with the degree and duration of viremia. Second, the presence of specific antibodies in the blood effectively halts viral spreading in the host and prevents invasion of the CNS (Bodian and Horstmann, 1965; Melnick, 1985). On the other hand, there is ample evidence in support of the neural spread hypothesis. For example, in monkeys, inoculation of poliovirus into the sciatic nerve results in virus first in the lumbar cord, and soon afterwards in the leg area of the right motor cortex, indicating that poliovirus can spread along nerve fibers in both peripheral nerves and the CNS (Hurst, 1936). Following intramuscular injection of monkeys with the highly neurotropic poliovirus type 2 MV strain, localization of initial paralysis in the injected limb occurred at high frequency (Nathanson and Bodian, 1961). Freezing the sciatic nerve blocked spread of this virus from muscle to the CNS. In the Cutter incident, in which children received incompletely inactivated poliovaccine, a high frequency of initial paralysis was observed in the inoculated limb (Nathanson and Langmuir, 1963). Polioviruses are found in peripheral ganglia of the alimentary tract prior to invasion of the CNS in monkeys fed poliovirus (Faber, 1956; Sabin, 1956). In addition, trauma or exercise tends to localize paralysis to specific limbs, while tonsillectomy markedly increases the incidence of bulbar poliomyelitis (Bodian and Horstmann, 1965). The mechanisms of action of these localization factors are not known, but they may increase access of poliovirus to nerve termini in the injured area, or increase the permeability of blood vessels in the corresponding areas of the CNS. An important fact is that most natural infections result in viral multiplication in the alimentary tract without any sign of invasion of the CNS. Even in immunodeficient 18 humans the incidence of paralysis is low during severe epidemics (Sabin, 1956). The factors which determine whether an infected individual will experienced CNS infection, and if this infection will be severe are not completely understood, but they include the nature of the infecting strain, whether highly neurovirulent or not; the age of the patient; virus dose, and certain host factors, such as tonsillectomy, pregnancy, recent inoculations and physical exertion (Bodian and Horstmann, 1965). Strains of poliovirus may exhibit striking differences in neurovirulence. Neurovirulence in general refers to the ability of poliovirus to replicate in and destroy cells of the CNS. Measurement of this property is influenced significantly by the animal host employed (e.g. monkeys, chimpanzees, and mice) and the different routes of inoculation used (e.g. intracerebral, intraspinal, intraperitoneal, intramuscular, intravenous, and oral) (Racaniello, 1988). Experimental infection of primates, by intraspinal and intracerebral inoculation, has been the primary source of information about the relative neurovirulence of poliovirus strains and is referred to neurotropism. The neurotropism of many experimentally modified and naturally occurring strains of poliovirus varies quantitatively over an extremely wide range (Sabin, 1957). Poliovirus strains isolated from the CNS of fetal human cases are highly neurotropic (Sabin, 1956). In addition to high neurotropism, a neurovirulent strain of poliovirus may have to possess certain other properties, such as a high capacity for multiplication in extraneural tissues other than the alimentary tract, which may be required for invading the CNS. Consistent with this idea, the virulence of different strains correlated with the degree and duration of viremia (Bodian, 1954b). It is interesting that most naturally occurring type 2 and type 3 viruses are as highly neurotropic as the type 1 viruses, and yet almost all epidemics and about 85% of all paralytic cases are caused by the type 1 virus (Sabin, 1959). d) Pathology. The histopathology of experimental poliomyelitis in the primate is well known (Hurst, 1929). CNS lesions in poliomyelitis consist of neuronal changes and inflammation. Viral replication results in destruction of neurons and the inflammatory 19 process follows as a secondary response (Bodian and Horstmann, 1965). There is little evidence of viral replication in other cell types in the CNS. The characteristic pattern of distribution of poliomyelitis lesions has been shown experimentally to be due to two principal factors: (1) the inherent variation of susceptibility of nervous centers to infection, and (2) the restricted movement of virus along certain nerve fiber pathways (Bodian and Horstmann, 1965). The motor neurons of the anterior horns of the cervical and lumbar intumescences are the most sensitive to the virus, followed by neurons in motor nuclei of cranial nerves in the brain stem. In the spinal cord, although some fatal cases exhibit a striking restriction of alterations to the anterior gray columns, other cases may exhibit lesions of varying severity, but usually of spotty distribution, in the intermediate, the intermediolateral and the posterior grey columns (Bodian, 1959; Bodian and Horstmann, 1965). Lesions may extend to the sensory spinal ganglia. The lesions in the brain are primarily in brain stem, extending from the spinal cord to the anterior hypothalamus. Lesions in the forebrain are usually mild and restricted to the precentral gyrus (motor cortex) and the neighboring cortex, the thalamus and the globus pallidus. Severe lesions are also often found in the cerebellar vermis and the deep cerebellar nuclei (Bodian and Horstmann, 1965). e) Tissue tropism. Viral infections are often localized to specific cells and tissues within the host. This cell and tissue tropism results in distinct disease patterns for different viruses. Because all viruses initiate infection by binding to a specific receptor on the cell surface, the virus-receptor interaction has long been considered the first determinant of tissue tropism. For some viruses, such as human immunodeficiency virus type 1 (Maddon et al., 1986), and Epstein-Barr virus (Ahearn et al., 1988) expression of cell receptors appears to control the pattern of virus infection in the host. However, viral replication in the host may be blocked at steps in the viral life cycle other than receptor binding. For example, the receptor for influenza virus, sialic acid, is ubiquitous, yet viral replication is largely limited to the respiratory tract. Tropism of influenza virus is most 20 likely determined by availability of cellular proteinases required for cleavage of the hemagglutinin (Gotoh et al., 1990; Webster and Rott, 1987). In the primate host, poliovirus infection is characterized by a restricted tissue tropism despite the presence of virus in many organs during the viremic phase of infection (Bodian, 1955; Sabin, 1956). It has long been believed that the cellular receptor is a major determinant of its tissue tropism for the following reasons: 1) Assays for virus binding activity in tissue homogenates revealed a correlation between poliovirus binding and susceptibility to poliovirus infection (Holland, 1961). 2) Although poliovirus shows restricted tissue tropism, cells from almost any primate tissue are susceptible to poliovirus infection after cultivation in vitro (Enders et al., 1949; Holland and McLaren, 1961; Kaplan, 1955). It was shown that the acquired susceptibility correlates with appearance of the poliovirus receptor (Couderc et al., 1990; Holland, 1961; Holland and Hoyer, 1962). However, restriction of viral replication in many tissues may not be due solely to a lack of receptor in these tissues. For example, occasional binding of virus to tissues that are not sites of poliovirus replication has been reported (Holland, 1961; Kunin and Jordan, 1961). Studies on the binding of radiolabeled poliovirus to human regional CNS tissue homogenates showed that the binding activity within the CNS is much more widespread than the restricted distribution of pathologic lesions would lead one to predict (Brown et al., 1987). This observation suggested that factors other than receptor distribution must play a role in determining poliovirus neurotropism (Tyler, 1987a; Tyler, 1987b). An answer to the question whether restricted tissue tropism due to expression of the PVR might be obtained by determining the tissue distribution of poliovirus receptors. However, existing monoclonal antibodies directed against the poliovirus receptor (Minor et al., 1984; Nobis et al., 1985) are not suitable for immunofluorescent studies. A human cell receptor for poliovirus has recently been identified as a novel member of the immunoglobulin superfamily of proteins (Mendelsohn et al., 1989) (see section 4, 21 poliovirus receptor). PVR RNA and protein are expressed in a wide range of human tissues, including those that are not sites of poliovirus infection (Freistadt et al., 1990; Mendelsohn et al., 1989). However, little is known about PVR gene expression in individual cell types. It is possible that cells expressing PVR within nonsusceptible tissues are not accessible to poliovirus, or perhaps only a small fraction of cells in these tissues express PVR, and as a result virus growth is not detected. Alternatively, poliovirus tissue tropism may not be governed solely by expression of PVR, but may depend on tissue- or cell-specific modification of the PVR, additional factors required for PVR function, or perhaps factors required for subsequent stages in virus replication. f) Host range. Humans are the only known natural host for poliovirus. Chimpanzees and certain species of monkeys are susceptible to poliovirus infection by the intracerebral, intraspinal, and oral routes. However, the susceptibility of the neurons among the primates varies. For example, P1/Mahoney produces paralysis when 1 to 10 tissue culture infective doses (TCID) are inoculated intracerebrally in cynomolgus monkeys, whereas 106 to 108 TCID viruses are not paralytogenic after intracerebral inoculation of chimpanzees (Sabin et al., 1954). These and similar experiments with in vitro modified and naturally occurring strains of all three types of poliovirus have established a hierarchy of the sensitivity of primate neurons to infection with poliovirus (Sabin, 1956; Sabin, 1957). The lower spinal cord neurons of the monkey are most susceptible to infection, followed by the brain stem neurons of monkeys, and then the lower neurons of chimpanzees. Since the susceptibility of chimpanzees to oral poliovirus infection is much higher than that observed in human populations, it is expected that human neurons are either as susceptible as those of chimpanzees or less susceptible. It is of interest that this ranking of neuron susceptibility is the opposite of the susceptibility of the alimentary tract of the primates to poliovirus infection (Sabin, 1956). P2/Lansing and P2/MEF1, which are highly neurotropic in monkeys by intracerebral inoculation, do not infect the alimentary tract of monkeys, but infect chimpanzees by the 22 oral route. Attenuated virus strains that had limited infectivity in the alimentary tract of monkeys appear to multiply well in the alimentary tract in chimpanzees and even better in humans (Sabin, 1956). Certain species of monkeys are not susceptible to poliovirus infection by the oral route (Hashimoto et al., 1984). Most poliovirus strains are host restricted and cause paralysis in primates but not nonprimates, such as the P1/Mahoney strain (La Monica et al., 1986). However, by a process of adaptation involving serial passage of viruses in nonprimates, strains of poliovirus, including P2/Lansing, P1/LSb, and a variant of P3/Leon, were adapted in mice and other animal hosts (Armstrong, 1939b; Li and Schaeffer, 1953). Some strains of poliovirus are naturally virulent in mice (Moss and Racaniello, 1991). Mice inoculated intracerebrally with P2/Lansing develop a disease with clinical, histopathological, and agedependent features resembling human poliomyelitis (Jubelt et al., 1980a; Jubelt et al., 1980b). But in contrast to the human disease, the virus is not infectious by the oral route, and no extraneural sites of viral replication have been described in mice. The genetic basis for host restriction of poliovirus has been studied using two strains: P2/Lansing and P1/Mahoney. A host-range determinant of P2/Lansing maps to amino acids 95-104 of capsid protein VP1, which contributes substantially to neutralization antigenic site 1(N-Ag1) and comprises a loop connecting β-strands B and C (the BC loop) (Martin et al., 1988; Murray et al., 1988). Recently two other host range determinants located in the interior of the poliovirus capsid were identified (Moss and Racaniello, 1991). The BC loop sequence of the capsid protein VP1 may be involved in receptor binding (Murray et al., 1988). The internal host range determinants, as well as BC loop sequence, may also be involved in conformational transitions of the virion during entry (Moss and Racaniello, 1991). Poliovirus can infect many cell lines derived from primates. In contrast, nonprimate cell lines are not susceptible to poliovirus infection (Holland and McLaren, 1959). This host range restriction in cultured cells is determined at the level of the cell 23 receptor. Primate cells contain virus-binding activity while non-primate cells do not (Holland and McLaren, 1959). However, one replicative cycle occurs and infectious virus is released when a variety of non-primate cells are transfected with purified viral RNA (Holland et al., 1959a; Holland et al., 1959b). In addition, introduction of human genomic DNA containing the poliovirus receptor gene, or cloned human poliovirus receptor cDNA into mouse L cells results in susceptibility to multicycle viral infection (Mendelsohn et al., 1986; Mendelsohn et al., 1989). The basis for the restricted host range of poliovirus in animals is not known. While poliovirus does not infect nonprimate hosts, inoculation of viral RNA intracerebrally into rabbits, chicks, guinea pigs, and hamsters results in production of virus in the absence of disease (Holland et al., 1959a). On the basis of these results, it was concluded that the block to poliovirus replication in these animals is at the level of entry. However, in vivo susceptibility may be affected by hormonal, immunological, and physiological factors that determine whether virus reaches susceptible cells, the ability of virus to enter cells, and permissiveness for subsequent replicative steps. 4. Poliovirus receptors PVR is an integral membrane protein. As measured by saturation virus-binding studies, there are 3000 binding sites per Hela cell, but estimates of receptor density using monoclonal antibodies suggest a higher receptor density, close to one hundred thousand per cell (Crowell et al., 1983; Nobis et al., 1985). The three serotypes of poliovirus share one receptor which differs from those used by other members of the picornavirus family (Colonno, 1986). PVR is encoded by a gene present on human chromosome 19 (Miller et al. 1974). Several monoclonal antibodies have been isolated which inhibit the binding of poliovirus to cultured cells (Minor et al., 1984; Nobis et al., 1985; Shepley et al., 1988). Monoclonal antibody D171 competes with the three poliovirus serotypes for a common high affinity binding site on permissive cells but not on cells that are resistant to 24 poliovirus infection (Nobis et al., 1985). A second monoclonal antibody, AF3, blocks infection with poliovirus type 2 and to a lesser extent with poliovirus type 1, but has little effect on type 3 binding (Shepley et al., 1988). Monoclonal antibody AF3 recognizes a 100 kd protein in the membrane of poliovirus susceptible cell lines, certain neurons in the human CNS, and peripheral mononuclear cells. It has not been possible to purify the receptor protein from membrane preparations using assays that require binding of virus or antibody, probably because of the lability of the respective binding sites. The strategy for obtaining a molecular clone of the poliovirus receptor was to employ DNA transformation to transfer susceptibility to poliovirus infection from Hela cells to mouse L cells (Mendelsohn et al., 1986). The human receptor genomic DNA was identified in the mouse genome by virtue of its linkage to a human Alu repetitive sequence (Mendelsohn et al., 1989). The PVR genomic DNA was used to isolate two cDNA clones encoding polypeptides with molecular weight of 43 and 45 kd, that differ only in the length of the cytoplasmic tail (Mendelsohn et al., 1989). That these cDNA clones encode a receptor for poliovirus was proven in two way: transformation of resistant mouse cells with the cDNA clone leads to susceptibility to poliovirus infection and soluble protein encoded by the cDNA, when overexpressed in insect cells infected with a recombinant baculovirus, can bind and alter poliovirus (Kaplan et al., 1990). The PVR is a novel member of the immunoglobulin (Ig) superfamily of proteins (Mendelsohn et al., 1989). It is an integral membrane protein with three Ig-like domains. Many immunoglobulin superfamily members mediate functions involving cellular recognition and adhesion (reviewed in (Williams and Barclay, 1988). PVR RNA and protein are expressed in a wide range of human tissues, including those that are not sites of poliovirus infection (Freistadt et al., 1990; Mendelsohn et al., 1989). Alternatively spliced forms of PVR transcripts have been described that encode two soluble forms of PVR that lack the transmembrane domain (Koike et al., 1990). Transcripts encoding both 25 membrane-bound and soluble PVR were detected in all human organs tested (Koike et al., 1991). Several other human virus receptor cDNAs have been molecularly cloned and characterized, including the HIV-1 receptor CD4 (Dalgleish et al., 1984; Klatzman et al., 1984; Maddon et al., 1986), the major rhinovirus group receptor ICAM-1 (Greve et al., 1989; Staunton et al., 1989), and the Epstein-Barr virus receptor CR-2 (Moore et al., 1987). CD4 and ICAM-1 are also members of the immunoglobulin superfamily of proteins. Expression of CD4 is thought to be a major determinant of HIV-1 tissue tropism (Maddon et al., 1986). Human CD4 negative cells, which are resistant to infection by HIV-1, can be rendered susceptible to infection by transfection with cDNA clones encoding the CD4. Expression of ICAM-1 or CD4 in rodent cells, however, is not sufficient to render these cells susceptible to rhinovirus or HIV-1 infection, respectively, due to a block at the level of entry (Greve et al., 1989; Maddon et al., 1986). CR-2 has been shown to be a determinant of Epstein-Barr virus host range in vitro (Ahearn et al., 1988). 5. Poliovirus attenuation. a) Isolation of attenuated virus strains. As discussed in section 3, pathogenesis of poliomyelitis, poliovirus strains display a wide range of neurovirulence. Neurovirulence for primates is a function, not only of the virus , but of the varying susceptibilitis of different hosts. Attenuated viruses may be naturally occurring or isolated by passage of the virus in a different animal host or in various cultured cells, or by a combination of both (Paul, 1971). The first attenuated poliovirus strain was isolated by Theiler (Theiler, 1941), who reported that after 50 rapid intracerebral passages in mice, the Lansing strain no longer produced signs of poliomyelitis in rhesus monkeys inoculated intracerebrally. Enders and his colleagues showed that cultivation of the P1/Brunhilde strain in human nonneural tissue resulted in a marked reduction of its neurovirulence in monkeys. Subsequently Sabin showed that serial propagation of wildtype poliovirus strains 26 (P1/Mahoney, P2/YSK, and P3/Leon) in cynomolgus kidney cultures had no effect on virulence for cynomolgus monkeys when single or small numbers of virus were used to initiate the cultures, while rapid passage with large inocula led to the appearance of avirulent variants, which ultimately were separated from the virulent variants by the terminal dilution technique (Sabin et al., 1954). Polioviruses have different reproductive capacities at various temperatures. The temperature at which viruses multiply plays an important role for isolating vaccine strains (Sabin, 1961). Highly attenuated poliovirus strains can be isolated by cold passage (23°C or 25°C) of virulent strains in monkey primary kidney cell culture (Carp et al., 1963; Sabin, 1960; Sabin, 1961). The monkey neurovirulence of the attenuated virus isolated at 25°C is lower than that of virus isolated at higher temperature (e.g. 36°C) (Sabin, 1960; Sabin, 1961). However, the attenuated virus isolated at 25°C either multiplies poorly in the human alimentary tract or multiplies and rapid reverts to virulent strains. The Sabin live oral polio vaccine strains were produced by controlled passage of viruses in animals and cultured cells until variants unable to cause paralysis in primates were obtained (reviewed in (Sabin and Boulger, 1973). The Sabin vaccine strains cause no paralysis when inoculated into the CNS of animals, yet after oral administration in humans replicate sufficiently in the alimentary tract to induce a protective immune response. The P1/Sabin strain (LSc, 2ab) and P3/Sabin strain (Leon 12a1b) were derived from neurovirulent strains, P1/Mahoney and P3/Leon respectively. The P2/Sabin strain (P712, Ch, 2ab), however, was derived from P712, an isolate of low intraspinal cynomlgous neurovirulence obtained from the faeces of healthy children (Sabin and Boulger, 1973). The three Sabin vaccine strains have been studied extensively to identify properties that correlate with their reduced neurovirulence (reviewed in (Racaniello, 1988). For example, the poliovirus vaccine strains were found to be temperature- sensitive mutants. The replication of the Sabin strains is greatly reduced at high 27 temperatures (39.5 or 40.1°C) as compared to wild-type viruses. In addition, vaccine strains in general have low stability at high temperature. The production and distribution of vaccine strains requires maintenance at low temperature (WHO, Programme for Vaccine Development and Transdisease Vaccinology, Activities & Prospects, 1989). The occurrence of epidemic poliomyelitis has been greatly reduced by extensive vaccination with attenuated strains and inactivated virus preparations. However, poliomyelitis has not been totally eradicated. The continuing occurrence of poliomyelitis, estimated to be at least 400,000 cases world wide annually by the WHO (Melnick, 1983), and a low level of vaccine-associated poliomyelitis (Assaad and Cockburn, 1982; Cann et al., 1984) makes continued vaccination and construction of completely safe, alternative vaccine strains essential. b) Determinants of attenuation. The live, attenuated poliovirus vaccine strains developed by Sabin have been extremely effective in controlling poliomyelitis. Since the Sabin strains were isolated, it has been of great interest to determine the molecular and functional basis for their attenuation phenotypes (reviewed in (Almond, 1987; Racaniello, 1988). This information has provided insight into the biology of poliovirus, enabled better understanding of vaccine-associated disease, and suggests ways to improve the existing vaccines. To identify determinants of attenuation in each of the three vaccine strains, genomic recombinants have been constructed between the attenuated viruses and closely related neurovirulent strains, and the ability of these recombinants to cause paralysis has been assayed in primates. Because the poliovirus genome is an RNA molecule, the recombinants have been constructed using cloned cDNAs from which virus can be derived by transfection (Racaniello and Baltimore, 1981a). Using this approach, two attenuation determinants have been identified in the type 3 vaccine strain, P3/Sabin: a uridine (U) residue at nucleotide 472 in the 5'-ncr (U-472), and a phenylalanine (phe) at amino acid 91 (phe-91) of capsid protein VP3, which is also responsible for the temperature sensitive 28 (ts) phenotype of the virus (Minor et al., 1989; Westrop et al., 1987). Similar studies have revealed that attenuation determinants occur in at least three regions of the type 1 vaccine strain, P1/Sabin, and include a guanine (G) residue at position 480 in the 5'-ncr (G-480) (Kohara et al., 1985; Nomoto et al., 1987), M. Bouchard and V. R. Racaniello, unpublished observations). An approach to identifying attenuation determinants in the type 2 vaccine strain, P2/Sabin, has involved construction of recombinants with P2/Lansing. This type 2 strain is able to cause poliomyelitis in mice, allowing the recombinants to be tested for virulence in mice instead of in primates. Although it was established that the determinants of poliovirus neurovirulence in mice and in human are not absolutely linked, it was found that some attenuation determinants of poliovirus identified in monkeys also result in attenuation in mice (La Monica et al., 1987a). For these studies, a cDNA clone of P2/P712, a viral strain that is nearly identical in nucleotide sequence to P2/Sabin, has been used (Moss et al., 1989; Pollard et al., 1989). Two regions that attenuate P2/P712 were mapped using this strategy: the 5'-ncr and a central region encoding capsid protein VP1, 2Apro, 2B and part of 2C (Moss et al., 1989). The rest of the P2/P712 genome does not contain determinants of attenuation. Recently, the nucleotide sequence of a neurovirulent type 2 strain, P2/117, which was isolated from a vaccine-associated case of poliomyelitis was determined (Pollard et al., 1989). Comparisons between attenuated type 2 strains and P2/117 may suggest candidate determinants of attenuation. 29 Chapter II. Materials and Methods 30 Cells, virus and antibody Cell lines. Hela S3 cells were grown in suspension cultures in Joklik minimal essential medium containing 5% horse serum (GIBCO). For growth in monolayers, Hela cells were plated in Dulbecco's modified Eagle's medium (DMEM) (GIBCO) containing 10% horse serum as described (Racaniello and Meriam, 1986). Mouse Ltk- fibroblast cells were maintained in DMEM containing 10% fetal bovine serum, 100 units of penicillin per ml, 100 µg of streptomycin per ml, 2.5 µg of amphotericin per ml. DNA transformants were grown in the same medium with 300 µg of G418 (GIBCO) per ml. Mouse primary kidney and muscle culture. Kidneys were dissected from 3 week old mice or suckling mice. The skeletal muscles were dissected from the legs of the suckling mice (2 to 5 days old). Tissues were washed three times with DMEM and minced with a scalpel blade. The minced tissues were then placed in sterile filtered saline that was free of Ca and Mg and contained 137 mM NaCl, 5mM KCl, 0.7 mM Na2HPO4, 25 mM HEPES, 5 mg/ml collagenase A (Sigma), and 10 µg/ml DNase I (Sigma). After 1 hour of enzyme treatment at 37°C, the suspension was centrifuged at room temperature at 1000 x g for 10 min. The cell pellet was resuspended in DMEM and triturated several times to dissociate the cells. The suspension was removed to a new tube leaving undigested tissue behind. The suspension was pelleted and resuspended in DMEM containing 10% fetal bovine serum, 100 units of penicillin per ml, 100 µg of streptomycin per ml and 10 µg/ml gentamicin for culture in monolayers, or cultured in suspension as described (Racaniello and Meriam, 1986). For culture in monolayer, the cells were placed in 25 cm2 tissue culture flasks pretreated with 0.1% gelatin at room temperature for 2 hours. The cells were incubated at 37°C in an atmosphere of 6% CO2. Viruses. Poliovirus P1/Mahoney (Racaniello and Baltimore, 1981b) and P1/Sabin (Burke, 1988) were derived from the infectious cDNA clones. Poliovirus type 2 viruses P2/Lansing (La Monica et al., 1986), P2/P712 (Moss et al., 1989), and P2/117 (Pollard et al., 1989), or variants of these generated as part of this study were derived from cloned 31 genomic cDNAs. In vitro transcription of cDNAs and transfection of RNA into HeLa cells to derive viruses were performed as described (Moss et al., 1989). Viruses were plaque purified twice. P2/MEF-1, a mouse adapted poliovirus, was provided by A. Sabin. P2/Rom, isolated from the gut of a fatal case in Rumania (Crainic et al., 1984), was provided by F. Horaud. Poliovirus type 3 viruses P3/Leon (KP3), P3/Sabin (Lederle Sab 3, polio monopool 3-508), and the P3/Leon and P3/Sabin recombinant viruses S5'/L and SV3/L (Westrop et al., 1989) were provided by A. Sabin, Lederle-Praxis Biologicals, and P. Minor respectively. Viral stocks were prepared by infecting Hela cells at 32°C (for P1/Sabin, P3/Sabin, S5'/L, and SV3/L) or 37°C for the rest of the viruses. Viral titres were determined by plaque assay on Hela cell monolayers at the temperature described above. Antibody. Mouse monoclonal antibody D171, directed against the human poliovirus receptor, was a generous gift of P. Nobis (Nobis et al., 1985). Virus growth and assay. Monolayers or suspension cultures of cells were infected with poliovirus at a multiplicity-of-infection (MOI) indicated. After 60 min adsorption, the cells were washed three times to remove unattached viruses and the medium was replaced. Aliquots of the supernatant were removed at different times after infection and virus titres were determined by plaque assay on Hela cell monolayers. Tenfold serial dilutions of viruses, prepared in phosphate-buffered saline (PBS) plus 0.2% horse serum, were used to inoculate 6-cm dishes of Hela cells, and adsorbed at 37°C. The cells were then covered with 5 ml of 0.9% Bacto-Agar (Difco Laboratories) in DMEM plus 5% horse serum. After incubation at 37°C in 5% CO2 for 2-3 days, plates were stained with crystal violet as described (Racaniello and Meriam, 1986). Growth at low and high temperatures was measured by plaque assay at 32°C and 39.5°C as described (Racaniello and Meriam, 1986). To prepare high titer viral stocks for inoculation of mice, Hela cell monolayers in 15-cm dishes were infected at a multiplicity of infection of 10 plaque-forming units 32 (PFU) per cell, and then incubated in medium at 37°C for 5-7 h. Infected cells were collected by centrifugation, resuspended in 1 ml of PBS, subjected to three cycles of freeze-thawing, clarified and stored at -70°C. RNA and DNA isolation Total RNA was isolated from cultured cells and tissues by homogenization in 4 M guanidine isothiocyanate followed by centrifugation through a cushion of CsCl (Chirgwin et al., 1979). High molecular weight Hela cell genomic DNA was isolated as previously described (Gross-Bellard, 1973). Cultured cells were pelleted by low speed centrifugation (1000 x g) for 10 min at room temperature. Following centrifugation, cell pellets were washed once in isotonic buffer (10mM Tris-HCl, pH8.0/ 140mM NaCl) and resuspended in digestion buffer (50mM Tris-HCl, pH8.0, 25mM EDTA, pH7.5, 100mM NaCl, 0.5% SDS and 0.2mg/ml proteinase K) to a concentration of about 1 X 107 cells per ml. The mixture was incubated overnight at 50°C. Following digestion with proteinase K, the mixture was gently extracted with an equal volume of phenol equilibrated with 500mM Tris-HCl, pH8.0, 10mM EDTA, 10mM NaCl. The aqueous and organic phases were separated by centrifugation at 3000 x g for 10 min at room temperature. The viscous aqueous phase was poured into a new tube and extracted again. Following extraction, the aqueous phase was dialysed twice against 50mM Tris-HCl, pH8.0, 10mM EDTA, and 100mM NaCl solution at 4°C. Following dialysis, the DNA was treated with 50µg/ml ribonuclease A (Boehringer Mannheim) at 37°C for 2 hours, followed by proteinase K (0.1mg/ml) and SDS (0.5%) treatment (3 hours) and phenol extraction as described above. The DNA solution was dialyzed twice against STE (10mMTris-HCl, pH8.0, 1mMEDTA, 10mM NaCl) at 4°C. DNA prepared with this method was greater than 150 kb in size as judged by electrophoresis in 0.3% agarose gels, using Herpes simplex virus DNA as a molecular weight marker. Construction of Hela cell genomic cosmid library 33 High molecular weight genomic DNA prepared from Hela cells was partially digested with Mbo I and fractionated by electrophoresis in a 0.4% low melting gel. DNA 35-48 kb in length was isolated from the gel by phenol, phenol/chloroform and chloroform extraction, ligated to BamH I cleaved, dephosphorylated pWE15 (Stratagene), and packaged with Gigapack Gold extract (Stratagene). This vector was chosen because it contains Not I restriction sites flanking the insertion site, which facilitates isolation of the insert DNA (Wahl, 1987); this is necessary because prokaryotic vector sequences may interfere with eukaryotic gene expression in transgenic mice (Palmiter, 1986). Packaged cosmids were transduced into E. coli NM554 (Stratagene). The library was plated on LB agar containing 50 µg/ml ampicillin and screened in duplicate with a 0.97 kb EcoR I fragment derived from PVR cDNA clone HeLa 1.5 (Mendelsohn et al., 1989), which contains most of the PVR coding sequences, using the hybridization procedure described for Southern blots below. DNA transformation Mouse Ltk- fibroblasts were seeded in plastic cell culture plates one day before use (2 x 106 cells per 6-cm plate for transient assay and 7.5 X 105 cells per 10-cm plate for stable transformants). Each plate was treated with either 0.5 ml (for transient assay) or 1 ml (for stable transformants) of a DNA-calcium phosphate coprecipitate containing 10 µg of either pWE15 vector or PRG cosmid constructs and 10 µg herring sperm DNA. After 18 hours of incubation at 37°C, the medium was replaced and incubation was continued for 24 hours. For transient receptor assays, cells were infected with poliovirus; for isolation of stable transformants, cells were grown in G418 containing medium for 2 weeks, and then G418 resistant colonies were subcultured. Microinjection and production of transgenic mice Transgenic mice were generated using previously described procedures (Hogan et al., 1986). Founders were derived by microinjection of DNA into (C57BL6/J X CBA/J) F2 zygotes. The founding transgenic mice and their initial offspring were identified by 34 Southern blot analysis of tail DNA as described (Hogan et al., 1986). Copy number of the transgene was estimated by comparison with known amounts of cosmid PRG1 and PRG3 DNAs. Transgenic lines TgPVR1-7, TgPVR1-17, and TgPVR3-9 contained 30, 10 and 30 copies of the transgene, respectively. Transgenic founders derived from microinjection with cosmids PRG-1 or PRG-3 were named TgPVR1 or TgPVR3, respectively, followed by a number assigned to different founders. Most transgenic offspring were identified by polymerase chain reaction (PCR) under the conditions recommended by Perkin Elmer Cetus, using two primers derived from PVR cDNA 20A 3' noncoding sequences: 20A-1W: 5'-AGAAGGACTCACTAGACTCAGG-3' 20A-1C: 5'-CTCACCACTGTACTCTAGTCTG-3' The PCR reaction was carried out for 30 cycles at 94°C for 1 min, 54°C for 2 min, and 72°C for 3 min. Lines were expanded by backcrossing against (C57BL6/J X CBA/J) F1 mice. All transgenic mice were housed within an isolator unit to prevent escape and possible spread of the human PVR gene to the wild mouse population. Poliovirus receptor binding assay Mouse brain, liver, kidney and lung were removed and washed once in PBS; intestine (about 5 cm) was removed and cut open, the contents were squeezed out, and the tissue was washed three times in PBS. These tissues were homogenized in 3 ml of ice cold PBS in a Polytron (Brinkmann). 5 X 106 PFU of poliovirus was incubated at 25°C for 2 hours in 1 ml of a 5% (w/v) tissue homogenate. After incubation for 2 hours, the virus-homogenate mixture was diluted in PBS containing 0.2% horse serum and virus titer was determined by plaque assay in HeLa cells. After 45 min adsorption at 37°C the cell monolayers were washed twice to remove tissue debris and unattached virus, and were overlaid with semisolid medium and incubated at 37°C for plaque development. Control 35 virus titer was determined after incubation of virus in PBS with 0.2% horse serum at 25°C for 2 hours. For virus binding assay of entire cells, dispersed kidney cells, from approximately 50 mg of fresh kidney, were mixed with 1 X 106 pfu of P1/Mahoney, and incubated at 37°C for 2 hr. Cells were removed by centrifugation, and the remaining infectious virus in the supernatant was determined by plaque assay. Percent binding was calculated by subtracting the amount of virus remaining in the supernatant from the input virus, determined after mock-incubation in phosphate-buffered saline. Neurovirulence assay. Viruses were tested for neurovirulence in normal Swiss-Webster mice and in TgPVR1-17 transgenic mice. Groups of eight 3-4 week-old mice, four male and four female, were inoculated intracerebrally with 50 µl of virus. Ten-fold dilutions of each virus were made in PBS plus 0.2% horse serum, and groups of mice each received one dilution, such that each virus was inoculated over a range approximately from 104 to 109 pfu. Mice were observed daily for 21 days for paralysis or death. Paralyzed mice were sacrificed and scored as dead. The amount of the virus which caused paralysis or death in 50% of mice, LD50, was calculated by the method of Reed and Muench (Reed and Muench, 1938). All values represent the average of two independent determinations, which did not vary by more than 0.5 log10 from the reported values. For determining LD50 by other routes of inoculation, mice were inoculated intracerebrally, intramuscularly, intraperitoneally and intravascularly with 50 µl of virus. For oral inoculation, 100 µl of virus was administered through a 21G animal feeding needle inserted into the stomach. When paralysis resulted, virus was isolated from the spinal cord of at least one infected mouse, and the virus identity was confirmed by nucleotide sequencing of genomic RNA. By this analysis it was found that all viruses isolated from the spinal cords of paralyzed mice resembled the inoculated virus (data not shown). 36 Assay for viral replication in mouse brain and spinal cord Transgenic and non-transgenic mice were inoculated intracerebrally with 1 X 105 PFU of P1/Mahoney. Three transgenic and two nontransgenic mice were sacrificed each day, and the brains and spinal cords were removed and homogenized in either 2 ml PBS (for brain) or 1 ml PBS (for spinal cord) using a Dounce tissue grinder. Viral titres in brain and spinal cord homogenates were determined by plaque assay on Hela cell monolayers. Animal inoculation and tissue sampling. Mice were inoculated intramuscularly with 50 µl of virus. Three mice were sacrificed each day and tissue samples including brain, superior and inferior spinal cord were removed and homogenized in 1 ml PBS using a Dounce tissue grinder. The cervical and upper thoracic cord segments were included in the superior spinal cord block, and the lower thoracic and lumbosacral cord segments were included in the inferior spinal cord block. Viral titres in tissue homogenates were determined by plaque assay on Hela cell monolayers. For examining virus in other tissues, mice were inoculated intramuscularly, intraperitoneally, or intravascularly with 50 µl of virus. Three mice were sacrificed each day and the hamstring muscle, kidney, heart, lung, thymus, or intestine were removed, homogenized, and the virus titer was determined by plaque assay. For oral inoculation of suckling mice, 1-2 day old mouse pups were anesthetized with Metofane (methoxyflurane) (Pitman-Moore, Inc.). The suckling mice were then inoculated perorally with poliovirus in 30 µl through a thread 6 in. of intramedic PE tubing #7400 (Becton Dickinson) passed into the esophagus as described (Morrison et al., 1991). Sciatic nerve transection. Transgenic mice were anesthetized by intraperitoneal injection with 15-17 µl of 2.5% Avertin in PBS per gram of body weight (Hogan et al., 1986). 37 Sciatic nerve transection was performed as described (Tyler et al., 1986). A small incision was made above the gastrocnemius muscle, and the muscle layers were cut to expose the sciatic nerve. Approximately 1mm of nerve was removed, and the incision was closed with autoclips (Hogan et al., 1986). After surgery, mice were checked for paresis. For sham operations, the same procedure was carried out except that the nerve was not cut. Virus was inoculated intramuscularly one day after nerve transection or sham operation. Neuropathology A 21 day neurovirulence test, similar to the intrathalamic safety test conducted in primates for the evaluation of oral poliovaccine (Code of Federal Regulations, 1988), was performed on a group of 5 1/2 week old TgPVR1-17 transgenic animals as well as age- and strain-matched nontransgenic control mice. Similar groups of transgenic and nontransgenic animals were also tested with the P1/Sabin vaccine strain. A maintenance media-inoculated age-matched control group using CD-1 conventional mice was also included. CD-1 animals were used because of insufficient numbers of transgenic mice. Each mouse was anesthetized with an intramuscular injection of ketamine/xylazine and injected intracerebrally into the left cerebral hemisphere using a 27 gauge X 3/16 inch hypodermic needle, with 0.05 ml of each test virus (containing 5.4 X 105 pfu) diluted in modified Eagles lactal maintenance medium. All mice were observed daily for 21 days for signs of illness. When CNS signs such as paralysis developed, the animals were sacrificed and necropsy performed. The brain and spinal cord were removed and fixed by immersion in 10% neutral buffered formalin. Any clinically normal animals remaining on day 21 were also sacrificed and their brains and spinal cords fixed in the same way. Tissues were processed by standard techniques and embedded in paraffin. Coronal sections were cut at 5-8 microns and stained with both hematoxylin and eosin (H&E) and Einarson's method for gallocyanin (Luna, 1968). Brain sections examined included cerebral cortex and hippocampus at the level of mid-thalamus and hypothalamus, midbrain at the level of red nucleus, and cerebellum and brain stem at the level of deep 38 cerebellar nuclei and the vestibular complex. Also examined were sections of both cervical and lumbar spinal cord. Triplicate sets of three sections each of both the cervical and lumbar intumescences were studied. The triplicate sets of sections were cut 20 microns apart. The midbrain and lower brain stem sites along with the spinal cord ventral horns are the commonly involved poliovirus target sites in human and nonhuman primate infections (Hurst, 1929). All sections were evaluated qualitatively. Hybridization probe synthesis. A 1263 bp SmaI-XhoI fragment of PVR cDNA H20B, which contains most of the PVR coding sequences, was subcloned into plasmid vector pBluescript KS (-) and designated pBS5R. For in situ hybridization, anti-sense probes labeled with [35S]UTP to a specific activity of 2 to 4 x 106 cpm/ng were generated using BamHI-digested pBS5R as template for transcription by T3 RNA polymerase. Alternatively, the control sense strand was synthesized with XhoI-digested pBS5R template and T7 polymerase. Similarly the 1452-bp BglII-PvuII fragment of P1/Mahoney cDNA, containing poliovirus polymerase coding sequences, was subcloned into plasmid vector pBluescript KS(-) and designated pBSMP. Anti-sense probe was generated by using SacI-digested pBSMP and T3 polymerase and sense probe was synthesized from ClaI-digested pBSMP by T7 polymerase. The RNA was hydrolyzed at pH 10.2 at 60°C as described (Jaffe et al., 1990) to yield probes approximately 100 bp in length. Probe length before and after hydrolysis was assessed on formaldehyde-agarose gels. In situ hybridization. Mice were sacrificed and the brain, spinal cord, thymus, lung, intestine, spleen, kidney, adrenal gland and hindlimb muscle or embryo were removed and fixed by overnight immersion in 4% paraformaldehyde at 4°C. The paraformaldehyde was replaced with phosphate-buffered saline, and tissues were washed, dehydrated and embedded in paraffin wax as described (Jaffe et al., 1990). Normal human tissues were provided by the Department of Pathology of Columbia University. Embedded tissues 39 were sectioned at 6 microns and collected on Tespa-treated slides. Hybridization, exposure to photoemulsion (3-6 days for PVR probes and 1 day for poliovirus probes), counterstaining, and microphotography were as described (Jaffe et al., 1990). Positive hybridization was determined by comparison with background levels, defined by using sense probes on transgenic mouse sections, and antisense probes on nontransgenic mouse sections. To determine the origin of the cells expressing PVR RNA in human placenta, the sections of placenta tissue, which are adjacent to that used for in situ hybridization with radiolabeled PVR RNA probes, were examined by immunochemical analysis using monoclonal antibody against cytokeratin and human placental lactogen (hPL). The immunochemistry study was carried out in Immunocytochemistry Lab of Met Path Inc. PCR amplification of cDNA. Preparation of total cell or tissue RNA, and oligo(dT)-primed cDNA synthesis, was as described previously (Mendelsohn et al., 1989). Quantitative PCR amplification of cDNA was carried out as described (Zack et al., 1990) using PVR-specific primers flanking the transmembrane domain (nt 1170-1191 and 1422-1443; numbered as in ref. (Mendelsohn et al., 1989). Construction of viral recombinants. Plasmid DNAs were grown in E. coli DH5α and purified by CsCl centrifugation (Ausubel et al., 1987). DNAs were cleaved with restriction endonucleases under conditions recommended by the manufacturers (Boehringer-Mannheim Biochemicals and New England Biolabs). Restriction fragments were purified by electrophoresis in low gelling-temperature agarose gels (Ausubel et al., 1987). Ligations were performed according to the instructions of the manufacturer of T4 DNA ligase (Boehriger-Mannheim Biochemicals). The constitution of each recombinant is diagrammed in Figure VII-1, 2. The construction of SRL, SLL, and LP1 has been reported (Moss et al., 1989). SPL, SVL, and 40 117LP were generated as part of this study. SVL and SPL are the result of a reciprocal exchange between P2/Lansing and SRL at Pst I sites introduced into their corresponding cDNAs by site-directed mutagenesis at nucleotides 3413 and 3416 respectively. 117LP was generated by replacing a Kpn I-Nar I fragment, nucleotides 66-751, of P2/Lansing cDNA with a corresponding fragment from P2/117 cDNA (Pollard et al., 1989). Mutagenesis of viral recombinants. Oligonucleotide-directed mutagenesis was performed on DNAs subcloned in M13 grown in E. coli CJ236 (Ausubel et al., 1987). Pst I restriction sites were introduced into the P2/Lansing sequence at nucleotide 3413, and into the P2/P712 sequence of recombinant SRL at nucleotide 3416. The antisense oligonucleotide used for site-directed mutagenesis in both cases was 5'-TAGCCTGCAGTGTACACAG-3'. The mutations caused by this oligonucleotide occur at analogous positions in the open reading frames of these strains but do not result in amino acid coding changes. Using the same methods, the ATT codon at nucleotide 2905 of the P2/P712 sequence in the SVL recombinant was changed to GTT to generate SVL-val, and to ACC to generate SVL-thr. The T residue at position 437 of the P2/P712 sequence in the cDNA of recombinant SLL was changed to C to generate the cDNA of SLL437. The A residue at position 481 in the SLL cDNA was changed to G to generate the cDNA of SLL481. The mutant/recombinant LP481 was derived from SLL481, has a G at position 481, and otherwise resembles recombinant LP1. Nucleotide sequencing. Sequencing of recombinant and mutant cDNAs to confirm their identity was performed using Sequenase™ according to the manufacturer's directions (U.S. Biochemical). The identity of viruses was confirmed by sequencing of genomic RNA at recombination junctions and mutation sites. Isolation of genomic RNA and chain- termination sequencing using oligonucleotide primers was performed as described (La Monica et al., 1987b). 41 Chapter III. Transgenic mice expressing a human poliovirus receptor: A new model for poliomyelitis With Frank Costantini, Edward J. Gorgacz, and James J. Lee 42 Isolation of a human poliovirus receptor gene To generate transgenic mice expressing PVR in a pattern as close as possible to that in humans, the human PVR gene and its promoter were isolated. A library of Hela cell genomic DNA was constructed using cosmid vector pWE15 and screened with a PVR cDNA probe containing most of the PVR coding region. Of six positive clones isolated from 1.1 million primary clones screened, two clones, PRG1 and PRG3, were shown by Southern blot analysis to contain sequences that hybridize with PVR cDNA clones encoding functional poliovirus receptors (Figure 4A). PRG1 and PRG3 contain DNA inserts of approximately 37 kb, with 26 kb of overlapping sequences. PRG1 contains about 11 kb more 3' sequence than PRG3, while the latter has approximately 11 kb of additional 5' sequence. To determine whether the cosmid clones encoded functional PVR, the DNAs were transiently expressed in mouse L cells. Forty eight hours after DNA transformation the cells were infected with poliovirus, and the cell culture medium was assayed for the presence of infectious virus twenty four hours later. When L cells transformed with either PRG1 or PRG3 were infected with poliovirus, large numbers of viral progeny were produced (Table 1). In contrast, L cells that had been transformed with pWE15 were not susceptible to poliovirus infection. In addition, stable L cell transformants containing PRG1 or PRG3 were selected in G418-containing media. About 75% of G418 resistant transformants expressed the PVR on the cell surface, as shown by a rosette assay with anti-PVR monoclonal antibody D171, and efficiently supported multicycle poliovirus infection (data not shown). These results demonstrate that cosmid clones PRG1 and PRG3 contain a functional PVR gene. Although both cosmids contain the SV40 early promoter, the DNA insert in PRG1 is in the opposite 43 orientation. The Table 1. Yields of poliovirus after infection of mouse cells transformed with poliovirus receptor cosmid clones. Ltk- cells were transformed with the indicated DNAs pfu/ml transforming DNA 0 hr 24 hr pWE15 pSVL-H20A PRG1 PRG1 PRG3 PRG3 120 80 90 80 30 110 180 4.3 X 104 2.4 X 104 4.3 X 104 7.1 X 103 4.6 X 103 and 48 hr later were infected with P1/Mahoney. pfu/ml in cell culture medium was determined at 0 and 24 hr after infection. 44 high frequency expression of the PVR in cosmid transformants suggests that the promoter of the PVR gene is present and functional. Generation of transgenic mice carrying a human poliovirus receptor gene The DNA inserts of PRG1 and PRG3 were purified and used to produce transgenic mice. Transgenic founder mice carrying the human PVR gene were identified by Southern blot analysis of tail DNA. Probe A, consisting of a 0.97 kb EcoR I fragment from PVR cDNA HeLa1.5 (Mendelsohn et al., 1989), hybridized to 10.4, 3.0 and 1.1 kb BamH I DNA fragments in both PRG1 and PRG3 transgenic mice (Figure 4B). Probe B, a 0.5 kb EcoR I-Sma I probe derived from the 5' end of PVR cDNA H20B (Mendelsohn et al., 1989) hybridized to an internal 6 kb BamH I fragment in cosmid PRG3 (Figure 4A) and in transgenic mice carrying this DNA (Figure 4B). The same probe hybridized with a 17.5 kb BamH I fragment in cosmid PRG1 (Figure 4B, lane 1), which consists of a 6 kb 5' end fragment and a 2.7 kb 3' fragment of PVR DNA linked to the 8.8 kb pWE15 vector. In transgenic mice carrying PRG1, a new 8.7 kb BamH I fragment (the 5' end 6 kb fragment plus the 3' end 2.7 kb fragment of PRG1) hybridized with probe B, indicating that multiple copies of intact PRG1 had integrated into the mouse genome in a head-totail array. Transgenic mice carrying PRG1 and PRG3 are designated TgPVR1 and TgPVR3, respectively. A total of 21 TgPVR1 mice were identified out of 55 born, and 13 TgPVR3 out of 35 born. Offspring of 2 TgPVR1 and 1 TgPVR3 founders were used for subsequent studies. Expression of poliovirus receptor RNA in transgenic mouse tissues Northern blot analysis with a PVR cDNA hybridization probe was used to examine the expression of PVR transcripts in tissues from different transgenic mouse lines (Figure 5). examined, A 3.3 kb major transcript was detected in all transgenic mouse tissues including brain, spinal 45 cord, intestine, liver, kidney, Figure 4. Identification of transgenic mice containing PVR DNA. (A) Southern blot analysis of tail DNAs from founder mice. The blot was hybridized with a mixture of probes A and B (see [B]). Numbers indicate different founder mice. 1C, one copy equivalent of DNA from cosmid PRG1. (B) Restriction map of cosmid clones PRG1 and PRG3. BamH I fragments that hybridize with probes A (0.97 kb EcoR I fragment derived from PVR cDNA clone HeLa1.5, (Mendelsohn et al., 1989) and B (0.5 kb EcoR I-Sma I fragment derived from PVR cDNA clone H20B, (Mendelsohn et al., 1989) are shown as stippled bars. Line indicates BamH I DNA fragments that hybridize with the entire PVR cDNA clone H20A or H20B. 46 48 B PRG-1 PRG-3 5' 3 kb B BB 5' BB BB B B hybridizes with probe A hybridizes with PVR cDNAs H20A/H20B hybridizes with probe B BB B B B B BB B heart, lung, thymus, spleen and muscle, as well as in HeLa cells as reported previously (Mendelsohn et al., 1989). No RNAs were detected in mouse L cells or in nontransgenic mouse liver or kidney under the hybridization conditions used. In addition to the predominant 3.3 kb transcript, a 2 kb RNA and higher molecular weight RNAs were detected, which are also found in human cells and tissues. The expression level of PVR transcripts roughly correlates with PVR DNA copy number. For example, TgPVR1-7 mice contain 30 copies of the transgene, and PVR RNA levels in this line are generally higher than in TgPVR1-17, which contains 10 copies of the transgene (Figure 5). When transgenic mice containing PRG1 or PRG3 DNA are compared, there do not appear to be significant differences in either the level or tissue distribution of PVR transcripts. The expression level of PVR transcripts in transgenic mice varied in different tissues. Brain, spinal cord, lung and thymus consistently expressed the highest levels of stable RNAs, while RNA levels in other tissues varied but were always lower (Figure 5). Low expression of PVR RNA was observed in intestine, which may be due in part to the poor quality of the RNA preparations, as judged by ethidium bromide staining. The expression of PVR transcripts in liver varied in different transgenic mouse lines. Liver expression was low but present in line TgPVR1-7, and barely detectable in lines TgPVR117 and TgPVR3-9 (Figure 5). Similar patterns of expression were observed in one other TgPVR1 and TgPVR3 line (data not shown). Poliovirus receptor binding activity in transgenic mouse tissues A poliovirus binding assay was used to identify transgenic mouse tissues expressing functional poliovirus binding sites. Tissues from the TgPVR1-17 line of transgenic mice or normal mice were homogenized and incubated with virus at 25°C, and the remaining infectivity was determined by plaque assay. Homogenates of TgPVR1-17 brain, intestine, liver, lung and kidney bound 49 Figure 5. Northern hybridization analysis of mouse tissue RNAs. Equal amounts of total cell RNA were used from tissues of offspring of founders TgPVR1-7, TgPVR1-17, and TgPVR3-9. With the exception of intestinal RNA, all lanes contained equal amounts of RNA as judged by ethidium bromide staining. The DNA probe is probe A, Fig. 1a. Positions of 28S and 18S RNA markers are shown. HeLa cells express 3.3 kb PVR transcripts, as described (Mendelsohn et al., 1989). ntg, nontransgenic. 50 significant levels of P1/Mahoney (Figure 6). No significant binding activity was detected in tissue homogenates of nontransgenic mice. Virus binding assays of tissue homogenates from TgPVR1-7 mice also demonstrated high levels of virus binding activity in a variety of tissues (data not shown). Infection of PVR transgenic mice with poliovirus Transgenic mice were inoculated intracerebrally with poliovirus P1/Mahoney to determine their susceptibility to infection. This virus strain was chosen because it is unable to infect normal mice but is neurovirulent in primates. Intracerebral inoculation of TgPVR1-17 mice with 5 x 105 pfu lead to paralysis in 3 of 3 animals. Paralyzed animals displayed typical signs of poliomyelitis, including one or more paralyzed limbs, ruffled fur, and tremulousness. None of four nontransgenic mice inoculated with 5 x 108 pfu showed any signs of disease. This result demonstrated that PVR transgenic mice are susceptible to poliovirus infection. Mice of the TgPVR3-6 line, which carry 4 copies of PRG3, also developed paralysis after inoculation with P1/Mahoney. The LD50 of representative neurovirulent poliovirus strains of all three serotypes, P1/Mahoney, P2/Lansing, and P3/Leon in transgenic and nontransgenic mice was determined (Table 2). P1/Mahoney infects only transgenic mice. P3/Leon is highly paralytogenic in transgenic mice, but only causes paralysis in nontransgenic mice with a high inoculum (e.g. one nontransgenic mouse developed paralysis when 2 X 107 pfu P3/Leon was inoculated intracerebrally). Both transgenic and nontransgenic mice are susceptible to P2/Lansing. Interestingly, TgPVR1-17 mice inoculated with any of the three serotypes of attenuated poliovirus, P1/Sabin, P2/P712, and P3/Sabin did not develop signs of disease. The flaccid paralysis of poliomyelitis is the result of virus multiplication in and destruction of motor neurons (Bodian, 1959; Hashimoto et al., 1984). 52 To 100 % BINDING 80 60 TgPVR1-17 nontransgenic 40 20 0 brain kidney liver lung intestine Figure 6. Poliovirus binding activity in mouse tissue homogenates. Tissue homogenates were prepared from transgenic mice of the TgPVR1-17 line or from nontransgenic mice, and assayed for the ability to deplete poliovirus infectivity. Virus input was 5 X 106 pfu, and 8 X 104 pfu remained after incubation with TgPVR1-17 brain homogenate. 53 Table 2. Susceptibility of mice to poliovirus infection pfu/LD50 Virus P1/Mahoney P1/Sabin P2/Lansing P2/712 P3/Leon P3/Sabin non-transgenic PVR transgenic >5X108* ND 1X105 >2X109* >2X107 ND 5.8x104 >1X108* 1X105 >2X109* <2X103 >1X108* * no mice paralyzed 54 determine the basis for the neurovirulence of P1/Mahoney in PVR transgenic mice, replication of P1/Mahoney in TgPVR1-17 brain and spinal cord was examined. P1/Mahoney replicated in the brain and spinal cord of transgenic but not nontransgenic mice (Figure 7). Virus replication was detected in the brain beginning on day 1 and one day later in spinal cord, when paralysis was first observed. The difference in viral titres at day 0 in nontransgenic versus transgenic mice probably reflects receptor-mediated eclipse of virus by PVR. It is interesting that replication in spinal cord was delayed, despite presence of virus in spinal cord as shown by the residual inoculum in nontransgenic mice on day 0. Viral titres peaked at day 3 in brain and day 4 in spinal cord, reaching a maximum in the spinal cord about 100 fold higher than in the brain. Virus in the CNS of wild type mice was completely cleared within 4 to 5 days; in the transgenic mice which did not develop paralytic disease, the virus titer began to decrease significantly after 5 days. To determine whether transgenic mice were susceptible to poliovirus infection when inoculated by different routes, TgPVR1-17 mice were inoculated intramuscularly, intraperitoneally, intravenously, or orally with P1/Mahoney and observed for signs of disease. Transgenic mice developed paralytic disease after inoculation with 5 X 108 pfu P1/Mahoney by all routes. The LD50 of P1/Mahoney by different routes of inoculation was determined and presented in chapter VI. Neuropathology of PVR transgenic mice infected with poliovirus The nature and degree of morphological changes associated with P1/Mahoney infection of PVR transgenic mice was determined. Clinical and histopathologic results of these studies are summarized in Table III. Clinical signs of paralysis were seen in 2/3 mice inoculated with P1/Mahoney virus, while all P1/Sabin-inoculated and maintenance media-inoculated CD-1 control animals remained clinically normal throughout the 21-day period. 55 Figure 7. Time course of paralysis and poliovirus replication in mice. Twenty-one transgenic mice of the TgPVR1-17 line were inoculated intracerebrally with 1 X 105 pfu of poliovirus P1/Mahoney. Mice were sacrificed daily, and virus titer in brain and spinal cord was determined by plaque assay on HeLa cell monolayers. Each time point represents the average of three transgenic or two non-transgenic mice. Top panel, total number of mice paralyzed (cumulative); middle and bottom panels, virus titres in brain and spinal cord, respectively. Closed circles, transgenic mice; open squares, nontransgenic mice. 56 MICE PARALYZED TOTAL 8 paralysis 7 6 5 4 3 2 1 0 0 1 2 3 4 5 6 2 3 4 5 6 2 3 4 5 6 5 brain PFU/MG 4 3 2 1 0 0 1 7 spinal cord PFU/MG 6 5 4 3 2 1 0 0 1 DAY POST-INFECTION 57 Table 3. Summary of clinical and neuropathological findings of 21 day neurovirulence test polio histopathology animal # strain virus paralysis brain spinal cord 1 TgPVR1-17 P1/Mahoney day 3 yes yes 2 3 4,5 6-9 10-13 14-17 TgPVR1-17 TgPVR1-17 TgPVR1-17 nontransgenic nontransgenic CD-1 P1/Mahoney P1/Mahoney P1/Sabin P1/Mahoney P1/Sabin medium day 6 no no no no no yes no no no no no yes no no no no no mice were inoculated intracerebrally with 5.4 X 105 pfu of the indicated virus, or with maintenance medium 58 Histopathologic changes consistent with poliovirus infection were seen in the two P1/Mahoney-inoculated transgenic mice which developed paralysis (Figure 8). Mouse #1 had slight edema (loosening of the neuropil) and gliosis of the vestibular nucleus, scattered perivascular cuffs in the ponto-medullary tegmentum and marked necrosis of neurons in the cervical and lumbar ventral horns. Mouse #2 had perivascular cuffing and focal proliferation of rod-shaped microglial cells in the oculomotor and trochlear nuclei, and focal proliferation of rod-shaped microglial cells in the red nuclei as well as in other areas of the midbrain and ponto-medullary tegmentum. In addition there was marked perivascular cuffing, gliosis and proliferation of rod-shaped microglial cells in both the vestibular nuclei and the deep cerebellar nuclei, and marked necrosis of neurons in the cervical and lumbar ventral horns. Mild to moderate perivascular cuffing and focal tissue infiltration associated with the neuronal necrosis was also seen in the spinal cord sections of both animals. Three additional changes were noted in both animals: first, there was a mild to moderate multifocal meningitis involving both the brain and spinal cord meninges. The meningeal inflammation in the brain consisted predominantly of lymphocytes while that in the spinal cord was a mixture of neutrophils and lymphocytes. The same cell type distribution was also noted in the inflammatory lesions (perivascular cuffs and focal tissue infiltrates) in the brain and spinal cord parenchyma. Second, there was multifocal proliferation of rod-shaped microglial cells and perivascular cuffing in the molecular and pyramidal cell layers of the hippocampus with focal necrosis of the pyramidal neurons. Third, there was mild edema of the cerebral deep white matter (loosening of the neuropil and pallor of staining) and mild to moderate nonsymmetrical dilation of the lateral ventricles. The hippocampal changes were bilateral in both animals with mouse #2 having more severe inflammation and focal necrosis of pyramidal neurons. 59 Figure 8. Neuropathology of poliovirus infected transgenic mice. H&E stained sections. a, Lumbar spinal cord, left ventral horn, PVR transgenic mouse #1. Acute necrosis of all neurons (arrow), mild vacuolation of the neuropil (Wallerian degeneration with swollen axons) (arrowhead), and slight inflammatory cell infiltrate along the border of gray and white matter. Magnified 113x. b, Cervical spinal cord, left ventral horn, PVR transgenic mouse #2. Acute neuronal necrosis with very few neuronal cell bodies remaining (arrow), marked loosening, pallor of staining, and vacuolation (edema) of the neuropil. Magnified 198x. c, Right vestibular nucleus, PVR transgenic mouse #2. Severe inflammation with perivascular cuffing, and diffuse and focal proliferation of microglial cells. Several dark neurons (arrow) present (probable artifact). Magnified 56x. d, Left deep cerebellar (fastigial) nucleus, PVR transgenic mouse #2. Moderate inflammation with perivascular cuffing, focal proliferation of microglial cells and two or three vacuoles in the neuropil. Adjacent to one vacuole is cellular debris, possibly a necrotic neuron (arrow). Magnified 71x. e, right red nucleus, PVR transgenic mouse #2. Focal area of proliferation of rodshaped microglial cells (arrow) and a capillary with possible slight exudation of inflammatory cells (arrowhead). Magnified 71x. f, left hippocampal formation, PVR transgenic mouse #2. Focal necrosis of the pyramidal cell layer (arrow) with slight perivascular cuffing and scattered proliferation of rod-shaped microglial cells. Magnified 35x. 60 There were no microscopic lesions characteristic of polio in either the third P1/Mahoney-inoculated mouse or the two P1/Sabin-inoculated animals. There also were no polio lesions in any of the nontransgenic control mice or the control animals inoculated with maintenance medium. The third P1/Mahoney inoculated mouse and the two P1/Sabin animals as well as nontransgenic control mouse #8 all had mild edema of the cerebral deep white matter and slight dilation of the left lateral ventricle. The maintenance medium-inoculated CD-1 controls did not appear to have this change. 62 Chapter IV. Human poliovirus receptor gene expression in humans and in PVR transgenic mice 63 Localization of PVR RNA in transgenic mouse tissues. Previous analysis of PVR gene expression by Northern blot hybridization showed that PVR transcripts are expressed at different levels in a wide range of transgenic mouse organs (chapter III). High stringency in situ hybridization with radiolabeled PVR RNA probes was used to examine the distribution of PVR RNA in specific cell types. Expression of PVR in transgenic mouse CNS was examined by hybridizing transverse sections of adult spinal cord and sagittal sections of the brain with antisense PVR RNA probes. Most neurons in all areas of the CNS expressed high levels of PVR transcripts (Figure 9F-H). All neurons in the spinal cord, including motor and autonomic neurons and interneurons, expressed high levels of PVR RNA. In the brain, PVR RNA was detected in neurons of all cortical layers of the cerebral cortex, cerebellum, nuclei in the brain stem, hippocampus, thalamus, hypothalamus, amygdala, pyriform cortex, basal ganglia and olfactory bulb. The signals observed in spinal cord neurons were consistently higher than those observed in the brain. Within the brain, high levels of PVR transcripts were detected in neurons of the cerebral cortex, brain stem, pyramidal cell layers of the hippocampus and pyriform cortex, amygdala, mitral cell layer of the olfactory bulb and the anterior olfactory nucleus (Figure 9H). Neurons in the peripheral nervous system, such as parasympathetic ganglia, also showed high levels of expression (data not shown). In the kidney, high levels of PVR transcripts were detected in renal corpuscles and in some tubular epthelial cells in the medulla, predominantly in the outer stripe of the outer zone and the renal papilla (Figure 9A-B). In renal corpuscle, high level of PVR RNA appears to be expressed in epithelial cells of the parietal layer of Bowman's capsule, possibly also in podocytes (the visceral layer of Bowman's capsule) in the glomerulus (Figure 9B). PVR transcripts were 64 Figure 9. PVR mRNA expression in transgenic mouse tissues. Sections were hybridized with an 35S-labeled antisense PVR RNA probe. Silver grains, indicating positive hybridization, appear bright white in dark field photomicrographs (a, c, f, and h) or bright green in polarized light epiluminescence photomicrographs (b, d, e, and g). (a). Kidney section showing cortex (C) and medulla (M). Strong hybridization signals localized specifically in renal corpuscles (RC) and some renal tubules. Magnified 71X. (b). Higher magnification of renal corpuscles. High level PVR mRNA expression in epithelial cells of the parietal layer of Bowman's capsule and possibly also in podocytes (the visceral layer of Bowman's capsule) in the glomerulus. Magnified 444X. (c). Section of adrenal gland showing cortex (C) and medulla (M). Strong hybridization signals localized specifically in the adrenal cortex. Magnified 71X. (d). Section of thymus showing cortex (C) and medulla (M). Strong hybridization signals in T-lymphocytes in the cortex and some cells in the medulla. Magnified 444X. (e). Section of lung, showing alveoli (A). Some alveolar lining cells show strong hybridization signals. Magnified 444X. (f). Transverse section of lumbar spinal cord. Strong hybridization signals in grey matter. Magnified 44X. (g). Higher magnification of spinal cord transverse section; grey (G) and white (W) matter are labeled. High levels of PVR mRNA in all neurons. Magnified 178X. (h). Sagittal section of brain. Regions of the brain that show PVR expression include olfactory bulb (OB), cerebral cortex (C), hippocampus (Hi), thalamus (Th), brain stem (BS) and cerebellum (Cb). Magnified 11X. 65 not detected in the renal pelvis, nor in lymph nodes, fatty tissue or blood vessels that surround the kidney. In the lung, PVR transcripts were detected in cells, tentatively identified as macrophages, lining the alveoli (Figure 9E). Bronchial epithelial cells expressed lower levels of PVR transcripts. PVR RNAs were also detected in T-lymphocytes in the cortex of the thymus, as well as in some cells in the medulla of the thymus, and in endocrine cells of the adrenal cortex (Figure 9C, D). PVR gene expression was detected at low levels in most cells of the intestine, spleen and skeletal muscle (data not shown). Expression of alternatively spliced forms of PVR transcripts in transgenic mouse tissues. Alternatively spliced forms of PVR transcripts have been described that encode two soluble forms of PVR that lack the transmembrane domain (Koike et al., 1990; Koike et al., 1991). Using PCR amplification of cDNA, PVR RNA encoding membrane bound and two soluble forms of PVR was detected in all organs examined in the PVR Tg mice studied here. For example, all three forms were detected in brain and kidney, as well as in HeLa cells (Figure 10). Nucleotide sequence analysis of PCR products indicated that the alternatively spliced forms are identical to those reported previously (Koike et al., 1990). Expression of PVR RNA in transgenic mouse embryo and placenta. Mid-sagittal sections of transgenic mouse fetus and placenta at days 12 and 16 of maturity were hybridized with radiolabeled PVR RNA probes. In the transgenic mouse fetus, PVR transcripts were expressed in most tissues at various levels. A high level of PVR RNA was detected in spinal cord, peripheral ganglia, and brain (Figure 11A, B). The strong hybridization signal was uniformly distributed in neurons of the spinal cord and peripheral ganglia. Within the brain, the level of expression is different in different neuron classes. High levels of PVR 67 Figure 10. Detection of alternatively spliced PVR RNA. PVR mRNAs were amplified by quantitative PCR of cDNA, using 5'-32P-labeled primers flanking the PVR transmembrane region, and fractionated on a 6% polyacrylamide gel. 10, 5 or 2.5 µg of total RNA was used for cDNA synthesis/amplification. Size markers (m) of 123 and 246 bp are indicated. p, products of PCR using PVR cDNA as template. The positions of amplified products from H20A, H20A∆1 and H20A∆2 are shown; nucleotide sequence analysis confirmed that these are derived from mRNAs encoding membrane bound and two soluble forms of PVR. H20A∆2 products are visible in tissues after long exposure of the autoradiograph. 68 Figure 11. PVR mRNA expression in the prenatal transgenic mouse. Sections were hybridized with an 35S-labeled antisense PVR RNA probe. Silver grains, indicating positive hybridization, appear bright white in dark field photomicrographs (b and d) or bright green in polarized light epiluminescence photomicrographs (e, and f). (a). Midsagittal section of fetus at day 16 of maturity showing morphology of the fetal tissues under bright field. Brain (B), spinal cord (sp), dorsal root ganglia (DRG), tongue (T), thymus (Th), heart (H), lung (Lu), liver (L), stomach (S), Intestine (I), adrenal gland (A), kidney (K), and ovary (O) are labeled. Magnified 10X. (b). Section (a) under dark field. Strong hybridization signals localized specifically in certain parts of the brain, spinal cord, dorsal root ganglia, tongue, lips, cortex of the adrenal gland, and kidney. Red blood cells in heart and blood vessels around heart, vertebra, and in liver and tail showed autofluorescence. (c). Sagittal section of placenta of the same conceptus showing morphology of the placenta and fetal membranes under bright field. The labyrinth (L), spongiotrophoblast (Sp) region and subchorionic clefts (SC) of the placenta and viseral yolk sac (VYS) are labeled. Magnified 32X. (d). Section (c) under dark field. Strong hybridization signals localized specifically in the labyrinth region of placenta. (e). Higher magnification of fetal kidney showing renal corpuscle (RC). High level PVR mRNA is present in possibly epithelial cells of the the visceral layer of Bowman's capsule (podocytes in the glomerulus). Magnified 159X. (f). Higher magnification of placenta showing the labyrinth (L) and spongiotrophoblast (Sp) region. A high level PVR mRNA is present in certain cells in the labyrinth of placenta. Magnified 159X. 69 transcripts were detected in neurons of the brain stem and olfactory bulb (data not shown). A strong hybridization signal was also detected in both kidney and adrenal gland (Figure 11B). In kidney, high levels of PVR transcripts appear to accumulate in the visceral layer of Bowman's capsule (Figure 11E). The parietal layer of Bowman's capsule and some tubular epithelial cells appear to have weak hybridization signals. In the adrenal gland, PVR transcripts are expressed in the cortex. A strong hybridization signal was also observed in tongue, hair follicles, lung, and cortex of thymus (Figure 11B). Expression of PVR transcripts was also detected in salivary glands, esophagus, nasal mucosa, brown adipose tissues, skin, skeletal muscles, and certain cells in bone. Expression of PVR transcripts in tissues such as heart, liver, stomach, intestine, spleen, and gonad was detected at low levels (data not shown). In transgenic mouse placenta at both day 12 and 16 of maturity, very high level expression of PVR transcripts was detected in the labyrinth (Figure 11C, D). Expression of PVR transcripts in the subchorionic clefts and the spongiotrophoblast was not detected. Within the labyrinth of the placenta, only certain cells express high levels of PVR transcripts (Figure 11F). The cell type which expresses PVR transcripts is not known. However, since maternal cells do not have the transgene, the cells expressing PVR transcripts must be of fetal origin. In addition to the placenta, expression of PVR RNA was also detected in transgenic mouse amnion and parietal endoderm. The endoderm of the viseral yolk sac also expresses low levels of PVR RNA, when examined at higher magnification (data not shown). Expression of PVR RNA in human adult and embryonic tissues. PVR gene expression was examined in human kidney and intestine by in situ hybridization with radiolabeled PVR RNA probes. In kidney, a specific hybridization 71 signal was detected in the glomerulus (Figure 12A,B) and in some tubular epthelial cells. The cells expressing PVR RNA in the glomerulus appear to be podocytes. In intestine, a high level of PVR RNA was expressed in the epithelium lining the intestinal villi and in the crypts of Lieberkuhn, which are a continuous supply of new cells for the epithelium of the villi (Figure 12E). Lymphocytes in the intestine appear to express only low levels of PVR RNA. Expression of PVR RNA was not detected in submucosa and muscular mucosa. PVR gene expression was also examined in tissues from human embryos at 6 to 10 weeks gestation. High levels of PVR RNA were detected in intestinal epithelium (Figure 12F). Stomach epithelium expresses a much lower level of PVR RNA (data not shown). PVR RNA was also detected in neurons of the spinal cord, trigeminal ganglion, dorsal root ganglia, and brain (Figure 12C, D). Expression of PVR RNA in spinal cord and peripheral ganglia is generally higher than in brain. The salivary gland, skeletal muscle, fetal liver, and certain cells in bone all accumulate low levels of PVR RNA. Significant expression of PVR RNA was not detected in kidney, lung, heart, and thymus in the embryo at the stages examined. Expression of PVR RNA in human placenta. PVR gene expression was examined in human placental tissues, ranging from 4 weeks to 16 weeks gestation and at term, by in situ hybridization with radiolabeled PVR RNA probes. A high level of PVR RNA was expressed in nonvillous parts of the placenta in all stages examined. The cells expressing high levels of PVR RNA are located in following structures: 1) Cell islands (Figure 13A,B), which are connected to either the villous tree or the chorionic plate. 2) Basal plate (Figure 13D,E), which is the contact zone of fetal and 72 maternal tissues. Figure 12. PVR mRNA expression in human adult and fetal tissues. Sections were hybridized with an 35S-labeled antisense PVR RNA probe. Silver grains, indicating positive hybridization, appear bright white in dark field photomicrographs (b, c, and d) or bright green in polarized light epiluminescence photomicrographs (e, and f). (a). A section of adult kidney showing the morphology of the renal corpuscle (RC) and renal tubules under bright field. Magnified 159X. (b). Section (a) under dark field. Specific hybridization signals over the high background are localized possibly in epithelial cells of the the visceral layer of Bowman's capsule (podocytes in the glomerulus). (c). Sagittal section of fetal hindbrain and part of the spinal cord under dark field. Specific hybridization signal localized in spinal cord and brain. Magnified 32X. (d). A section of vertebra showing dorsal root ganglia (DRG), vertebral body (below the DRG), and skin (above the DRG) under dark field. Strong hybridization signals localized specifically in the DRG and possibly in the anular epiphyses of vertebral body and skin. Magnified 64X. (e). Cross section of adult intestine. High level PVR mRNA was shown in intestinal epithelia (E) lining the villi and the crypts of Lieberkuhn (C). Magnified 159X. (f). Cross section of fetal intestine. High level PVR mRNA is present in intestinal epithelia. Magnified 159X. 73 Figure 13. PVR mRNA expression in human placenta. Sections (a-b and d-e) were hybridized with an 35S-labeled antisense PVR RNA probe. Silver grains, indicating positive hybridization, appear bright white in dark field photomicrographs (b and e). Sections (c and f) were immunostained with monoclonal antibody against cytokeratin. The distribution of cytokeratin was indicated by the peroxidase reaction product (dark brown). (a). A section of placenta at 16 weeks gestation showing cell island (CI) and villus (V) under bright field. Magnified 58X. (b). Section (a) under dark field. Strong hybridization signals localized specifically in cell islands but not in villi. (d). A section of the same placenta showing the morphology of the extravillous trophoblastic cells (ET), decidual cells (D), and a layer of Nitabuch's fibrinoid (N) separating the extravillous trophoblast and decidua in basal plate under bright field. Magnified 145X. (e). Section (d) under dark field. Strong hybridization signals localized specifically in the extravillous trophoblastic cells. (c). Immunochemistry of the same placenta (sections adjacent to ab). Magnified 58X. (f). Immunochemistry of the same placenta (sections adjacent to de). Magnified 145X. 75 3) Septa, which arise from the basal plate and protrude into the intervillous space and are composed of fibrinoid and various fetal and maternal cells. 4) Cell columns, which anchor the placenta to the endometrium. Cell islands, cell columns, and septa are usually included in the basal plate. 5) Placental bed, where fetal trophoblasts infiltrate into maternal endometrium and myometrium. Some PVR RNA expressing cells are mononuclear, and others are multinucleated. 6) The spiral arteries at the placental site, in which maternal blood vessels are invaded by trophoblast. 7) Underlying the chorioamnion in the chorionic plate. The morphology and location of the PVR expressing cells suggested that they are extravillus trophoblastic cells. However, it is difficult to distinguish fetal extravillus trophoblastic cells from maternal decidual cells and myometrial cells in certain places, as there is great structural variability of the placenta. To determine the origin of the cells expressing PVR RNA, the sections of placenta tissue, which are adjacent to that used for in situ hybridization with radiolabeled PVR RNA probes, were examined by immunochemical analysis using monoclonal antibody against cytokeratin and human placental lactogen (hPL). Keratin is one of the most sensitive markers for the distinction of trophoblast cells from decidual cells (Yeh et al., 1990). Figure 13C and 13F show that cells expressing high levels of PVR RNA contain keratin. However, expression of PVR RNA was not detected in villous cytotrophoblasts and syncytial trophoblasts, which also show prominent staining for keratin. Most PVR RNA expressing cells also contain hPL, whereas decidual and myometrial cells do not (data not shown). The PVR RNA expressing cells, therefore, are identified as extravillus trophoblasts. Both mononuclear trophoblastic cells and multinucleated placental site giant cells express high levels of PVR RNA. In addition to the extravillus trophoblastic cells, the decidual cells in the basal plate also express a lower levels of PVR RNA (Fig. 13E). 77 Chapter V. Poliovirus tissue tropism in transgenic mice 78 Poliovirus replication sites in the CNS of transgenic mice. To identify the sites of virus multiplication in the CNS, mice were infected intraperitoneally or intracerebrally, and tissues from paralyzed animals were examined by in situ hybridization with viral RNA probes. In paralyzed transgenic mice, spinal cord lesions were the most severe in the neuraxis, with inflammation and neuronal degeneration localized largely to the ventral horns. Infected neural cells were detected by in situ hybridization in all areas of the grey matter, including the ventral horn, intermediate and intermediolateral columns, and dorsal horn (Figure 14). Viral RNA was detected in the cytoplasm of neurons, in both the cell bodies and their axonal and dendritic processes. Diffuse hybridization was often observed around lysed neurons, in areas containing inflammatory cells (Figure 14A). Viral RNA was not detected in vascular endothelial cells or glial cells. Sites of lesions in the spinal cord of paralyzed animals corresponded with the distribution of clinical paralysis. For example, in mice with left leg paralysis, viral RNA was detected in most neurons in both sides of the lumbar spinal cord, but neuronal destruction was observed only in the left ventral horn of the cord. As infection progressed, other neurons on the same side of the cord were infected; at later stages, neuronal destruction was present in the affected side, and virus spread to both motor neurons and interneurons of the unaffected side and to upper segments of the spinal cord and brain stem. In the brain of paralyzed mice that had been inoculated intraperitoneally, most sites of viral replication were in the brain stem, accompanied by marked microglial proliferation and perivascular cuffing. Viral replication was not detected in the cerebral cortex, cerebellum, hippocampus, thalamus, hypothalamus, or olfactory bulb. It was not clear if neurons in these regions were not susceptible to poliovirus, or whether virus was unable to reach 79 these areas Figure 14. In situ detection of poliovirus RNA in spinal cord of PVR transgenic mice infected intraperitoneally with poliovirus. Sections were hybridized with an 35S-labeled antisense P1/Mahoney viral RNA probe. Silver grains, indicating positive hybridization, appear bright green in the polarized light epiluminescence photomicrographs. (a). Lumbar spinal cord, left ventral horn. Strong hybridization signals are present in neurons. Some neurons have lysed (filled arrowhead). Severe inflammation with perivascular cuffing (open arrow head) and diffuse and focal proliferation of microglial cells. Hybridization is not detected in neuroglial cells in both the white and grey matter, nor in the central canal (CC). Magnified 155X. (b). Higher magnification of poliovirus infected neuron in the ventral horn of the spinal cord. Poliovirus RNA present in both the cell body and axonal and dendritic processes. Magnified 624X. (c). Lumbar spinal cord, left dorsal horn. Neurons infected with poliovirus (arrowhead) in the absence of significant inflammatory changes. Magnified 155X. 80 after intraperitoneal inoculation. To address this question, transgenic mice were infected intracerebrally, and brains from paralyzed mice were examined by in situ hybridization (Figure 15). The results indicated that neurons in the brain stem were extensively infected, and viral replication was also detected in neurons in many other areas of the brain, including the cerebral cortex, pyramidal layer of the hippocampus, olfactory bulb, thalamus, hypothalamus and deep cerebellar nuclei. Poliovirus susceptibility of transgenic mouse nonneural tissues. PVR transgenic mice developed paralytic disease after intramuscular inoculation with poliovirus strain P1/Mahoney (chapter VI). To determine whether poliovirus could replicate in muscle, mice were inoculated intramuscularly with 5 X 105 PFU of poliovirus P1/Mahoney, and at different times after infection, the hamstrings of three mice were removed and homogenized, and virus titres were determined by plaque assay. Levels of virus in muscle rose to 106 PFU/mg by day 4 post-infection (Figure 16A), at which time paralysis was observed. nontransgenic mice. In contrast, poliovirus did not replicate in muscle of Examination of infected PVR Tg mouse muscle by in situ hybridization, using a viral RNA probe, revealed poliovirus replication in muscle cells (Figure 16B). Intravenous and intraperitoneal inoculation of PVR transgenic mice with poliovirus leads to development of poliomyelitis (data not shown). To determine whether nonneural tissues which express PVR transcripts can support poliovirus infection, the ability of poliovirus to replicate in kidney and thymus was determined. PVR transgenic mice were inoculated intravenously with 1 X 106 PFU of poliovirus type 1 Mahoney strain, and at different times after infection organs were removed and homogenized, and virus titer was determined by plaque assay. Virus titer in both kidney and thymus declined within the 82 first three days after Figure 15. In situ detection of poliovirus RNA in infected PVR transgenic mouse brain. Sections were hybridized with an 35S-labeled antisense P1/Mahoney viral RNA probe. Silver grains, indicating positive hybridization, appear bright white in the dark field photomicrograph of sagittal (a and c) and coronal (b) sections of brain. (a). Strong hybridization signals localized specifically in the brain stem (BS). Infected neurons are in the deep nuclei of the cerebellum (Cb), cerebral cortex (C), hippocampus (Hi), thalamus (Th) and olfactory bulb (OB). Magnified 10X. (b). Strong hybridization in neurons in the pyramidal layer (P) of the hippocampus, thalamus (Th), cerebral cortex (C), but not in the dentate gyrus (DG) of the hippocampus. Magnified 20X. (c). Higher magnification of the olfactory bulb (OB) from the infected brain. Strong hybridization signals form a patch of neurons in the olfactory bulb. The cerebral cortex (C) is identified. Magnified 20X. 83 Figure 16. Poliovirus replication in PVR transgenic mouse skeletal muscle. Top). Time course of poliovirus replication in skeletal muscle. Transgenic (TgPVR, closed circles) or nontransgenic (nTg, open circles) mice were inoculated with 5 X 105 PFU of P1/Mahoney in the left hamstring muscle. At the indicated times, the hamstring was removed, homogenized, and the virus titer was determined by plaque assay. Each point represents the mean of values obtained for three mice. Bottom). In situ detection of poliovirus RNA in infected PVR transgenic mouse skeletal muscle. Cross-section prepared from hamstring of a paralyzed mouse inoculated intramuscularly as described above. The tissue section was hybridized with an 35S-labeled antisense P1/Mahoney viral RNA probe. Silver grains, indicating positive hybridization, appear dark green in the polarized light epiluminescence photomicrographs. Strong hybridization signals localized in muscle cells. Note extensive tissue infiltration in the infected area. Magnified 364X. 85 PFU/MG MUSCLE 10 6 10 4 10 2 TgPVR nTg 10 0 0 1 2 3 4 DAY POST-INFECTION 5 6 infection, suggesting that virus replication did not occur at these sites (data not shown). Poliovirus also failed to replicate in the liver and spleen after intravenous inoculation, and the intestine after oral inoculation (data not shown). However, it was possible that the inoculated virus did not have the opportunity to infect susceptible cells, because virus in the blood is quickly removed by the reticuloendothelial system (Sabin, 1956). To examine this possibility, transgenic mice were inoculated intraperitoneally with 5 X 107 PFU of P1/Mahoney, and virus levels in the kidney were determined by plaque assay. Virus titres in both transgenic and nontransgenic kidney declined rapidly after inoculation, and no virus was detected in this organ after day 2 post-infection (Figure 17). Paralysis was observed in many of these animals, indicating that virus had reached the CNS. Because only a small fraction of kidney cells express PVR, virus growth in these cells might not be detected by plaque assay of kidney homogenates. Therefore poliovirus replication in transgenic mouse kidney and surrounding tissues was examined by in situ hybridization, using an antisense viral RNA probe. Viral replication was not detected in the kidney, adrenal gland, connective tissue, fatty tissue, lymph node, parasympathetic ganglia, blood cells or blood vessels for one week after intraperitoneal inoculation, while paralytic disease developed in some of these animals (data not shown). Susceptibility of cultured PVR transgenic mouse kidney cells to poliovirus. Although poliovirus tropism in primates is restricted, cells from almost any tissue become susceptible to poliovirus infection after cultivation in vitro (Enders et al., 1949; Holland, 1961; Kaplan, 1955). To examine the susceptibility of cultured PVR transgenic mouse kidney cells to poliovirus, kidneys were dispersed with collagenase and cultivated either in monolayers or in suspension, and infected with poliovirus at an MOI of 10. Although freshly isolated 87 transgenic 1000 PFU PER MG Tg kidney ntg kidney 100 10 1 0 1 2 3 4 5 DAY POST-INFECTION Figure 17. Poliovirus replication in PVR transgenic mouse kidney. 6 Transgenic and nontransgenic mice were inoculated intraperitoneally with 5 X 107 PFU P1/Mahoney. Mice were sacrificed daily, and virus titer in the kidney was determined by plaque assay on Hela cell monolayers. Each time point represents the average of three mice. Closed circles, transgenic mice; open circles, nontransgenic mice. 88 mouse kidney cells were resistant to poliovirus infection, after 24 hours of growth in culture, the cells became highly susceptible to poliovirus infection (Table 4). Cultured kidney cells from normal mice did not develop susceptibility to poliovirus infection. Freshly dispersed, poliovirus-resistant transgenic kidney cells expressed PVR at the cell surface, as determined by a poliovirus binding assay (Table 5). Despite expression of poliovirus binding sites on the cell surface, freshly dispersed PVR transgenic mouse kidney cells are resistant to poliovirus infection. Poliovirus infection in suckling mice following oral inoculation. Adult transgenic mice develop paralytic disease inefficiently following oral inoculation of poliovirus P1/Mahoney (see Table 7). However, virus replication was not detected in the intestine from TgPVR1-17 and TgPVR3-6 transgenic mice (data not shown). Some of the possibilities to explain the failure of poliovirus replication in intestine are: 1) interference with poliovirus replication by other viruses in mouse intestine; 2) poliovirus replication was inhibited by some components in the food; 3) the susceptibility of mouse intestine to poliovirus infection is lost in the adult. To examine these possibilities, suckling transgenic and nontransgenic mice were inoculated with P1/Mahoney perorally. The suckling transgenic, but not nontransgenic, mice can develop paralytic disease following peroral inoculation of poliovirus (Table 6). Most transgenic mice developed forelimb paralysis first. To identify the sites of virus multiplication in the infected suckling mice, sections of paralyzed animals were examined by in situ hybridization with viral RNA probes. Poliovirus multiplies extensively in skeletal muscles and neurons in the CNS (Figure 18). Following peroral inoculation, poliovirus spreads to most parts of the body and replicates in skeletal muscles of tongue, face, back, chest, and those underlying the skin. Poliovirus replication was also detected 89 to a much Table 4. Susceptibility of PVR transgenic mouse kidney cells after in vitro cultivation PVR Tg kidney poliovirus susceptibility Day post-dispersion monolayer suspension 0 ND R 1 S S 2 S S 3 S ND a determined by plaque assay in HeLa cell monolayers; R, resistant to infection: no increase in poliovirus titer observed 24 hr post infection; S, susceptible to infection: poliovirus titres increased 2-6 log10 pfu 24 hr post infection, with nearly complete destruction of cell monolayers; ND, not done. Cultured kidney cells from normal mice did not develop susceptibility to poliovirus infection. Collagenase treatment did not affect poliovirus susceptibility of HeLa cells or 24-hour cultures of PVR Tg kidney cells. Table 5. Poliovirus binding to dispersed PVR transgenic mouse kidney cells origin of cells PFU in supernatant % binding PVR Tg kidney 1.5 X 105 90 nTg kidney 1.5 X 106 0 90 Table 6 Susceptibility of suckling mice to poliovirus infection following peroral inoculation virus P1/Mahoney " " transgenic line TgPVR1-17 " nontransgenic pfu inoculated 2 x 108 2 x 107 2 x 108 91 paralyzed/inoculated 12/18 4/9 0/10 Figure 18. In situ detection of poliovirus RNA in suckling PVR transgenic mouse orally infected with P1/Mahoney. Sections were hybridized with an 35S-labeled antisense P1/Mahoney viral RNA probe. Silver grains, indicating positive hybridization, appear bright white in the dark field photomicrograph. (a). Sagittal section of the cervical segment of poliovirus infected suckling mouse showing morphology of tissues under bright field. The spinal cord (Sp), lung (Lu), Heart (H), thymus (T), skeletal muscle (M), and brown adipose tissue (BF) are labeled. Magnified 18X. (b). Section of (a) under dark field. A strong hybridization signal localized specifically in skeletal muscles, spinal cord, and, to a much lesser extent, in brown adipose tissues. 92 lesser extent in brown adipose tissue, nasal epithelium, and neurons of peripheral ganglia. In the CNS, poliovirus multiplies extensively in neurons in the spinal cord and brain stem. Replication of poliovirus in Purkinje cells in the cerebellum, neurons in the hippocampus, cerebral cortex, and olfactory bulb was also detected (data not shown). Viral replication was not detected in other tissues such as the intestine, heart, liver, thymus, kidney, and lung. 94 Chapter VI. Poliovirus spreads from muscle to the central nervous system by neural pathways 95 Efficiency of induction of poliomyelitis by different inoculation routes. To understand how poliovirus reaches the CNS, the efficiency by which poliovirus caused paralytic disease by different routes of inoculation, as measured by the LD50 value, was compared (Table 7). The LD50 for peroral inoculation was the highest, followed by intravenous, intracerebral, and intramuscular inoculation. Surprisingly, the LD50 values for intramuscular and intracerebral inoculation were similar. This result suggested that virus might enter the CNS directly from the injected muscle by neural routes, or alternatively, because poliovirus replicates in muscle cells, virus shed into the bloodstream might invade the CNS through the BBB. The following experiments were carried out to distinguish between these possibilities. Paralysis following intramuscular inoculation of poliovirus. If virus spreads from the muscle to the CNS through nerves innervating the injected muscle, it would be predicted that the inoculated limb would be the first to become paralyzed. As predicted, in TgPVR mice inoculated intramuscularly with poliovirus P1/Mahoney in the right or left hindlimb or the left forelimb, 100% localization of initial paralysis to the inoculated limb was observed (Table 8). Similar localization of paralysis was also observed after intrafootpad inoculation of TgPVR mice with P1/Mahoney (data not shown). The typical course of disease in mice inoculated in the hindlimb was first hindlimb paralysis, followed by forelimb paralysis and then death. In mice inoculated in the forelimb, paralysis of that limb was first noted, followed rapidly by death, with little involvement of the hindlimbs (data not shown). These observations are consistent with neural spread of poliovirus to the CNS from the inoculation site. Poliovirus P2/Lansing, which causes poliomyelitis in normal mice (Armstrong, 1939a), did not result in localization of paralysis when inoculated intramuscularly in nontransgenic mice (Table 8). However, 96 when P2/Lansing was Table 7. Effect of route of inoculation on LD50 of poliovirus P1/Mahoney in PVR transgenic mice. Route of inoculation LD50 intracerebral 5.8x104 intramuscular 4.5x104 intraperitoneal 8.2x105 intravenous 1.5x106 oral >2x108* * 3 out of 12 PVR transgenic mice paralyzed after oral inoculation. 97 Table 8. Localization of initial paralysis in mice inoculated intramuscularly with poliovirus. Virus Mouse Limb inoculated First limb paralyzed P1/Mahoney1 TgPVR LH LH (40/40)3 " " RH RH (36/36) " " LF LF (14/14) " " LH virus, RH PBS LH (16/16) P2/Lansing2 " LH LH (12/12) " normal LH RH (1), RF (1), RLH (2) LH, left hindlimb; RH, right hindlimb; LF, left forelimb 1 5 x 107 pfu inoculated 2 7 x 107 pfu inoculated 3 # of mice paralyzed/# of mice inoculated 98 inoculated into the hindlimb of TgPVR mice, 100% localization of initial paralysis was observed, indicating that the high frequency of localization of paralysis is determined by the presence of PVR. High frequency localization of initial paralysis in the injected limb has also been observed in humans and monkeys (Nathanson and Bodian, 1961; Nathanson and Langmuir, 1963). It has been suggested that localization might result if virus from the inoculated muscle enters the circulation and invades the CNS through the BBB. Localization of paralysis would occur if the injection enhances the permeability of the capillary bed in the corresponding part of the spinal cord, a phenomenon called the "provoking effect" (Nathanson and Bodian, 1961). To test this possibility, virus was inoculated into the left hindlimb of TgPVR mice, and then the same volume of PBS was inoculated into right hindlimb. The left hind limb inoculated with virus was still the first to develop paralysis (Table 8), indicating that localization cannot be explained by a "provoking effect" of the inoculation trauma. Spread of poliovirus to the CNS. To understand how poliovirus invades the CNS, it is important to identify which part of the CNS the virus enters first. To address this question, TgPVR mice were inoculated intramuscularly with P1/Mahoney, and at each day after infection the inferior and superior segments of the spinal cord and brain were isolated. Tissues were homogenized and virus titer was determined by plaque assay on Hela cells. After inoculation, virus is first detected in the inferior spinal cord, where the highest levels of replication also occur (Figure 19). Virus is next detected in the superior spinal cord and later in the brain. These results indicate that after intramuscular inoculation, poliovirus first enters the lower spinal cord and then spreads to upper segments of the spinal cord and brain. This pattern of invasion is consistent with the localization of paralysis to the injected hindlimb. 99 LOG 10 PFU PER MG 5 X 107 PFU P1/Mahoney inoculated i.m. left leg 8 isc 7 ssc brain 6 5 4 3 2 1 0 MOUSE #: 1 DISEASE: DAY P.I.: 2 0 3 1 2 3 1 1 2 L 3 L 2 1 L 2 L 3 L, R 3 Figure 19. Time course of poliovirus replication in the CNS. Twelve TgPVR mice were inoculated intramuscularly with poliovirus P1/Mahoney. At the indicated day post infection, three mice were sacrificed, and the virus titer in the inferior spinal cord (isc), superior spinal cord (ssc) and brain of each animal was determined. Presence of paralytic disease is indicated by L or R (left or right leg paralysis, respectively). 100 Effect of nerve transection on poliovirus infection. To further differentiate between neural and humoral routes of virus spread, a differential block of the neural route was produced by transecting the sciatic nerve. Because the sciatic nerve is the principal neural pathway from the hindlimb to the spinal cord, nerve transection should prevent spread of virus from the hindlimb through neural pathways, but should not affect its capacity to spread through the bloodstream. To carry out this experiment, the sciatic nerve of TgPVR mice was transected, and one day later the animals were inoculated in the hindlimb footpad with P1/Mahoney (Table 9). In two separate experiments, transgenic mice with sciatic nerve transection did not develop lethal disease, while mice given a sham operation developed paralytic disease and died. In experiment 2, two out of twenty three transgenic mice with sciatic nerve transection developed paralytic disease. In these two mice the injected limb, which showed paresis following sciatic nerve transection, was the first to develop flaccid paralysis, suggesting that virus may enter the termini of nerves whose pathway to the CNS is not blocked by sciatic nerve transection. Indeed, when high levels of P1/Mahoney (1 X 107 PFU) were inoculated, there was no protection afforded by sciatic nerve transection (data not shown). 101 Table 9. Effect of sciatic nerve transection on poliovirus-induced lethality in TgPVR mice #mice dead/inoculated Cut Uncut exp. 1 0/12 12/12 exp. 2 2/23 18/23 Mice were inoculated in the footpad with 2 x 105 PFU P1/Mahoney one day after sciatic nerve transection 102 Chapter VII. Attenuation determinants in a vaccine-related type 2 poliovirus 103 Mapping an attenuation determinant in the coding region of P2/P712. Using a strategy of constructing recombinants with the mouse-virulent P2/Lansing strain of poliovirus, and testing the recombinants for neurovirulence in mice, an attenuation determinant of the vaccine-related strain P2/P712 has been mapped to a region that encodes capsid protein VP1 and nonstructural proteins 2Apro, 2B and part of 2C (Moss et al., 1989). To more precisely map this determinant, recombinants were generated between P2/Lansing and SRL, a strain that contains the attenuating central region of P2/P712 in an otherwise P2/Lansing background (Figure 20). Pst I restriction sites were introduced into the cDNAs of P2/Lansing and SRL near the boundary of sequences encoding VP1 and 2Apro, and two reciprocal recombinant cDNAs were constructed using the Pst I sites, from which viruses SVL and SPL were derived by transfection (Figure 20). SVL encodes VP1 of P2/P712 in a P2/Lansing background, and SPL encodes part of the P2 coding region (P2') in a P2/Lansing background. The recombinant viruses resembled the parental strains in both plaque size and growth at high and low temperatures (data not shown). SVL and SPL were inoculated into Swiss-Webster mice intracerebrally for determination of LD50 values. SPL was as neurovirulent as P2/Lansing, indicating that the P2' region of P2/P712 carries no attenuation determinants (Figure 20). However, recombinant SVL was attenuated to the same degree as SRL, indicating that the attenuation determinant is located in VP1 (Figure 20). Identification of the major attenuation determinant in capsid protein VP1 of P2/P712. P2/P712 and the neurovirulent strain P2/117 differ at only one nucleotide in the region encoding VP1, which results in a difference at amino acid position 143 (Pollard et al., 1989). P2/P712 encodes an isoleucine 104 (ile) at this position, and Figure 20. Constitution and mouse neurovirulence of P2/P712-P2/Lansing capsid protein VP1 coding sequences in recombinant and mutant viruses. The genomic RNA of each virus derived from the recombinant and mutant viral cDNA is represented below a genetic map of viral genomic RNA. The name of each virus is shown at the left, and its corresponding LD50 value in nontransgenic (nTg) mice and transgenic (Tg) mice expressing human poliovirus receptors (TgPVR1-17) is shown at the right. Black, sequences derived from cDNA of P2/Lansing; white, sequences derived from cDNA of P2/P712. The amino acid and nucleotide substitutions are shown at the relevant position of the viral RNA. 105 nt SVL-thr SVL-val SVL P712 Lansing RNA 5'nc VP4 VP2 VP3 thr val ile ile thr VP1 2900 106 2A 2B 3411 2269 2C 3A 3B 3C 3D 1 X 105 1 X 105 3 X 107 1 X 105 3 X 105 2 X 109 3 X 106 3 X 106 ND Tg nTg pfu/LD 50 >2 X 109 3'nc AAA P2/117 encodes valine (val). The amino acid at this position in P2/Lansing, as well as in several other poliovirus strains, is threonine (thr). To determine the role of amino acid 143 of VP1 in neurovirulence and attenuation, site-directed mutagensis of SVL cDNA was performed to generate two mutant cDNAs, which upon transfection into HeLa cells gave rise to viruses SVL-val and SVL-thr. SVLval has a val at VP1-143, as in P2/117, and SVL-thr has a thr at that position, as in P2/Lansing (Figure 20). The plaque size and growth at high temperature of the mutant viruses in HeLa cells resembled that of SVL (data not shown). In neurovirulence assays, both SVL-thr and SVL-val were approximately 1000-fold more paralytogenic than SVL (Figure 20). Therefore, ile-143 is the primary determinant of attenuation in VP1 of P2/P712. Identification of the major attenuation determinant in the 5' noncoding region of P2/P712. Previous neurovirulence analysis of the recombinants between P2/Lansing and P2/P712 indicated the presence of a strong attenuation determinant in the 5'-ncr of the P2/P712 genome (Moss et al., 1989). P2/P712 and P2/117 sequences differ in the 5' noncoding region by three nucleotides, at positions 437, 481, and 685 (Pollard et al., 1989). To determine which of these positions are important for the attenuation phenotype, mutations were made in the cDNA of attenuated recombinant SLL, which consists of the 5'-ncr and a portion of the coding region from P2/P712 in a P2/Lansing background. The coding region of P2/P712 in this recombinant confers little or no attenuation (Moss et al., 1989). SLL cDNA was mutagenized to change the nucleotides at 437 or 481 to the corresponding residues in P2/117, and mutant viruses SLL437 and SLL481 were derived by transfection with the altered cDNAs. SLL437 has a C at position 437 where SLL and P2/P712 have U. SLL437 is no more virulent in mice than is SLL (Figure. 21). SLL481 has a G at position 481 where SLL and P2/P712 have an A. SLL481 is at least 100-fold more paralytogenic than SLL, though it is approximately 100- 107 fold less paralytogenic than P2/Lansing (Figure 21). To confirm that the coding region of P2/P712 plays no role in the attenuation phenotype, LP481 was constructed. It was derived from SLL481, and has the coding region of P2/P712 replaced with that of P2/Lansing. LP481 was as neurovirulent as SLL481 in mice (Figure 21). These results indicate that A-481 is a major determinant of attenuation in the 5'-ncr of P2/P712. To determine the effect on neurovirulence of all three differences between P2/P712 and P2/117 in the 5' noncoding region, the noncoding region of P2/Lansing was replaced with that of P2/117 to generate recombinant 117LP. The neurovirulence of 117LP is slightly higher than that of LP481 (Figure 21). Therefore, either A-685, or an interaction among the three nucleotides, contributes partially to the attenuation of P2/P712. Neurovirulence of recombinant viruses in transgenic mice expressing human poliovirus receptors. To determine the degree of host range restriction caused by ile-143, viruses SVL, SVL-val and SVL-thr were assayed for neurovirulence in transgenic mice expressing human poliovirus receptors (PVR). Host-restricted viruses such as P1/Mahoney that do not cause disease in mice but are able to induce paralysis in primates, are neurovirulent in PVR transgenic mice. The LD50 of SVL was about 100-fold lower in TgPVR1-17 mice compared to normal mice (Figure 20). Both SVL-val and SVL-thr were approximately 10fold more neurovirulent in PVR transgenic mice than in normal mice, and as virulent as P2/Lansing. These results indicate that ile-143 is primarily a general attenuation determinant. To rule out the possibility that all attenuated polioviruses are nonspecifically more paralytogenic in transgenic mice expressing PVR, the neurovirulence of recombinants LP1 and LP481 were tested in TgPVR1-17 108 mice. LP1 was as Figure 21. Constitution and mouse neurovirulence of P2/P712-P2/Lansing 5'-ncr recombinant and mutant viruses. The genomic RNA of each virus derived from the recombinant and mutant viral cDNA is represented below a genetic map of viral genomic RNA. The name of each virus is shown at the left, and its corresponding LD50 value in nontransgenic (nTg) mice and transgenic (Tg) mice expressing human poliovirus receptors (TgPVR1-17) is shown at the right. Black, sequences derived from cDNA of P2/Lansing; white, sequences derived from cDNA of P2/P712. The amino acid and nucleotide substitutions are shown at the relevant position of the viral RNA. 109 pfu/LD 50 nt LP481 SLL437 SLL 117LP LP1 G C CG U UA A 6 X 107 >1 X 10 6 X 107 ND ND >6 X 108 9 ND >2 X 109 3 X 106 >2 X 109 ND UA A 3'nc AAA >2 X 109 3D P712 3A 3B 3C 1 X 105 2C 1 X 105 2A 2B Lansing VP1 Tg VP3 nTg RNA 5'nc VP4 VP2 437 481 685 752 110 attenuated in normal and in PVR transgenic mice (Figure 21). As expected, LP481, which contains G-481 as in the neurovirulent P2/117, was equally neurovirulent in transgenic and nontransgenic mice (Figure 21). Poliovirus replication in PVR transgenic mouse skeletal muscle. It was shown that P1/Mahoney can replicate in transgenic mouse skeletal muscle (chapter IV). To determine whether type 2 poliovirus could replicate in muscle, mice were inoculated intramuscularly with 1 X 107 PFU of poliovirus P2/MEF-1, a mouse adapted poliovirus which is neurovirulent after intramuscular injection in both transgenic and nontransgenic mice (data not shown). At different times after infection, the hamstrings of three mice were removed and homogenized. Virus titres were determined by plaque assay on Hela cell monolayers. Levels of virus in PVR transgenic mouse muscle rose to about 3 X 105 PFU/mg by day 3 post-infection (Figure 22), before paralysis was observed. In contrast, the virus titer in muscle of nontransgenic mice decreased rapidly in the same period of time. Interestingly, when PVR transgenic mice were inoculated intramuscularly with 7.5 X 107 PFU of attenuated poliovirus P2/P712, which is non-paralytogenic by both intracerebral and intramuscular inoculation in PVR transgenic mice, the virus titer of P2/P712 in muscle decreased rapidly and most viruses were cleared in 3 days.(Figure 22). Temperature sensitivity of polioviruses in transgenic mouse primary muscle culture. Attenuated poliovirus strain P2/P712 is not phenotypically different from the virulent strains on Hela cells. attenuation in vitro. This makes it difficult to study the mechanism of However, it was shown recently that P2/Sabin is temperature sensitive (ts) above 38°C in Vero and BGM cell lines but not in the Hep-2 cells (Macadam et al., 1991b). Since P2/P712 does not replicate in PVR transgenic mouse muscles, it was of interest to examine its capacity to multiply in a primary 111 106 Log 10 PFU/MG 105 104 P2/MEF-1 (Tg) P2/MEF-1 (ntg) P2/P712 (Tg) 103 102 101 0 1 2 3 DAY POST-INFECTION 4 Figure 22. Time course of poliovirus replication in skeletal muscle. Transgenic (TgPVR, closed circles) or nontransgenic (nTg, open circles) mice were inoculated with 1 X 107 PFU of P2/MEF-1 or 7.5 X 107 PFU of P2/P712 (for transgenic mice, open diamond) in the left hamstring muscle. At the indicated times, the hamstring was removed, homogenized, and the virus titer was determined by plaque assay. Each point represents the mean of values obtained for three mice. 112 culture (MPMC) from transgenic mice. MPMC monolayers were infected with poliovirus P2/Lansing, P2/P712, and P2/Rom at MOI of 10 at both 37°C and 32°C. Infection by these viruses had different cytopathological effects (CPE) at different temperature (Figure 23). At 37°C, cells infected with P2/Lansing (Figure 23B) and P2/Rom (Figure 23F) developed over 90% CPE in 24 hours. Cells infected with P2/Rom developed a little less CPE than those infected with P2/Lansing at 37°C. Infection of P2/P712 (Figure 23A) did not result in significant CPE in the same period of time. Cells infected with P2/P712 developed over 50% CPE after 2 days. Often a significant number of resistant cells grew out in the ensuing days (data not shown). Similar differences between P2/P712 (Figure 23C) and P2/Lansing (Figure 23D) were observed at 32°C. Interestingly, cells infected with the neurovirulent strain P2/Rom (Figure 23E) developed CPE more slowly than those infected with P2/Lansing. When MPMC monolayers were infected with P1/Sabin (Figure 24A,C), the cells developed significant CPE at 32°C (Figure 24C) but not at 37°C (Figure 24A) after 24 hours. In contrast, cells infected with P1/Mahoney (Figure 5B, D) developed almost complete CPE at 37°C (Figure 24B) but not at 32°C (Figure 24D) in the same time period. Similar experiments were carried out with poliovirus type 3 strains and type 2 and type 3 attenuated/virulent recombinant viruses. The temperature sensitivity of poliovirus strains, as measured by the capacity of virus to cause CPE on MPMC monolayers, are summarized in Table 10. The viruses which are ts include P1/Sabin, P2/P712, P2/LP1, P3/Sabin, and S5'/L. The viruses which are cs include P1/Mahoney, P2/P712, P2/Rom, P2/SVL, P3/Leon, and S5'/L. SV3/L is slightly ts at 37°C in MPMC cells. Since the P2/P712/P2/Lansing recombinant virus P2/LP1, which has the 5'-ncr of P2/P712 in a P2/Lansing background, is ts, and P2/SVL, which encodes VP1 of P2/P712 in a P2/Lansing background, is cs, the determinant of the ts phenotype of P2/P712 should be in 113 Figure 23. Cytopathic changes in MPMC cells infected with type 2 polioviruses. MPMC monolayers were infected with P2/P712 (a and c), P2/Lansing (b and d), and P2/Rom (e and f) at 37°C (a, b, and f) and 32°C (c, d, and e). Photographs were taken at 24 hours post-infection. Magnified 155X. Figure 24. Cytopathic changes in MPMC cells infected with type 1 polioviruses. MPMC monolayers were infected with P1/Sabin (a and c), P1/Mahoney (b and d), at 37°C (a and b) and 32°C (c and d). Photographs were taken at 24 hours post-infection. Magnified 227X. 115 Table 10. The temperature sensitivity of poliovirus strains on MPMC monolayers phenotypes on MPMC viruses temperature sensitivea cold sensitiveb P1/Mahoney ts + cs P1/Sabin ts cs+ P2/Lansing ts + cs+ P2/Rom ts + cs P2/P712 ts cs P2/LP1 ts cs+ P2/SVL ts + cs P3/Leon ts + cs P3/Sabin ts cs+ S5'/Lc ts cs SV3/Ld ts + cs+ a. Virus infection caused (ts+) or did not (ts) cause significant CPE at 37°C in 24 hours. b. Virus infection caused (cs+) or did not (cs) cause significant CPE at 32°C in 24 hours. c. The type 3 recombinant virus with 5'-ncr of P3/Sabin in a P3/Leon background. d. The type 3 recombinant virus with VP3 of P3/Sabin in a P3/Leon background. 117 the 5'-ncr and the cs determinant in the capsid protein VP1. Similarly, the ts determinant of P3/Sabin maps in the 5'-ncr and the cs determinant of P3/Leon in the capsid protein VP3, where P3/Sabin differs from P3/Leon only at amino acid residue 91 (Phe in P3/Sabin and Ser in P3/Leon). 118 Chapter VIII. Discussion 119 Determinant of poliovirus host range in mice. Most poliovirus strains infect only primates. In this study, transgenic mice containing the human PVR gene in the germ line were established. The transgenic mice express PVR transcripts and poliovirus binding sites in a wide range of tissues. Inoculation of PVR transgenic mice with all three serotypes of poliovirus leads to development of a fatal paralytic disease that clinically and histopathologically resembles human poliomyelitis. Absence of PVR is therefore the determinant of poliovirus host range restriction in mice. Certain type 2 strains of poliovirus, such as P2/Lansing, have been adapted to infect mice and cause poliomyelitis after intracerebral inoculation (reviewed in (Racaniello, 1988). Replacement of the 8 amino acid sequence of the VP1 BC loop of P1/Mahoney, which infects only primates, with the corresponding sequences from P2/Lansing confers infectivity in mice (Martin et al., 1988; Murray et al., 1988). Recently two other host range determinants, located in the interior of the poliovirus capsid, have been identified (Moss and Racaniello, 1991). How these determinants extend the host range of the virus is not known. The fact that the human receptor can overcome the host range restriction of polioviruses indicates that they are normally blocked at a stage in virus-receptor interaction. This finding suggests that the viral host range determinants may confer infectivity of P1/Mahoney in mice by suppressing the failure of the virus to either bind to a mouse receptor or undergo receptor mediated conformational transition which is required for entry. Several other human virus receptor cDNAs have been molecularly cloned and characterized, including the HIV-1 receptor CD4 (Dalgleish et al., 1984; Klatzman et al., 1984; Maddon et al., 1986), the major rhinovirus group receptor ICAM-1 (Greve et al., 1989; Staunton et al., 1989), and the Epstein-Barr virus receptor CR-2 (Moore et al., 1987). CD4 and ICAM-1 are also members of the immunoglobulin superfamily of proteins. Expression of CD4 is thought to be a major determinant of HIV-1 tissue 120 tropism (Maddon et al., 1986). CD4 negative human cells, which are resistant to infection by HIV-1, can be rendered susceptible by transfection with cDNA clones encoding the CD4. Expression of ICAM-1 or CD4 in rodent cells, however, is not sufficient to render these cells susceptible to rhinovirus or HIV-1 infection, respectively, due to a block at the level of entry (Greve et al., 1989; Maddon et al., 1986). CR-2 has been shown to be a determinant of Epstein-Barr virus host range in vitro (Ahearn et al., 1988). The cell receptor for poliovirus is the first virus receptor proven to be a host range determinant in animals. PVR gene expression in transgenic mice. The PVR is a novel member of the immunoglobulin superfamily of proteins (Mendelsohn et al., 1989). Many immunoglobulin superfamily members mediate functions involving cellular recognition and adhesion (Williams and Barclay, 1988). PVR transgenic mice show no obvious phenotypes other than susceptibility to poliovirus infection, indicating that expression of the PVR in mice is not deleterious. Mice contain a homolog of the PVR that is expressed in many tissues (M. Morrison and V.R.R., unpublished). Apparently expression of the human PVR gene in mice does not interfere with the function of the endogenous gene product. Northern blot analysis showed that PVR transcripts are expressed in a wide range of transgenic mouse tissues. The expression pattern of PVR RNA is similar in 5 different transgenic mouse lines examined. A similar pattern is observed in humans (Mendelsohn et al., 1989), indicating that the PVR gene used to establish the transgenic lines contains sequence elements necessary for PVR expression. In addition, the multiple spliced forms of PVR mRNAs observed in human tissues are also present in transgenic mouse tissues. Since the overall pattern of RNA expression in PRG1 and PRG3-containing transgenic mice was similar, the poliovirus receptor gene and the cis-acting element(s) that control this pattern must be located within the 26 kb region of overlap between the two genomic clones. 121 The results of in situ hybridization of tissues from adult and embryonic transgenic, TgPVR1-17 (contains 10 copies of PRG1 transgene), mice show that the expression pattern of PVR RNA in the transgenic mouse mimics that in humans. A similar pattern was observed in TgPVR3-6 (contains 4 copies of PRG3 transgene) transgenic mouse tissues. A significant difference, however, is the expression of PVR RNA in the intestine. Both adult and fetal human intestinal epithelia accumulate high levels of PVR RNA, whereas PVR RNA is present at only low levels in both adult and fetal transgenic mouse intestine. The low expression of PVR RNA in transgenic mouse intestine may result from the absence of positive cis-acting regulatory element(s) in the PVR gene used to establish transgenic lines, or the presence of tissue-specific negative trans-acting factor(s) for regulatory elements shared between the PVR gene and mouse intestine epithelial cells. Study of the expression of the murine cognate of the human PVR may address these possibilities. It was not possible to use polyclonal antibody against PVR to study the expression of PVR protein, since the antibody cross reacts with mouse proteins as well (Freistadt et al., 1990). Expression of PVR protein was studied by a poliovirus binding assay. Results presented here show that poliovirus binding sites are expressed in a wide range of transgenic mouse tissues. This finding correlates with the wide range of expression of PVR proteins in human tissues (Freistadt et al., 1990). The finding that poliovirus binding sites are widely expressed in PVR transgenic mouse tissues, however, differs from observations with primate tissues. There are several possible explanations for this difference. Our preparation of tissue homogenates and assay conditions may differ significantly from those previously used (Holland, 1961). The receptor protein in nonsusceptible human tissues may be masked or shielded by its natural ligand, preventing binding of virus; in transgenic mice the receptor protein might either be expressed in excess or may not interact with an endogenous ligand, permitting poliovirus binding. Alternatively, there may be small numbers of cells in non-susceptible primate tissues that 122 express active poliovirus binding sites which are difficult to detect. For example, poliovirus binding activity has been irregularly detected in primate liver (Holland, 1961), where poliovirus replication is not observed. PVR gene expression in humans. The PVR is a novel member of the immunoglobulin superfamily of proteins (Mendelsohn et al., 1989). The normal function and ligand of PVR are not known. The study of PVR gene expression presented here provides a basis for understanding the normal function of PVR and the location of the ligand. PVR gene expression in the adult and the embryo. PVR RNA is expressed in the glomerulus in human adult kidney but not in embryonic kidney before the renal corpuscle is formed. The nephrons are blind-ended tubules consisting of a single layer of epithelium before the renal corpuscle is formed. Subsequently the ends of the tubules dilate and become invaginated by a tiny mass of tissue which differentiates to form the glomerulus. The layer of invaginated epithelium differentiates into podocytes which become closely applied to the surface of the knot of glomerular capillaries. A small amount of connective tissue remains to support the capillary loops and differentiates to form the mesangium. In PVR transgenic mice, high levels of PVR RNA appear in podocytes in fetal kidney. High levels of PVR RNA also appear in the parietal layer of Bowman's capsule in adult kidney. In human adult kidney high levels of PVR RNA appear to be expressed in podocytes, although the results did not rule out the possibility that PVR RNA is expressed in the mesangial cells. The exact cell type needs to be identified by specific antibodies. Nevertheless, expression of the PVR gene appears to occur during differentiation of the renal corpuscle in which renal tubular epithelial cells, masangial cells, and endothelial cells of the glomerular capillary interact with each other. PVR, may therefore function in formation of the renal corpuscle, and its ligand may be on these cells. Further study of PVR expression during human development is required to confirm this hypothesis. 123 High levels of PVR RNA accumulate in both adult and fetal human intestinal epithelia. The expression of PVR RNA in fetal intestinal epithelia is consistent with the finding that human fetal intestine has poliovirus binding activity (Holland, 1961), suggesting that PVR protein is also expressed in intestinal epithelia. However, the epithelia of the fetal stomach express much less PVR RNA. The functional basis of differential expression of PVR RNA in different parts of the gastrointestinal tract is not known. New intestinal epithelial cells in crypts of Lieberkuhn progress up the villi along the lamina propria, and cells are continually shed from the tip of the villi. Considering the possible role of PVR in kidney and placenta (see below), a similar scenario may occur in the intestine, where PVR may be required for cell-cell communication or "rolling" along the lamina propria, a process mimicing lekocyte-endothelial cell recognition (Butcher, 1991). The PVR in the CNS and peripheral ganglia and other tissues may serve similar functions. The molecular basis of poliovirus tissue tropism and the sites of poliovirus replication in the human intestine are not known. That PVR is expressed in embryonic intestinal epithelia has shed some light on these questions. It was believed that expression of poliovirus binding sites determines tissue tropism (Holland, 1961). According to this "rule", intestinal epithelia should be susceptible to poliovirus infection. If poliovirus infects only Peyer's patches (Bodian, 1955), this would indicate that poliovirus tissue tropism is not governed solely by expression of the poliovirus binding site (see section on poliovirus tissue tropism in this chapter). PVR gene expression in extravillous trophoblast of placenta. The implanting blastocyst contains a trophoblastic shell, a particular syncytiotrophoblastic layer. The trophoblastic shell of the blastocyst actively invades the endometrial stroma containing capillaries and glands at implantation, and the blastocyst slowly sinks into the endometrium. The decidual cells degenerate in the region of the penetrating syncytiotrophoblast. During implantation the trophoblastic shell consists only of 124 syncytiotrophoblast. Subsequently, the cytotrophoblast reaches the shell and splits the syncytiotrophoblast into an apical layer that faces the intervillous space and a basal layer which contacts the maternal tissues. The latter becomes incomplete and may invade deeply into the endometrium and myometrium. With human implantation, the syncytiotrophoblastic cells invading into the maternal tissues appear as multinucleated trophoblast giant cells (Boyd and Hamilton, 1970), although the latter may also be derived from invading cytotrophoblast that later fused (Pijnenborg et al., 1981). These multinucleated trophoblast giant cells accumulate high levels of PVR RNA. It is not known at present if the early trophoblastic shell expresses PVR RNA. As the cytotrophoblastic cells come into contact with the endometrium, they invade the stroma as single cells or in small groups. On day 22 post-conception, the term "trophoblastic shell" is usually replaced by the term "basal plate", which is composed of various tissues during development, such as extravillous trophoblast, endometrial stroma with its pregnancy-specific specialization, fibrinoid, residues of degenerating villi, and maternal vessels. The majority of the cytotrophoblast forms the villous cytotrophoblast, which is the inner layer of the villous. The villous cytotrophoblast and syncytiotrophoblast do not express PVR RNA. The remaining cytotrophoblastic cells differentiate into extravillous trophoblastic cells, these cells accumulate high levels of PVR RNA. The nomenclature for the trophoblast residing outside the villi is confusing (Benirschke and Kaufmann, 1990). The term "intermediate trophoblast" describes a distinctive form of trophoblastic cells with specific morphological, biochemical and functional features (Kurman et al., 1984). Intermediate trophoblast cells are mononuclear and located in overlying the chorionic villi, in the trophoblastic columns, basal plate and the trophoblastic shell. Accordingly, the mononuclear PVR RNA expressing cells can be identified as intermediate trophoblasts. One of the primary functions of these cells is in implantation, and in the establishment of the uteroplacental circulation, since it extensively invades the spiral arteries at the placental site (Kurman et al., 1984). 125 Expression of PVR RNA in the invasive trophoblastic cells is consistent with a possible role of PVR in cell adhesion and/or cell-cell communication as a member of immunoglobulin superfamily. Although it is not known if PVR protein is expressed in these trophoblatic cells, finding this would shed some light on the function of PVR and the role of PVR in implantation. Moreover, since the classification and differentiation of the placental trophoblast is not completely understood, PVR might be used as a cell marker for trophoblast differentiation. Maternal decidual cells express low levels of PVR RNA. It is not known if this is due to pregnancy-specific specialization of the endometrium. The full significance of decidual cells is not understood, but it has been suggested that they may provide some nourishment for the embryo and protect the maternal tissues against uncontrolled invasion by the trophoblast (Ramsey, 1965). It is also not known what alternatively spliced form of PVR is expressed in the decidual cells, e.g. the membrane bound form, the secreted form or both. It would be interesting to study the role of PVR in the interaction between trophoblastic cells and decidual cells. Poliovirus tissue tropism. Although PVR expression was widespread, when transgenic mice were inoculated with poliovirus, viral replication was limited to skeletal muscle, neurons in the central nervous system, and to a lesser extent in brown adipose tissue, peripheral ganglia, and nasal mucosa. Poliovirus RNA was detected in adult mouse skeletal muscle cells of the hamstring after intramuscular inoculation. In addition, poliovirus replicates extensively in skeletal muscle of suckling transgenic mice after oral administration of virus. Replication of poliovirus has also been demonstrated in monkey skeletal muscle after intramuscular inoculation (Wenner and Kamitsuka, 1957). Poliovirus RNA was detected in all neurons of the spinal cord and in most neurons in the brain stem. In the brain, infected neurons were detected in several areas, including the cerebral cortex, pyramidal layer of the hippocampus, olfactory bulb, thalamus, hypothalamus and deep cerebellar nuclei. In 126 suckling transgenic mice, poliovirus replication in Purkinje cells in the cerebellum, and neurons in peripheral ganglia was also observed. Poliovirus replication in brown adipose tissue and renal mucosa can be occasionally detected. Poliovirus replication was not detected in kidney, adrenal gland, thymus or intestine. Therefore, susceptibility of cells to poliovirus appears to correlate with PVR RNA expression in the CNS, muscle, brown adipose tissues, and renal mucosa but not in other tissues. There are several possible explanations for the failure of poliovirus to replicate in transgenic mouse tissues that express PVR. It is unlikely that the PVR transcripts detected by in situ hybridization are not translated. Organ homogenates from PVR transgenic mice contain poliovirus binding activity, and freshly dispersed transgenic mouse kidney cells express PVR on the cell surface, as shown by their ability to bind poliovirus. Virus may be unable to reach some cells which express PVR RNA, such as developing T-lymphocytes of the thymus and epithelial cells of Bowman's capsule. However, tubular epithelial cells in the kidney and endocrine cells in the adrenal cortex should be exposed to circulating viruses. Poliovirus replication was not detected in these cells, indicating that poliovirus tissue tropism is not governed solely by expression of the PVR gene or by accessibility of cells to virus. Alternative splicing of PVR transcripts might control susceptibility of tissues to poliovirus infection. Human tissues contain both membrane-bound and secreted PVR isoforms, that are generated by alternative splicing of PVR mRNA (Koike et al., 1990). Expression of secreted PVRs in transgenic mouse tissues might result in neutralization of poliovirus infectivity (Kaplan et al., 1990). The results presented here, together with observations made with another PVR transgenic mouse line (Koike et al., 1991), reveal that RNAs encoding both membrane-bound and secreted PVRs are present in susceptible and nonsusceptible tissues, as is found in human tissues (Koike et al., 1990). These results, together with our observation that dispersed kidney cells in culture, from which soluble PVR would have been removed by washing, are still resistant to poliovirus 127 infection, support the idea that secreted PVRs are not likely to restrict poliovirus in vivo. Alternatively, virus binding to nonsusceptible tissues might be blocked in vivo by the natural ligand of the PVR, or poliovirus entry might require factors in addition to the PVR that are lacking in nonsusceptible tissues. For example, expression of human CD4 in rodent cells is not sufficient to render these cells susceptible to HIV-1 infection, due to a block at the level of entry (Maddon et al., 1986). Poliovirus replication in nonsusceptible tissues might be controlled at stages beyond virus entry, such as translation, replication or assembly. This possibility has been generally discounted in the past, as inoculation of viral RNA intracerebrally into rabbits, chicks, guinea pigs, and hamsters results in one cycle of replication and production of infectious virus (Holland et al., 1959b). However, these experiments indicate only that poliovirus host range restriction (species tropism) is determined at the level of entry in neural cells. Whether or not there is an internal block to poliovirus infection in nonsusceptible tissues remains unresolved. Although poliovirus infection in primates is restricted, cells from almost any tissue develop susceptibility to infection after cultivation in vitro (Enders et al., 1949; Holland, 1961; Kaplan, 1955). PVR transgenic mouse kidney cells express poliovirus binding sites but are initially resistant to poliovirus infection. When cultured in vitro, kidney cells develop susceptibility to poliovirus infection after 24 hours. The basis of the acquired susceptibility to poliovirus infection in these cells is not known, but might involve the induction of factors required for virus entry or replication. Study of the changes that occur in cultured PVR transgenic mouse kidney cells that permit poliovirus infection would provide information on the block to poliovirus infection in these cells, and might reveal the mechanism of poliovirus tissue tropism in primates. An important question is whether studying poliovirus tropism in PVR transgenic mice provides information on tropism of the virus in primates. The pattern of expression of many human genes in transgenic mice is often similar to that observed in humans 128 (reviewed in (Palmiter, 1986). Indeed, PVR gene expression in transgenic mice generally mimics that in humans. One difference, however, is that poliovirus binding sites are expressed in all PVR transgenic mouse tissues examined, while binding sites in humans have been detected largely in neural tissues and intestine and occasionally in kidney and liver (Holland, 1961; Kunin and Jordan, 1961). It is possible that the basis of poliovirus tropism in humans differs from that in PVR transgenic mice and is determined by factors that control the ability of the receptor to bind virus. Alternatively, poliovirus binding sites might be expressed in all human tissues, but at low levels or in unstable forms, making their detection difficult in some tissues. Consistent with this idea, poliovirus binding activity can be detected in human fetal liver, which is shown to express PVR RNA in this study. The PVR transgenic mice studied to date contain multiple copies of the PVR gene and express high levels of PVR mRNA, perhaps facilitating detection of binding sites in all organs. Resolution of this question awaits development of more sensitive assays to detect poliovirus binding sites in human tissues. Histopathology of experimental poliomyelitis in PVR transgenic mice. The histopathology of experimental poliomyelitis in the primate is well known (Hurst, 1929). The infection in monkeys has generally been considered to accurately reflect the microscopic features of the disease in humans. The motor neurons of the ventral (anterior) horns of the cervical and lumbar intumescences are the most sensitive to the virus, followed by neurons in motor nuclei of cranial nerves in the brain stem. Infection of PVR transgenic mice resulted in a paralytic disease closely resembling human and nonhuman primate polio. The lesions in these mice occurred, for the most part, in the expected poliovirus target sites of the spinal cord and brain stem. This included spinal cord ventral horns, vestibular nuclei, deep cerebellar nuclei (dentate, interpositus and fastigial nuclei), red nuclei, oculomotor nuclei and other areas in the midbrain and pontomedullary tegmentum, and the hypothalamus. The infection resulted in acute neuronal necrosis in the spinal cord associated with mild to moderate inflammation, whereas 129 inflammation was the principal change seen in the brain stem with necrosis of neurons being much less obvious. This type and distribution of change is also seen in acute poliomyelitis of primates (Hurst, 1929). An additional significant microscopic change seen in the PVR transgenic animals was inflammation and focal neuronal necrosis of the hippocampus. The sites of poliovirus replication in the CNS of PVR transgenic mice closely parallel those observed in primates. As in primates, the motor neurons of the ventral horns of the cervical and lumbar intumescences are the most sensitive to the virus, followed by neurons in motor nuclei of cranial nerves in the brain stem. One difference in transgenic mice is that poliovirus replicates in the hippocampus of PVR transgenic mice. This result provides an explanation for the observed pathological changes. It is not clear why involvement of the hippocampus is not observed in primates. Indeed, it is interesting that in both transgenic mice and in primates, poliovirus replication is limited to specific areas of the brain. Intracerebral inoculation of PVR transgenic mice resulted in viral replication at brain sites not observed after intraperitoneal inoculation, such as the olfactory bulb. These results suggest that the restricted movement of virus along certain nerve fiber pathways, and the progression of the disease, may determine which neurons become infected. In the case of intraperitoneal inoculation, virus may enter the spinal cord, and mice develop paralysis or die due to destruction of neurons in the brain stem before virus spreads to brain neurons. However, the progression of the disease is not likely to be the only determinant of tropism, since even after intracerebral inoculation, replication was not observed in all brain neurons and extensive viral replication was still observed in the spinal cord and brain stem. Other determinants of neurotropism might include susceptibility of specific cell types to infection, or physical restrictions to virus spread within the brain. In this study, poliovirus replication was detected in neurons in both the ventral and dorsal horns of the spinal cord. However, inflammation and neuronal degeneration 130 was localized largely to the ventral horns, suggesting that neurons in the dorsal horn were either infected at a late stage of the disease, or that poliovirus replication in these neurons does not lead to cell destruction. Poliovirus histopathology is observed in the posterior horn (analogous to the dorsal horn of mice) in human and monkey spinal cord, although less frequently than in the anterior horn (Bodian, 1959). Poliovirus pathogenesis. The primary replication sites of poliovirus in the alimentary tract are still an unsolved question. High levels of PVR RNA accumulate in the human intestinal epithelia, suggesting that the intestinal epithelia is a primary site of poliovirus multiplication. The entire epithelial lining of the intestine is replaced every 3-5 days by the continual shedding of cells from the tips of the villi into the lumen. This fact may explain the failure to find significant pathological lesions in the alimentary tract of primates (Bodian and Horstmann, 1965; Sabin, 1956). Viral replication in lymphoid tissues has been a subject of controversy (Bodian and Horstmann, 1965; Sabin, 1956). High concentrations of virus are present in sections of intestine containing the Peyer's patches (Bodian, 1955). Virus can be isolated from tonsilopharyngeal tissue and lymph nodes of humans and chimpanzees (Sabin and Ward, 1941; Wenner and Rabe, 1951). On the other hand, virus multiplication was found to be as extensive in the throats of the human volunteers without tonsils or adenoids as in those who still had these tissues (Sabin, 1956). In the present study, viral replication was not observed in lymphoid tissues, suggesting that lymphocytes are not susceptible to poliovirus infection. The presence of virus in lymphoid tissues of primates may reflect viral replication in these tissues or the absorption of virus into the regional lymphoid tissues after replication in epithelial cells. Resolution of this question awaits examination of poliovirus infection in primates by in situ hybridization. Studies on reoviruses have shown that virus enters the host from the intestinal lumen through M cells overlying ileal Peyer's patches, and undergoes primary replication 131 in mononuclear cells and in neurons of the adjacent myenteric plexus (Morrison et al., 1991; Wolf et al., 1981). Poliovirus was also shown to adhere to, and be endocytosed by intestinal epithelial M cells with a low efficiency (Sicinski et al., 1990). The exact mode of initial poliovirus infection needs further investigatation. Replication of P1/Mahoney, however, was not detected in the transgenic mouse intestine . It was noted that the alimentary tract of the monkey is less susceptible to poliovirus infection than that of chimpanzees and humans (Sabin, 1956). Certain species of monkeys are not susceptible to oral infection of poliovirus (Hashimoto et al., 1984). The alimentary tract of the mouse is expected to be more insensitive to poliovirus infection according to this evolutionary hierarchy of the sensitivity of alimentary tract to infection with poliovirus. In addition, high levels of PVR RNA were not detected in PVR transgenic mouse intestine. However, replication of P1/Mahoney in the intestine of another line of PVR transgenic mice was detected (Koike, personal communication). This transgenic line was derived by microinjection of a PVR genomic DNA into CD1 mouse zygotes (Koike et al., 1991). The LD50 of P1/Mahoney in this line of transgenic mice is about 1 X 102 by intracerebral inoculation (Koike et al., 1991) and 1 X 107 by peroral inoculation (Koike, personal communication), whereas the LD50 of P1/Mahoney in the PVR transgenic mice used in this study is about 1 X 105 by intracerebral inoculation. It is possible that the differences between these two transgenic mouse lines are due to different mouse strains (CD1 compared with (C57BL6/J X CBA/J) F2 mice) used to generate transgenic lines. Alternatively, the P1/Mahoney strain used in the experiments reported here may be slightly attenuated. It will be interesting to examine the susceptibility of transgenic mouse alimentary tract to poliovirus infection using highly neurovirulent strains. Although viral replication in the PVR transgenic mouse intestine was not detected, transgenic mice developed paralytic disease after oral administration of virus. This phenomenon was also observed in a proportion of monkeys infected orally with 132 poliovirus (Sabin, 1956). The paralytic disease may result from infection by olfactory pathways, since viral replication was detected in nasal mucosa and the olfactory bulb in suckling transgenic mice, or from spreading of virus into the body through inoculation trauma. It is believed that viral replication in extraneural tissues results in maintenance of the persistent viremia which is required for viral invasion of the CNS. In transgenic mice poliovirus replication was detected in skeletal muscle, brown adipose tissues, and nasal mucosa. Poliovirus replicates extensively in skeletal muscle of suckling transgenic mice after oral administration of virus, whereas replication in brown adipose tissues and nasal mucosa was detected to a much lesser extent. The basis for the different extent of viral replication in these tissues is not known, but might include tissue specific sensitivity or accessibility to poliovirus infection. The mechanism by which poliovirus enters the CNS from the blood is of great interest. Two possibilities have been suggested which are not mutually exclusive: the virus might enter the CNS from blood by crossing the blood-brain barrier (BBB), or enter the neuromuscular junction and spreads via nerve fibers to the CNS. Virus antigen has been detected by immunofluorescence in vascular endothelial cells of monkeys infected with poliovirus (Blinzinger et al., 1969; Kanamitsu et al., 1967), and PVR has been detected in a small percentage of freshly dispersed endothelial cells (Couderc et al., 1990). These observations suggest that poliovirus may use receptors on endothelial cells to gain access to the CNS from capillaries. However, neither PVR expression nor virus replication was detected in endothelial cells in PVR transgenic mice. Poliovirus spread in transgenic mice is therefore not likely to involve receptors on endothelial cells. The presence of poliovirus RNA in axonal and dendritic processes of neurons suggests that virus could spread though neural pathways. The study of viral spread following intramuscular or intrafootpad injection in PVR transgenic mice demonstrates that poliovirus enters the CNS through peripheral nerves. 133 This conclusion is based on three experimental observations: first, after intramuscular inoculation, the first limb paralyzed was always the limb that was inoculated; second, following inoculation of virus into the hindlimb, virus was first detected in the lower spinal cord; and third, sciatic nerve transection blocked poliovirus infection after footpad inoculation. A possible scenario for the spread of poliovirus through peripheral nerves is shown in Figure 25. Following intramuscular or intrafootpad inoculation, virus replicates in muscle cells, binds to poliovirus receptors at the neural-muscular junction and enters the 2° motor axon. Virus then spreads, by retrograde axonal transport, to the 2° motor neuron in the spinal cord. Replication in the neuron cell body results in cell destruction and release of new virus particles, which infect neighboring neurons. When sufficient numbers of neurons are destroyed, paralysis of the innervated limb results. Our results suggest that virus may also spread to upper levels of the spinal cord and the brain, and this spread, which may occur through neural pathways, may lead to paralysis of other limbs and death. At present, it is not known if PVR is expressed on the surface of the synapses, although it has been reported that human synaptosomes contain poliovirus binding sites (Brown et al., 1987). Localization of initial paralysis by P2/Lansing depends on expression of PVR in mice, suggesting that PVR might be expressed at the synapse. 134 Figure 25. Possible route of poliovirus spread from muscle to the CNS in mice. An enlargement of the leg is shown at the left. Virus inoculated intramuscularly may enter the secondary motor axon via PVR at the synaptic cleft. Virus then spreads, by retrograde axonal transport, to the 2° motor neuron in the spinal cord. Once in the spinal cord, virus replicates in neurons, causing cell lysis and release of virus which spreads laterally to other neurons, and caudally to primary motor neuron cell bodies. 135 The finding that poliovirus spreads to the CNS through peripheral nerves in transgenic mice agrees with observations made in humans and monkeys. The best evidence for neural spread in humans comes from the Cutter incident of 1955, in which children developed poliomyelitis after administration of incompletely inactivated poliovaccine (Nathanson and Langmuir, 1963). Of 65 vaccinees who developed paralysis, 71% had initial paralysis in the inoculated limb. Experiments in monkeys showed that poliovirus replicates in muscle after intramuscular inoculation (Wenner and Kamitsuka, 1957). Blocking the sciatic nerve by freezing prevented CNS invasion by the neurotropic poliovirus P2/MV in monkeys (Nathanson and Bodian, 1961). In contrast to our findings in mice, sciatic nerve block did not prevent the spread of the pantropic P1/Mahoney strain in monkeys (Nathanson and Bodian, 1961). This difference may be due to the use of different levels of inocula in different animal models. For example, when a small amount of virus is injected into a limb, most of the virus will initially remain, replicate in situ and enter the peripheral nerve. In this case, localization of initial paralysis is expected irrespective of the animal model. However, the effect of a large inoculum will vary according to the animal model. A large inoculum will produce an immediate viremia, which will carry virus to all parts of the body. Although virus at the initial site will still travel to the CNS along nerve pathways, virus at other sites, some of which may have shorter nerve pathways to the CNS, will replicate almost simultaneously (Wyatt, 1990). In large animals such as monkeys, virus require more time to reach the CNS, and therefore virus entering nerves with shorter paths to the CNS may spread to the CNS before virus which enter the CNS along nerves with longer paths. As a result, when monkeys are inoculated with high levels of virus, a lower frequency of initial localization of paralysis would be predicted. In mice, the differences in transmission time from different nerve pathways are not as significant as in large animals. Since the injected limb will always receive a higher initial concentration of virus than other sites, more virus will enter the CNS from the inoculation point than from secondary sites, and the net 137 result in mice would still be localization of initial paralysis to the injected limb. Because of these considerations, failure to observe protection after sciatic nerve transection is not a conclusive result. For example, when large amounts of virus are inoculated, sciatic nerve transection does not protect against disease in TgPVR mice (data not shown). Based on our studies of poliovirus pathogenesis in transgenic mice, combined with observations on the disease in humans, chimpanzees, and monkeys (Bodian, 1955; Sabin, 1956), we suggest a hypothetical scheme for the pathogenesis of the disease in humans (Figure 26). Ingested poliovirus initially replicates in the alimentary tract, possibly in epithelial cells lining the alimentary tract. Replication leads to release of virus into the throat and gut lumen and establishment of viremia. Disseminated virus then replicates in skeletal muscle cells, enters peripheral nerves and spreads to the CNS. Virus replication in skeletal muscle maintains persisting viremia, which may disseminate infection to multiple sites from which virus may also enter the CNS. Poliovirus replication was also detected in brown adipose tissues and neurons in peripheral ganglia to a lesser extent in suckling transgenic mice after oral administration of virus. This finding is consistent with observations made in monkeys (Bodian, 1955; Faber, 1956). Brown adipose tissue might be another extraneural tissue that supports poliovirus replication and maintains the persisting viremia in human. Transmission of virus along nerve fibers from peripheral ganglia may provide an additional route for entry into the CNS. However, because poliovirus replicates more extensively in skeletal muscles, viral replication 138 in the ingested virus oropharyngeal mucosa intestinal mucosa virus in throat ? virus in feces ? ? CNS BLOOD central nervous system axonal transport skeletal muscle Figure 26. Possible scheme of poliovirus pathogenesis in humans. This figure is based on previous observations made in humans and monkeys and the results reported here in TgPVR mice. Dotted line, other possible routes of virus spread. 139 muscle and subsequent spreading along nerves innervating the muscle to the CNS may still be the major route for virus entry into the CNS. There are other pathways which poliovirus may use to gain entry to the CNS. It was demonstrated that reovirus can spread directly from the intestinal lumen to the CNS through vagal autonomic nerve fibers (Morrison et al., 1991). Because the initial site of poliovirus replication is the alimentary tract (Sabin, 1956), it is possible that poliovirus may spread via a similar pathway in humans. Indeed, the observation that 5-30% of poliovirus infections involve the brain stem is consistent with this route of spread. Despite these considerations, in the majority of paralytic infections in humans, virus appears to initially infect the lower motor neurons of the spinal cord, which is consistent with the hypothesis that poliovirus spreads from the muscle to the CNS. The persisting viremia that precedes paralytic infection is important for virus spread to the CNS (Bodian and Horstmann, 1965). This observation has been used as evidence in support of the hypothesis that virus enters the CNS from the blood. However, persisting viremia may be a result of successful viral replication in skeletal muscle, which leads to release of virus into the blood and virus spreading to the CNS. Factors which increase access of poliovirus to muscle cells and nerve terminals would therefore be expected to increase the incidence of poliomyelitis. Consistent with this idea are the observations that in many cases with initial lumbar or cervical involvement there appears to be a temporal association with recent vigorous exertion or injury of the lower or upper limbs, and bulbar poliomyelitis often develops in patients with previous tonsillectomy (Bodian and Horstmann, 1965). The observation that passive or active immunization against poliovirus terminates viremia and prevents CNS infection has been considered strong evidence that poliovirus enters the CNS through the BBB (Bodian and Horstmann, 1965; Melnick, 1985). Studies on the pathogenesis of reovirus serotype 3 in mice indicate that virus spreads by nerves and not by the bloodstream to the CNS, despite the presence of viremia (Flamand et al., 140 1991; Tyler et al., 1986). Anti-viral antibody decreases viremia and prevents appearance of virus in the CNS after inoculation of virus in the hindlimb footpad (Tyler et al., 1989). Recently it was shown that antibody can mediate clearance of alphavirus infection from neurons by restricting viral gene expression (Levine et al., 1991). These studies demonstrate that blocking virus entry to the CNS by the BBB is not the only mechanism by which antibody prevents CNS infection. The mechanism by which anti-poliovirus antibody prevents CNS infection clearly requires further study. Attenuating determinants of a vaccine-related type 2 poliovirus. Using a strategy of constructing recombinants between the poliovirus vaccinerelated strain P2/P712 and the neurovirulent P2/Lansing, two regions from P2/P712 that attenuate neurovirulence in mice were previously identified: the 5' noncoding region and a central region of the genome (Moss et al., 1989). All other regions of the P2/P712 genome cause little or no attenuation. In this study, the attenuation determinant in the central region was mapped to capsid protein VP1. Candidate nucleotides involved in attenuation were then identified by comparing the sequences of the 5'-ncr and VP1 of P2/P712 with those of the neurovirulent strain P2/117 (Pollard et al., 1989). By changing residues in the attenuated recombinants to the residues that occur in virulent viruses, it has been possible to identify nucleotide A-481 in the 5' noncoding region and ile-143 of capsid protein VP1 as the major attenuation determinants in P2/P712. Reduced poliovirus neurovirulence in mice may be the result of general attenuation determinants, such as those that also attenuate poliovirus in primates (La Monica et al., 1987a), or due to host range restriction, which specifically prevents P1/Mahoney from causing disease in mice (La Monica et al., 1986). It is possible to bypass potential host range restriction by testing viruses for virulence in transgenic mice that express human poliovirus receptors. All polioviruses tested to date that are virulent in primates are also virulent in PVR transgenic mice, regardless of their ability to cause paralysis in normal mice. This indicates that interaction with the mouse poliovirus receptor is the primary 141 determinant of host range. The three Sabin vaccine strains do not cause disease in transgenic mice, indicating that the attenuation determinants of these viruses function in this animal model. The two viruses described here that carry the attenuating regions from P2/P712, SVL and LP1, are also attenuated in PVR transgenic mice. Therefore, A-481 and ile-143 of P2/P712 are primarily general attenuation determinants. It is interesting to note that SVL is approximately 100-fold more neurovirulent in transgenic mice compared to nontransgenic mice, while SVL-val and SVL-thr are 10-fold more neurovirulent in transgenic mice. The difference in neurovirulence in transgenic mice compared with normal mice is probably in part due to the 17 amino acid differences between P2/P712 and P2/Lansing in VP1, which probably result in host range restriction. However, it is interesting that the difference in LD50 values is greater for SVL than SVLval and SVL-thr. Although this difference is probably too close to draw conclusions, the result suggests that ile-143 might also impart host range restriction. To test this possibility, it will be necessary to introduce ile-143 into P2/Lansing, and determine the neurovirulence of the virus in normal and transgenic mice.. Although A-481 and ile-143 account for most of the attenuation phenotype of P2/P712, changes at these positions to the sequences found in the virulent P2/117 did not fully restore neurovirulence. While LP481 and SLL481 are able to cause paralysis, they are 100-fold less virulent than P2/Lansing. In addition 117LP, which carries the 5' noncoding region of P2/117, is 10-fold more virulent than LP481. Therefore, either A685, or an interaction among U-437, A-481 and A-685 in a RNA secondary structure, contributes partially to the attenuation of P2/P712. Because SLL437 carries the major attenuation determinant A-481, it is difficult to assess the minor contribution of U-437 to attenuation. It remains unclear why 117LP is about 10-fold less virulent than P2/Lansing. It is possible that P2/117 still contains additional weak attenuation determinants in the 5'ncr. Studies of other vaccine-associated type 2 isolates suggest that nucleotide 398 might 142 also be a weak determinant of attenuation (Agol, 1990), but in P2/117 this nucleotide is identical to that in P2/Sabin. All three Sabin vaccine strains contain strong attenuation determinants in the 5' noncoding region of the viral genome. A mutation from C to U at position 472 is partly responsible for attenuation of the P3/Sabin strain (Westrop et al., 1987), and neurovirulent revertants of P3/Sabin have mutated to C at this position (Almond et al., 1984; Cann et al., 1984). An A to G change at position 480 partially accounts for the attenuation phenotype of P1/Sabin (Nomoto and Wimmer, 1987). Based on sequence changes occurring upon passage of P2/Sabin in the human gut, it was suggested that a base change from A to G at position 481 may accompany acquisition of neurovirulence (Minor and Dunn, 1988). A neurovirulent revertant of P2/Sabin, P2/117, has three base changes in the 5' noncoding region, including an A to G change at base 481 (Pollard et al., 1989). The fact that A-481 attenuates poliovirus P2/P712 in both normal and transgenic mice is consistent with the observation that 5'-ncr determinants that attenuate polioviruses in primates also attenuate these viruses in normal mice (La Monica et al., 1987a). The position of ile-143 in the structure of the poliovirus capsid may suggest the mechanism by which it attenuates neurovirulence. Amino acid residue 143 of VP1 is exposed on the external surface of the native virion very near the five-fold axis of icosahedral symmetry, in the loop connecting β-strands D and E (DE loop) of VP1 (Hogle et al., 1985). Five copies of the DE loop encircle the five-fold axis, and together with five HI and BC loops, form a prominent protrusion on the particle surface (Figure I2). A role for the DE loop in host range has been suggested by examination of the atomic structure of a poliovirus variant in which host range restriction has been overcome. Substitution of the BC loop of P1/Mahoney with that of P2/Lansing confers upon this chimeric virus the ability to infect mice (Martin et al., 1988; Murray et al., 1988). When the structure of the P2/Lansing-P1/Mahoney chimeric virus was solved by X-ray crystallography, in addition to the expected conformational changes in the heterologous 143 BC loop, significant conformational changes in the DE loop were observed although there are no amino acid sequence differences in this area (Yeates et al., 1991). This observation suggests that there may be structural interactions between the BC and the DE loops of VP1. This suggestion is supported by the fact that both the BC and DE loops of VP1 comprise a discontinuous neutralization antigenic site (Wiegers et al., 1989). The BC loop is believed to influence host range through receptor-mediated early events in infection (Moss and Racaniello, 1991). It is possible that the general attenuation caused by ile-143 is also receptor-mediated, but cannot be completely overcome by the presence of the human poliovirus receptor. Investigation of the interactions between SVL, SVL-thr and SVL-val with the human receptor in vitro should resolve these questions. The only other structural determinant of attenuation identified in a vaccine strain is VP3-phe-91 in P3/Sabin. This determinant has been extensively characterized with regard to its effect on the temperature-sensitivity of the P3/Sabin strain, and is found to have an assembly defect in the infectious cycle (Macadam et al., 1991a). P2/P712 and P2/Sabin are highly related in nucleotide sequence and resemble each other phenotypically (Moss et al., 1989). Nucleotide sequences of two variants of P2/Sabin have been determined. P2/P712 differs from P2/Sabin described by Toyoda et al (Toyoda et al., 1984) by 22 nucleotides and from P2/Sabin of Pollard et al (Pollard et al., 1989) by only two silent nucleotide changes. Both variants of P2/Sabin harbor the two attenuation determinants identified here. At least in the case of the P2/Sabin strain described by Pollard et al (Pollard et al., 1989) the nucleotide differences between it and P2/P712 are not likely to alter the attenuation affects of A-481 and ile-143 in that strain. Unless the differences suppress the effects of A-481 and VP1-ile-143, the two attenuation determinants identified in P2/P712 are also primary contributors to the attenuation of the Sabin type 2 vaccine strain. Molecular basis of poliovirus temperature sensitivity and attenuation. 144 In this study, polioviruses are shown to have different capacities to cause CPE on MPMC cells at different temperatures. The reproductive capacity of polioviruses, measured by one-step growth experiments, is currently being examined. P3/Leon was also shown to have a slightly cold sensitive phenotype in Hep-2 cells (Minor et al., 1989). This study shows that all naturally occurring poliovirus strains examined, including P1/Mahoney, P2/P712, P2/Rom, and P3/Leon, are cold sensitive. P2/Lansing is a mouse adapted strain and can not be considered as naturally occurring virus. Interestingly, the determinants of the cold sensitive phenotype in the type 2 and type 3 viruses map to capsid protein (VP1 in P2/P712 and VP3 in P3/Leon). This finding suggests that the growth of these viruses may be blocked at stages of disassembly and/or assembly of the virus at low temperature. It has been demonstrated that the attenuation mutation of P3/Sabin in VP3 at amino acid residue 91 (VP3-091) is the major ts determinant which imposes a block to virus assembly (Macadam et al., 1991a). This ts determinant is also believed to act by controlling the structural transitions that virus must undergo at other points (e.g. virus entry) in the infectious cycle (Filman et al., 1989). The crystal structures of P3/Sabin and P1/Mahoney have been determined to high resolution (Filman et al., 1989; Hogle et al., 1985). The positions of VP3-091 and the ts suppressor mutations suggest a structural basis for the ts phenotype. In P1/Mahoney, and presumably in P3/Leon, the side chain of the Ser at VP3-091 points into a pocket where the serine hydroxyl-group hydrogen bonds with a buried water molecule. In P3/Sabin, however, the large aromatic side chain of the Phe residue points outward, making unfavorable contacts with the solvent (Filman et al., 1989). This situation might predispose the virion to thermal transitions, resulting in burial of the Phe residue such that the virus particle is rendered unstable or defective in some way. Suppressor mutations have been found in several locations (Filman et al., 1989; Minor et al., 1989), including the interface between fivefold related protomers, the hydrophoic pocket, and the seven-stranded beta sheet where they could act by stabilizing 145 the virion against such transitions. The study reported here show that the serine residue at VP3-091 imparts a cold sensitive phenotype, suggesting that the serine residue at this position stabilizes the virion such that it is difficult to undergo conformational transition at a low temperature. It is not known why naturally occurring poliovirus strains are cold sensitive. Viral capsid can be considered to be a vehicle for delivery of the RNA genome from one host to another. Assembly and disassembly of the vehicle is an important event in delivery. Polioviruses can grow over a wide range of temperatures (from 23°C to 40°C), and different viruses have different reproductive capacities at various temperatures (e.g. a virus which grows at 25°C usually multiplies poorly at 40°C, and vice versa) (Carp et al., 1963; Sabin, 1960; Sabin, 1961). The adaptation (selection of certain mutations) of a poliovirus strain to grow from one temperature to another is accompanied by genetic changes (Carp et al., 1963). These findings suggest that the poliovirus capsid has a great capacity to tolerate structural changes in order to carry out its vehicular function under different conditions. Consistent with this notion, many amino acid residues were shown to affect the conformational transition that virus undergoes during assembly and disassembly. Virus adapted to growth at low temperatures may require a lower energy to undergo the conformational transition during assembly and disassembly than virus grown at higher temperatures. Naturally occurring virus strains are selected by passage at body temperature and survival in a harsh environment. These viruses are expected to grow well at high temperature but not low temperature. The results reported here also show that all attenuated poliovirus strains are ts and the determinants of ts in type 2 and type 3 viruses mapped to the 5'-ncr. As previously noted, VP3-091 is the major ts determinant of P3/Sabin (identified in Hep-2 cells at 40°C) (Minor et al., 1989). The attenuating determinants found in the Sabin vaccine strains are strongly selected against in the human gut but not in most tissueculture systems (Dunn et al., 1990; Minor and Dunn, 1988). It is possible that transgenic 146 MPMC cells are more sensitive at detecting the temperature sensitive phenotype of the virus. The ts phenotype of the virus may correlate with viral attenuation. The 5'-ncr of P3/Sabin differs from that of P3/Leon by two nucleotides at position 220 and 472. The latter is an attenuation determinant of P3/Sabin. Nucleotide 481 of P2/Sabin (closely related to P2/P712) is a determinant of both attenuation and temperature-sensitivity (in BGM cells, a continuous cell line derived from African Green Monkey Kidney) (Macadam et al., 1991b). Nucleotide 472 in the 5'-ncr of P3/Sabin is believed to disrupt the RNA secondary structure in the 470 to 485 region (Skinner et al., 1989). This mutation was found to decrease in vitro translation efficiency in a Krebs-2 cell extract (Svitkin et al., 1990). In a neuroblastoma cell line the Leon/Lansing virus with an attenuating allele at 472 gave 10-fold lower titres than the virus with a virulent allele at 472. Attenuation resulted from a reduction in protein synthesis (La Monica and Racaniello, 1989). The attenuated poliovirus strains were isolated by multiple rapid passage in monkey primary kidney cell cultures (Sabin et al., 1954). The selective pressures that lead to the appearance of attenuated variants by continued propagation in cultures of nonnervous cells under certain conditions are still not known. The temperature at which viruses multiply has been an important factor in isolating the vaccine strains. Highly attenuated poliovirus strains can be isolated by cold passages of virulent strains in monkey primary kidney cell culture. However, these virus strains may also have decreased reproductive capacity in the gut. The molecular basis of live oral vaccine strains may include the dissociation between the capacity of polioviruses to multiply in the alimentary tract and their capacity to multiply in the CNS of the same host (Sabin, 1961). This study demonstrated that attenuated strains (P1/Sabin and P3/Sabin) and their parental virulent strains (P1/Mahoney and P3/Leon) have differential reproductive capacities at different temperatures. The ts phenotype of attenuated strains is not 147 expressed at 37°C in other cell lines, such as Hela cells. The cell-type specific temperature sensitivity of poliovirus may have implications on the biological basis for isolating the Sabin vaccine strains. It is interesting that all Sabin vaccine strains have attenuation mutations in the 5'ncr. These appear to affect translational efficiency at high temperature. They also have attenuation mutations in their capsid proteins. P1/Sabin also has attenuation mutations in 3CD (RNA polymerase) (Nomoto et al., 1987), M. Bouchard and V. R. Racaniello, unpublished observations). The P2/Sabin vaccine strain was isolated from a naturally occurring strain of poliovirus (P712) possessing low neurovirulence for cynomlgous monkeys by the intraspinal route (Sabin and Boulger, 1973). Consistent with the fact that P712 is a naturally occurring strain, it has low reproductive capacity at low temperature (32°C) in MPMC cells. Conclusions and Perspects. The results presented here establish the transgenic mouse expressing human poliovirus receptors as a new model for studying poliovirus neurovirulence, attenuation and pathogenesis. The susceptibility of PVR transgenic mice to poliovirus infection demonstrates that the PVR is the determinant of poliovirus host range in mice. The transgenic mice express PVR transcripts and poliovirus binding sites in a wide range of tissues. The overall pattern of RNA expression in PRG1 and PRG3-containing transgenic lines is similar. Within the tissues, PVR RNA is expressed in a cell specific manner. For example, PVR RNA is expressed in neurons of the CNS and peripheral ganglia, epithelial cells of the renal corpuscle and some of the tubular cells. The expression of the PVR gene in transgenic mice generally mimics that in human. Poliovirus replication, however, is limited to neurons of both the CNS and peripheral skeletal muscle cell and brown adipose tissues. ganglia, The sites of viral replication are consistent with those found in primates. Poliovirus tissue tropism, therefore, is not governed solely by expression of PVR RNA and poliovirus binding sites. 148 An important question, which needs to be addressed to understand the mechanism of poliovirus tissue tropism, is where poliovirus replication in nonsusceptible tissues is blocked. It is not clear if the block is at stages of entry or beyond entry, such as translation, replication or assembly. This question could be addressed by introducing viral RNA into nonsusceptible cells which express PVR. This might be achieved by isolating nonsusceptible PVR expressing cells (e.g. epithelial cells of the renal corpuscle or T lymphcytes in the thymus) using a monoclonal antibody in conjunction with fluorescence-activated cell sorting. Poliovirus RNA then can be transfected into these freshly isolated cells. Alternatively, poliovirus cDNA might be carried into transgenic mouse cells by vaccinia virus. The replication of poliovirus in nonsusceptible cells could be detected by immunochemistry. The study of viral spread following intramuscular or intrafootpad injection in PVR transgenic mice demonstrates that poliovirus enters the CNS through peripheral nerves and PVR may play an important role in poliovirus spread. This finding, combined with the observation that poliovirus replicates extensively in skeletal muscles, suggests that viral replication in the muscle and subsequent spreading along nerves innervating the muscle to the CNS constitutes the major route for virus entry into the CNS. The observation that the transgenic mice do not develop clinical disease after inoculation with poliovirus vaccine strains indicates that mutations known to attenuate poliovirus neurovirulence in humans also attenuate neurovirulence in this mouse model. The transgenic mice have been used for identifying the attenuating determinants in poliovirus vaccine strains. The major determinants of attenuation of P2/P712 have been identified. They are an A at nucleotide 481 in the 5'-ncr and Ile at position 143 of capsid protein VP1. The transgenic mouse model for poliomyelitis could constitute an alternative host for the preliminary identification of new attenuated poliovirus strains. PVR transgenic mice may also be suitable for safety testing of poliovirus vaccines, which is currently done in monkeys. 149 PVR transgenic mouse MPMC cells were shown to be more sensitive at detecting the temperature-sensitivity phenotypes of poliovirus vaccine strains. The MPMC cells could be used as an in vitro system to study the mechanism of poliovirus attenuation. It would also be interesting to study the cs phenotype of naturally occurring strains. This study might include examining the effects of the cs determinants on viral replication in MPMC cells, isolating cs suppressors by adapting virus to grow at the low temperature, and characterizing the cs suppressors in terms of thermolability, PVR binding and alteration properties and three dimensional structure. These studies should provide information on virus-host interactions. The mechanism of poliovirus attenuation is a question that remains unsolved. Attenuating determinants have been identified in 5'-ncr, capsid proteins, and RNA polymerase, suggesting that poliovirus vaccine strains have defects in viral translation, replication, assembly and disassembly. It is not clear whether viruses are generally attenuated because they are defective in some neural-specific function, or are simply reduced in their overall efficiency of replication. The attenuating determinants found in the Sabin vaccine strains are strongly selected against in the human gut, suggesting that they are defective in their overall efficiency of replication. However, the dissociation between the capacity of polioviruses to multiply in the alimentary tract and their capacity to multiply in other extraneural tissues and in the CNS of the same host is believed to be the basis of the attenuated polioviruses that are used in the live oral vaccines. It is not known of the differential reproductive capacity in distinct cell types is a qualitative or quantitative difference. The different reproductive capacity of polioviruses in cells may be due to different amounts of factors required for poliovirus replication in these cells. Identifying the genetic determinants that govern the differential reproductive capacity of different viruses awaits further characterization of the cellular factors involved in poliovirus infection. 150 References Acharya, R., Fry, E., Stuart, D., Fox, G., Rowlands, D. and Brown, F. (1989). The threedimensional structure of foot-and-mouth disease virus at 2.9 Å resolution. Nature. 337, 709-716. Agol, V. (1990). Current approaches to the problem of poliovirus attenuation. In New aspects of positive-strand RNA viruses, M.A. Brinton and F.X. Heinz, ed). (American Soc. for Microbiology, Washington, D.C.), 311-318. Ahearn, J., Hayward, S., Hickey, J. and Fearon, D. (1988). Epstein-Barr virus (EBV) infection of murine L cells expressing recombinant human EBV/C3d receptor. Proc. Natl. Acad. Sci. USA. 85, 9307-9311. Almond, J.W. (1987). The attenuation of poliovirus neurovirulence. Ann. Rev. Microbiol. 41, 153-180. Almond, J.W., Stanway, G., Cann, A.J., Westrop, G.D., Evans, D.M.A., Ferguson, M., Minor, P.D. and Schild, G.C. (1984). New poliovirus vaccines: a molecular approach. Vaccine. 2, 177-184. Ambros, V., Pettersson, R.F. and Baltimore, D. (1978). An enzymatic activity in uninfected cells that cleaves the linkage between poliovirion RNA and the 5'-terminal protein. 15, 1439-1446. Armstrong, C. (1939a). The experimental transmission of poliomyelitis to the Eastern cotton rat, Sigmodon hispidus hispidus. Pub. Health Rep. 54, 2302-2305. 151 Armstrong, C. (1939b). Successful transfer of the Lansing strain of poliomyelitis virus from the cotton rat to the white mouse. Pub. Health Rep. 54, 2302-2305. Assaad, F. and Cockburn, W.C. (1982). The relation between acute persisting spinal paralysis and poliomyelitis vaccine--results of a ten-year enquiry. Bull. W.H.O. 60, 231242. Ausubel, F.M., Brent, R., Kingston, R.E., Moore, D.D., Smith, J.A., Seidman, J.G. and Struhl, K. (1987). Current Protocols in Molecular Biology. ed). (John Wiley & Sons, New York), Pages. Badger, J., Minor, I., Kremer, M.J., Lliveira, M.A., Smith, T.J., Griffith, J.P., Guerin, D.M.A., Krishnaswamy, S., Luo, M., Rossmann, M.G., McKinlay, M.A., Diana, G.D., Dutko, F.J., Fancher, M., Rueckert, R.R. and Heinz, B.A. (1988). Structural analysis of a series of antiviral agents complexed with human rhinovirus 14. Proc. Natl. Acad. Sci. USA. 85, 3304-3308. Bass, D.M., Trier, J.S., Dambrauskas, R. and Wolf, J.L. (1988). Reovirus type 1 infection of small intestinal epithelium in suckling mice and its effect on M cells. Lab. Invest. 58, 226-235. Benirschke, K. and Kaufmann, P. (1990). Nonvillous part of the placenta. In Pathology of the human placenta, K. Benirschke and P. Kaufmann, ed). York), Pages. 152 (Spinger-Verlag, New Blinzinger, K. and Anzil, A.P. (1974). Neural route of infection in viral disease of the central nervous system. Lancet. ii, 1374-1375. Blinzinger, K., Simon, J., Magrath, D. and Boulger, L. (1969). Poliovirus crystals within the endoplasmic reticulum of endothelial and mononuclear cells in the monkey spinal cord. Science. 163, 1336-1337. Bodian, D. (1954a). Viremia in experimental poliomyelitis. 1. General aspects of infection after intramuscular inoculation with strains of high and of low invasiveness. Am. J. Hyg. 60, 339-357. Bodian, D. (1954b). Viremia in experimental poliomyelitis. 2. Viremia and the mechanism of the "provoking" effect of injections or trauma. Am. J. Hyg. 60, 359-370. Bodian, D. (1955). Emerging concept of poliomyelitis infection. Science. 12, 105-108. Bodian, D. (1959). Poliomyelitis: pathogenesis and histopathology. Rickettsial Infections of Man, T.M. Rivers and F.L. Horsfall, ed). In Viral and (Lippincott, Philadelphia), 479-498. Bodian, D. and Horstmann, D.H. (1965). Polioviruses. In Viral and Rickettsial Infections of Man, F.L. Horsfall and I. Tamm, ed). (Philadelphia., Lippincott), 430-473. Boyd, J.D. and Hamilton, W.J. (1970). The human placenta. ed). Cambridge), Pages. 153 (Heffer and Sons, Brown, R.H., Johnson, D., Ogonowski, M. and Weiner, H.L. (1987). Type 1 human poliovirus binds to human synaptosomes. Ann. Neurol. 21, 64-70. Burke, K.L., Dunn, G., Ferguson, M., Minor, P.D. and Almond, J.W. . (1988). Antigen chimaeras of poliovirus as potential new vaccines. Nature. 332, 81-82. Butcher, E.C. (1991). Leukocyte-endothelial cell recognition: three (or more) steps to specificity and diversity. Cell. 67, 1033. Caliguiri, L.A., McSharry, J.J. and Lawrence, G.W. (1980). Effect of arildone on modifications of poliovirus in vitro. Virol. 105, 86-93. Cann, A.J., Stanway, G., Hughes, P.J., Minor, P.D., Evans, D.M.A., Schild, G.C. and Almond, J.W. (1984). Reversion to neurovirulence of the live-attenuated Sabin type 3 oral poliovirus vaccine. Nucl. Acids Res. 12, 7787-7792. Carp, R.I., Plotkin, S.A., Norton, T.W. and Koprowski, H. (1963). Modification of an attenuated type 2 polio vaccine following passage at 23o C. Proc. Soc. Exp. Biol. Med. 112, 251-256. Chirgwin, J., Przybyla, A., MacDonald, R. and Rutter, W. (1979). Isolation of biologically active ribonucleic acid from sources enriched in ribonuclease. Biochem. 18, 5294-5299. Chow, M., Newman, J.F.E., Filman, D., Hogle, J.M., Rowlands, D.J. and Brown, F. (1987). Myristylation of picornavirus capsid protein VP4 and its structural significance. Nature. 327, 482-486. 154 Colonno, R.J. (1986). Cell surface receptors for picornaviruses. BioEssays. 5, 270-274. Couderc, T., Barzu, T., Horaud, F. and Crainic, R. (1990). Poliovirus permissivity and specific receptor expression on human endothelial cells. Virol. 174, 95-102. Crainic, R., Blondel, B., Aubert-Combiescu, A., Beytout, D., Couillin, P., Candrea, A., Boue, A. and Horaud, F. (1984). Neutralization epitope patterns of poliovirus strains isolated from paralytic cases. Develop. biol. Standard. 57, 165-170. Crowell, R.L., Krah, D.L., Mapoles, J. and Landau, B.J. (1983). Methods for assay of cellular receptors for picornaviruses. In Methods in Enzymology, ed). (Academic Press, NewYork), 443-452. Dalgleish, A.G., Beverley, P.C.L., Clapham, P.R., Crawford, D.H., Greaves, M.F. and Weiss, R.A. (1984). The CD4 (T4) antigen is an essential component of the receptor for the AIDS retrovirus. Nature. 312, 763-766. Del Angel, R.M., Papavassiliou, A.G., Fernández-Tomás, C., Silverstein, S.J. and Racaniello, V.R. (1989). Cell proteins bind to multiple sites within the 5'-untranslated region of poliovirus RNA. Proc. Natl. Acad. Sci. USA. 86, 8299-8303. Dunn, G., Begg, N.T., Cammack, N. and Minor, P.D. (1990). Virus excretion and mutation by infants following primary vaccination wirh live oral poliovaccine from two sources. J. Med. Virol. 32, 92-95. 155 Enders, J.F., Weller, T.H. and Robbins, F.C. (1949). Cultivation of the Lansing strain of poliomyelitis virus in cultures of various human embryonic tissues. Science. 109, 85-87. Everaert, L., Vrijsen, R. and Boeyé, A. (1989). Eclipse products of poliovirus after coldsynchronized infection of HeLa cells. Virol. 171, 76-82. Faber, H.K. (1956). The evolution of poliomyelitic infection. Pediatrics. 17, 278-286. Filman, D.J., Syed, R., Chow, M., Macadam, A.J., Minor, P.D. and Hogle, J.M. (1989). Structural factors that control conformational transitions and serotype specificity in type 3 poliovirus. EMBO J. 8, 1567-1579. Flamand, A., Gagner, J., Morrison, L.A. and Fields, B.N. (1991). Penetration of the nerves systems of suckling mice by mammalian reoviruses. J. Viral. 65, 123-131. Freistadt, M.F., Kaplan, G. and Racaniello, V.R. (1990). Heterogeneous expression of poliovirus receptor-related proteins in human cells and tissues. Mol. Cell Biol. 10, 57005706. Fricks, C.E. and Hogle, J.M. (1990). The cell-induced conformational change of poliovirus: Externalization of the amino terminus of VP1 is responsible for liposome binding. J. Virol. 64, 1934-1945. Ginsberg, H.S. (1988). Picornaviruses. In Virology, R. Dulbecco and H.S. Ginsberg, ed). (J. B. Lippincott, Philadelphia), Pages. 156 Gotoh, B., Ogasawara, T., Toyoda, T., Inocencio, N.M., Hamaguchi, M. and Nagai, Y. (1990). An endoprotease homolgous to the blood clotting factor X as a determinant of viral tropism in chick embryo. EMBO J. 9, 4189-4195. Greve, J.M., Davis, G., Meyer, A.M., Forte, C.P., Yost, S.C., Marlor, C.W., Kamarck, M.E. and McClelland, A. (1989). The major human rhinovirus receptor is ICAM-1. Cell. 56, 839-847. Gromeier, M. and Wetz, K. (1990). Kinetics of poliovirus uncoating in HeLa cells in a nonacidic environment. Virol. 64, 3590-3597. Gross-Bellard, M., P. Oduet, and P. Chambon. (1973). Isolation of High-MolecularWeight DNA from Mammalian Cells. Eur. J. Biochem. 36, 32-38. Guttman, N. and Baltimore, D. (1977). A plasma membrane component able to bind and alter virions of poliovirus type 1: Studies on cell-free alteration using a simplified assay. Virol. 82, 25-36. Hambidge, S. and Sarnow, P. (1991). Terminal 7-methyl-guanosine acp structure on the normally 5' noncoding region of poliovirus mRNA inhibits its translation in mammalian cells. J. Virol. 65, 6312-6315. Harrison, S.C. (1990). Principles of virus structure. In Virology, B.N. Fields, D.M. Knipe, R.M. Chanock, J.L. Melnick, B. Roizman and R.E. Shope, ed). New York), Pages. 157 (Raven Press, Hashimoto, I., Hagiwara, A. and Komatsu, T. (1984). Ultrastructural studies on the pathogenesis of poliomyelitis in monkeys infected with poliovirus. Acta Neuropathol. (Berl). 64, 53-60. Hogan, B., Costantini, F. and Lacy, E. (1986). Manipulating the mouse embryo: A laboratory manual. ed). (Cold Spring Harbor Laboratory, Cold Spring Harbor), Pages. Hogle, J.M., Chow, M. and Filman, D.J. (1985). Three-dimensional structure of poliovirus at 2.9 Å resolution. Science. 229, 1358-1365. Hogle, J.M., Syed, R., Fricks, C.E., Icenogle, J.P., flore, O. and Filman, D.J. (1990). Role of conformational transitions in poliovirus assembly and cell entry. In New aspects of positive strand RNA viruses., M.A. Brinton and F.X. Heinz, ed). (American Society for Microbiology, Washington D.C.), Pages. Holland, J.J. (1961). Receptor affinities as major determinants of enterovirus tissue tropisms in humans. Virology. 15, 312-326. Holland, J.J. and Hoyer, B.H. (1962). Early stages of enterovirus infection. Cold Spring Harb. Symp. Quant. Biol. 27, 101- 111. Holland, J.J., McLaren, J.C. and Syverton, J.T. (1959a). The mammalian cell virus relationship III. Production of infectious poliovirus by non-primate cells exposed to poliovirus ribonucleic acid. Proc. Soc. Exp. Biol. Med. 100, 843-845. 158 Holland, J.J., McLaren, J.C. and Syverton, J.T. (1959b). The mammalian cell virus relationship IV. Infection of naturally insusceptible cells with enterovirus ribonucleic acid. J. Exp. Med. 110, 65-80. Holland, J.J. and McLaren, L.C. (1959). The mammalian cell-virus relationship II. Adsorption, reception, and eclipse by HeLa cells. J. Exp. Med. 109, 487-504. Holland, J.J. and McLaren, L.C. (1961). The location and nature of enterovirus receptors in susceptible cells. J. Exp. Med. 114, 161-171. Horstmann, D.M. (1952). Poliomyelitis virus in blood of orally infected monkeys and chimpanzees. Proc. Soc. Exp. Biol.Med. 79, 417. Horstmann, D.M., McCollum, R.W. and Mascola, A.D. (1954). Viremia in human poliomyelitis. J. Exp. Med. 355. Hurst, E.W. (1929). The histology of experimental poliomyelitis. J. Path. 32, 457-477. Hurst, E.w. (1936). The newer knowledge of virus diseases of the nervous system: A review and an interpretation. Brain. 59, 1-34. Jaffe, L., Jeannotte, L., Bikoff, E.K. and Robertson, E. (1990). Analysis of β2microglobulin gene expression in the developing mouse embryo and placenta. J. Immunol. 145, 3474-3482. Jang, S.K., Krausslich, H.-G., Nicklin, M.J.H., Duke, G.M., Palmenberg, A.C. and Wimmer, E. (1988). A segment of the 5' nontranslated region of encephalomyocarditis 159 virus RNA directs internal entry of ribosomes during in vitro translation. J. Virol. 62, 2636-2643. Jang, S.K. and Wimmer, E. (1990). Cap-independent translation of encephalomyocarditis virus RNA: structural elements of the internal ribosomal entry site and involvement of a cellular 57-kd RNA-binding protein. Genes Dev. 4, 1560-1572. Jubelt, B., Gallez-Hawkins, B., Narayan, O. and Johnson, R.T. (1980a). Pathogenesis of human poliovirus infection in mice. I. Clinical and pathological studies. J. Neuropathol. Exp. Neurol. 39, 138-148. Jubelt, B., Narayan, O. and Johnson, R.T. (1980b). Pathogenesis of human poliovirus infection in mice. II. Age- dependency of paralysis. J. Neuropathol. Exp. Neurol. 39, 149158. Kanamitsu, M., Kasamaki, A., Ogawa, M., Kasahara, S. and Imumura, M. (1967). Immunofluorescent study on the pathogenesis of oral infection of poliovirus in monkey. Japan. J. Med. Sci. Biol. 20, 175-191. Kaplan, A.S. (1955). Comparison of susceptible and resistant cells to infection with poliomyelitis virus. Ann. N.Y. Acad. Sci. 61, 830-839. Kaplan, G., Freistadt, M.S. and Racaniello, V.R. (1990). Neutralization of poliovirus by cell receptors expressed in insect cells. J. Virol. 64, 4697-4702. Kitamura, N., Semler, B.L., Rothberg, P.G., Larsen, G.R., Adler, C.J., Dorner, A.J., Emini, E.A., Hanecak, R., Lee, J.J., van der Werf, S., Anderson, C.W. and Wimmer, E. 160 (1981). Primary structure, gene organization and polypeptide expression of poliovirus RNA. Nature. 291, 547-553. Klatzman, D., Champagne, E., Chamaret, S., Gruest, J., Guetard, D., Hercend, T., gluckman, J.-C. and Montagnier, L. (1984). T-lymphocyte T4 molecule behaves as the receptor for human retrovirus LAV. Nature. 312, 767-768. Kohara, M., Omata, T., Kameda, A., Semler, B.L., Itoh, H., Wimmer, E. and Nomoto, A. (1985). In vitro phenotypic markers of a poliovirus recombinant constructed from infectious cDNA clones of the neurovirulent Mahoney strain and the attenuated Sabin 1 strain. J. Virol. 53, 786-792. Koike, S., Horie, H., Dise, I., Okitsu, H., Yoshida, M., Iizuka, N., Takeuthi, K., Takegami, T. and Nomoto, A. (1990). The poliovirus receptor protein is produced both as membrane-bound and secreted forms. EMBO J. 9, 3217-3224. Koike, S., Taya, C., Kurata, T., Abe, S., Ise, I., Yonekawa, H. and Nomoto, A. (1991). Transgenic mice susceptible to poliovirus. Proc. Natl. Acad. Sci. USA. 88, 951-955. Kozak, M. (1989). The scanning model for translation: an update. J. Cell Biol. 108, 229241. Kuhn, R., Luz, N. and Beck, E. (1990). Functional analysis of the internal translation initiation site of foot-and-mouth disease virus. J. Virol. 64, 4625-4631. Kunin, C.M. and Jordan, W.S. (1961). In vitro adsorption of poliovirus by noncultured tissues. Effect of species, age and malignancy. Am. J. Hyg. 73, 245-257. 161 Kurman, R.J., Main, C.S. and Chen, H.-C. (1984). Intermediate trophobalst: a distictive form of trophoblast with specific morphological, biochemical and functional features. Placenta. 5, 349-370. La Monica, N., Almond, J.W. and Racaniello, V.R. (1987a). A mouse model for poliovirus neurovirulence identifies mutations that attenuate the virus for humans. J. Virol. 61, 2917-2920. La Monica, N., Kupsky, W. and Racaniello, V.R. (1987b). Reduced mouse neurovirulence of poliovirus type 2 Lansing antigenic variants selected with monoclonal antibodies. Virology. 161, 429-437. La Monica, N., Meriam, C. and Racaniello, V.R. (1986). Mapping of sequences required for mouse neurovirulence of poliovirus type 2 Lansing. J. Virol. 57, 515-525. La Monica, N. and Racaniello, V.R. (1989). Differences in replication of attenuated and neurovirulent polioviruses in human neuroblastoma cell line SH-SY5Y. J. Virol. 63, 23572360. Levine, B., Hardwick, J.M., Trapp, B.D., O., C.T., Bollinger, R.C. and Griffin, D.E. (1991). Antibody-mediated clearance of alphavirus infection from neurons. Science. 254, 856-859. Li, C.P. and Schaeffer, M. (1953). Adaptation of type 1 poliomyelitis virus to mice. Proc.Soc.Exp.Biol.Med. 82, 477-481. 162 Lonberg-Holm, K., Gosser, L.B. and Kauer, J.C. (1975). Early alteration of poliovirus in infected cells and its specific inhibition. J. Gen. Virol. 27, 329-342. Lonberg-Holm, K. and Philipson, L. (1974). Early interaction between animal viruses and cells. Monogr. Virol. 9, 1-148. Luna, L.G. (1968). Manual of histologic staining methods of the Armed Forces Institute of Pathology. p. 212. Luo, M., Vriend, G., Kamer, G., Minor, I., Arnold, E., Rossmann, M.G., Boege, U., Scraba, D.G., Duke, G.M. and Palmenberg, A.C. (1987). The atomic structure of mengovirus at 3.0 A resolution. Science. 235, 182-191. Macadam, A.J., Ferguson, G., Arnold, C. and Minor, P.D. (1991a). assembly defect as a result of an attenuating mutation in the capsid proteins of the pliovirus type 3 vaccine strain. J. Virol. 65, 5225-5231. Macadam, A.J., Pollard, S.R., Furguson, G., Dunn, G., Skuce, R., Almond, J.W. and Minor, P.D. (1991b). The 5' non-coding region of the type 2 poliovirus vaccine strain contains determinants of attenuation and temperature-sensitivity. Virol. 181, 451-458. Maddon, P.J., Dalgleish, A.G., McDougal, J.S., Clapham, P.R., Weiss, R.A. and Axel, R. (1986). The T4 gene encodes the AIDS virus receptor and is expressed in the immune system and the brain. Cell. 47, 333-348. Madshus, I.H., Olsnes, S. and Sandvig, K. (1984a). Mechanism of entry into the cytosol of poliovirus type 1: requirement for low pH. J. Cell. Biol. 98, 1194-1200. 163 Madshus, I.H., Olsnes, S. and Sandvig, K. (1984b). Requirements for entry of poliovirus RNA into cells at low pH. EMBO J. 3, 1945-1950. Martin, A., Wychowski, C., Benichou, D., Crainic, R. and Girard, M. (1988). Construction of a chimaeric type 1/type 2 poliovirus by genetic recombination. Ann. Inst. Pasteur/Virol. 139, 79-88. McSharry, J.J., Caliguiri, L.A. and Eggers, H.J. (1979). Inhibition of uncoating of poliovirus by arildon, a new antiviral drug. Virol. 97, 307-315. Meerovitch, K., Pelletier, J. and Sonenberg, N. (1989). A cellular protein that binds to the 5'-noncoding region of poliovirus RNA: implications for internal translation initiation. Genes & Devel. 3, 1026-1034. Melnick, J.L. (1983). Portraits of viruses: the picornaviruses. Intervirology. 20, 61-100. Melnick, J.L. (1985). Enteroviruses: Polioviruses, coxsackieviruses, echoviruses and newer enteroviruses. In Virology, B.N. Fields, D.M. Knipe, R.M. Chanock, J.L. Melnick, B. Roizman and R.E. Shope, ed). (Raven Press, New York), 705-738. Melnick, J.L. (1990). Enteroviruses: Polioviruses, coxsackieviruses, echoviruses and newer enteroviruses. In Virology, B.N. Fields, D.M. Knipe, R.M. Chanock, J.L. Melnick, B. Roizman and R.E. Shope, ed). (Raven Press, New York), 549. 164 Mendelsohn, C., Johnson, B., Lionetti, K.A., Nobis, P., Wimmer, E. and Racaniello, V.R. (1986). Transformation of a human poliovirus receptor gene into mouse cells. Proc. Natl. Acad. Sci. USA. 83, 7845-7849. Mendelsohn, C., Wimmer, E. and Racaniello, V.R. (1989). Cellular receptor for poliovirus: Molecular cloning, nucleotide sequence and expression of a new member of the immunoglobulin superfamily. Cell. 56, 855-865. Mendelsohn, C.L. (1989). Isolation of molecular clones encoding human poliovirus receptors. Minor, P.D. and Dunn, G. (1988). The effect of sequences in the 5'-noncoding region on the replication of polioviruses in the human gut. J. Gen. Virol. 69, 1091-1096. Minor, P.D., Dunn, G., Evans, D.M.A., Magrath, D.I., John, A., Howlett, J., Phillips, A., Westrop, G., Wareham, K., Almond, J.W. and Hogle, J.M. (1989). The temperaturesensitivity of the Sabin type 3 vaccine strain of poliovirus: Molecular and structural effects of a mutation in the capsid protein VP3. J. Gen. Virol. 70, 1117-1123. Minor, P.D., Pipkin, P.A., Hockley, D., Schild, G.C. and Almond, J.W. (1984). Monoclonal antibodies which block cellular receptors of poliovirus. Virus Res. 1, 203212. Moore, M.D., Cooper, N.R., Tack, B.F. and Nemerow, G.R. (1987). Molecular cloning of the cDNA encoding the Epstein-Barr virus/C3d receptor (complement receptor type 2) of human B lymphocytes. Proc. Natl. Acad. Sci. USA. 84, 9194-9198. 165 Morrison, L.A. and Fields, B.N. (1991). Parallel mechanisms in the neuropathogenesis of enteric virus infections. J. Virol. 65, 2767-2772. Morrison, L.A., Sidman, R.L. and Feilds, B.N. (1991). Direct spread of reovirus from the intestinal lumen to the central nervous system through vagal autonomic nerve fibers. Proc. Natl. Acad. Sci. USA. 88, 3852-3856. Moss, E.G., O'Neill, R.E. and Racaniello, V.R. (1989). Mapping of attenuating sequences of an avirulent poliovirus type 2 strain. J. Virol. 63, 1884-1890. Moss, E.G. and Racaniello, V.R. (1991). Host range determinants located on the interior of the poliovirus capsid. EMBO J. 5, 1067-1074. Murray, M.G., Bradley, J., Yang, X.F., Wimmer, E., Moss, E.G. and Racaniello, V.R. (1988). Poliovirus host range is determined a short amino acid sequence in neutralization antigenic site I. Science. 241, 213-215. Nathanson, N. and Bodian, D. (1961). Experimental poliomyelitis following intramuscular virus injection. 1. The effect of neural block on a neurotropic and a pantropic strain. Bulletin of Johns Hopkins Hospital. 108, 308-319. Nathanson, N. and Langmuir, A. (1963). The Cutter incident. Poliomyelitis following formaldehyde-inactivated poliovirus vaccination in the United States during the spring of 1955. Am. J. Hyg. 78, 16-81. 166 Nobis, P., Zibirre, R., Meyer, G., Kuhne, J., Warnecke, G. and Koch, G. (1985). Production of a monoclonal antibody against an epitope on HeLa cells that is the functional poliovirus binding site. J. Gen. Virol. 6, 2563-2569. Nomoto, A., Detjen, B., Pozzati, R. and Wimmer, E. (1977). The location of the polio genome protein in viral RNAs and its implication for RNA synthesis. Nature. 268, 208213. Nomoto, A., Kohara, M., Kuge, S., Kawamura, N., Arita, M., Komatsu, T., Abe, S., Semler, B.L., Wimmer, E. and Itoh, H. (1987). Study on virulence of poliovirus type 1 using in vitro modified viruses. In Positive Strand RNA Viruses, M.A. Brinton and R.R. Rueckert, ed). (Alan R. Liss, Inc., New York), 437-452. Nomoto, A., Lee, Y.F. and Wimmer, E. (1976). The 5' end of poliovirus mRNA is not capped with m7G(5')ppp(5')np. Proc. Natl. Acad. Sci. USA. 73, 375-380. Nomoto, A. and Wimmer, E. (1987). Genetic studies of the antigenicity and the attenuation phenotype of poliovirus. In Molecular Basis of Virus Disease, W.C. Russell and J.W. Almond, ed). (Cambridge Univ. Press, Cambridge, England), 107-134. Palmiter, R.D. (1986). Germ-line transformation of mice. Ann. Rev. Genet. 20, 456-499. Paul, J.R. (1971). A History of Poliomyelitis. ed). (Yale University Press, New Haven and London.), Pages. 167 Pelletier, J., Flynn, M.E., Kaplan, G., Racaniello, V. and Sonenberg, N. (1988a). Mutational analysis of upstream AUG codons of poliovirus RNA. J. Virol. 62, 44864492. Pelletier, J., Kaplan, G., Racaniello, V. and Sonenberg, N. (1988b). Cap-independent translation of poliovirus messenger RNA is conferred by sequence elements within the 5' noncoding region. Mol. Cell Biol. 8, 1103-1112. Pelletier, J., Kaplan, G., Racaniello, V.R. and Sonenberg, N. (1988c). Translational efficiency of poliovirus messenger RNA: Mapping inhibitory cis-acting elements within the 5'-noncoding region. J. Virol. 62, 2219-2227. Pelletier, J. and Sonenberg, N. (1988). Internal initiation of translation of eukaryotic mRNA directed by a sequence derived from poliovirus RNA. Nature. 334, 320-325. Pelletier, J. and Sonenberg, N. (1989). Internal binding of eucaryotic ribosomes on poliovirus RNA: translation in Hela cell extracts. J. Virol. 63, 441-444. Pijnenborg, R., Robertson, W.B., Brosens, I. and Dixon, G. (1981). Trophoblast invasion and the establishment of haemochorial placentation in man and laboratory animals. Placenta. 2, 71-92. Pilipenko, E.V., Gmyl, A.P., Maslova, S.V., Svitkin, Y.V., Sinyakov, A.N. and Agol, V.I. (1992). Prokaryotic-like cis elements in the cap-independent internal initiation of trnslation on piconavirus RNA. Cell. 68, 119-131. 168 Pollard, S.R., Dunn, G., Cammack, N., Minor, P.D. and Almond, J.W. (1989). Nucleotide sequence of a neurovirulent variant of the type 2 oral poliovirus vaccine. J. Virol. 63, 4949-4951. Racaniello, V.R. (1988). Poliovirus neurovirulence. In Advances in Virus Research, K. Maramorosch, F.A. Murphy and A.J. Shatkin, ed). (Academic Press, San Diego), 217246. Racaniello, V.R. and Baltimore, D. (1981a). Cloned poliovirus complementary DNA is infectious in mammalian cells. Science. 214, 916-919. Racaniello, V.R. and Baltimore, D. (1981b). Molecular cloning of poliovirus cDNA and determination of the complete nucleotide sequence of the viral genome. Proc. Natl. Acad. Sci. USA. 78, 4887-4891. Racaniello, V.R. and Meriam, C. (1986). Poliovirus temperature-sensitive mutant containing a single nucleotide deletion in the 5'-noncoding region of the viral RNA. Virology. 15, 498-507. Ramsey, E.L. (1965). The placenta and fetal membranes. In Obstetrics, J.P. Greenhill, ed). (Saunders, W. B. Co., Philadelphia), Pages. Reed, L.J. and Muench, H. (1938). A simple method of estimating fifty per cent end points. Am. J. Hyg. 27, 493-497. Robinson, I.K. and Harrison, S.C. (1982). Structure of the expanded state of tomato bushy stunt virus. Nature. 297, 563-568. 169 Rossmann, M.G., Arnold, E., Erickson, J.W., Frankenberger, E.A., Griffith, J.P., Hecht, H.-J., Johnson, J.E. and Kamer, G. (1985). Structure of a human common cold virus and functional relationship to other picornaviruses. Nature. 317, 145-153. Rueckert, R.R. (1990). Picornaviridae and their replication. In Virology, B.N. Fields, D.M. Knipe, R.M. Chanock, J.L. Melnick, B. Roizman and R.E. Shope, ed). (Raven Press, New York), Pages. Sabin, A.B. (1956). Pathogenesis of Poliomyelitis: Reappraisal in light of new data. Science. 123, 1151-1157. Sabin, A.B. (1957). Properties of attenuated polioviruses and their behavior in human beings. In Cellular biology, nucleic acids and viruses, T.M. Rivers, ed). (New York Academy of Science, New York), 113-133. Sabin, A.B. (1959). Present position of immunization against poliomyelitis with live virus vaccines. Brit. med. J. 3, 5123. Sabin, A.B. (1960). Behavior of cold mutants of poliovirus in human beings. Live poliovirus vaccines, second international conference on live poliovirus vaccines. 101-108. Sabin, A.B. (1961). Reproductive capacity of polioviruses of disease origins at various temperatures. In Prospectives in virology, M. Pollard, ed). company, Minneapolis, Minn.), Pages. 170 (Burgess publishing Sabin, A.B. and Boulger, L.R. (1973). History of Sabin attenuated poliovirus oral live vaccine strains. J. Biol. Stand. 1, 115-118. Sabin, A.B., Hennessen, W.A. and Winsser, J. (1954). Studies on variants of poliomyelitis virus: I. Experimental segregation and properties of avirulent variants of three immunologic types. J. Exp. Med. 9, 551-576. Sabin, A.B. and Ward, R. (1941). The natural history of human poliomyelitis. I. Distribution of virus in nervous and non-nervous tissues. Sarnow, P. (1989). Translation of the glucose-regulated protein GRP78/BiP mRNA is increased in poliovirus infected cells at a time when cap-dependent translation of cellular mRNAs is inhibited. Proc. Natl. Acad. Sci. USA. 86, 5795-5799. Sarnow, P., Jacobson, S.J. and Najita, L. (1990). Poliovirus genetics. In Picornaviruses, V.R. Racaniello, ed). (Springer-Verlag, Berlin), 155-188. Semler, B.L., Kuhn, R.J. and Wimmer, E. (1988). Replication of the poliovirus genome. In RNA-directed virus replication., E. Domingo, J.J. Holland and P. Ahlquist, ed). (CRC Press, Inc., 23-48. Shepley, M.P., Sherry, B. and Weiner, H.L. (1988). Monoclonal antibody identification of a 100-kDa membrane protein in HeLa cells and human spinal cord involved in poliovirus attachment. Proc. Natl. Acad. Sci. USA. 85, 7743-7747. 171 Sicinski, P., Rowinski, J., Warchol, J.B., Jarzabek, Z., Gut, W., Szczygiel, B., Bielecki, K. and Koch, G. (1990). Poliovirus type 1 enters the human host through intestinal M cells. Gastroent. 98, 56-58. Skinner, M.A., Racaniello, V.R., Dunn, G., Cooper, J., Minor, P.D. and Almond, J.W. (1989). A new model for the secondary structure of the 5' noncoding RNA of poliovirus is supported by biochemical and genetic data which also show that RNA secondary structure is important to neurovirulence. J. Mol. Biol. 207, 379-392. Sonenberg, N. (1990). Poliovirus Translation. In Picornaviruses, V.R. Racaniello, ed). (Springer-Verlag, New York), 23-48. Sonenberg, N. (1991). Picornavirus RNA translation continues to surprise. TIG. 7, 105106. Staunton, D.E., Merluzzi, V.J., Rothlein, R., Barton, R., Marlin, S.D. and Springer, T.A. (1989). A cell adhesion molecule, ICAM-1, is the major surface receptor for rhinoviruses. Cell. 56, 849-853. Svitkin, Y.V., Cammack, N., Minor, P.D. and Almond, J.W. (1990). Translation deficiency of the Sabin type 3 poliovirus genome: association with an attenuating mutation C472--->U. Virol. 175, 103-109. Theiler, M. (1941). Studies on poliomyelitis. Medicine. 20, 443- 462. Toyoda, H., Kohara, M., Kataoka, Y., Suganuma, T., Omata, T., Imura, N. and Nomoto, A. (1984). Complete nucleotide sequences of all three poliovirus serotype genomes. 172 Implication for genetic relationship, gene function and atigenic determinants. J. Mol. Biol. 174, 561-585. Trono, D., Andino, R. and Baltimore, D. (1988). An RNA sequence of hundreds of nucleotides at the 5' end of poliovirus RNA is involved in allowing viral protein synthesis. J. Virol. 62, 2291-2299. Tyler, K.L. (1987a). Host and viral factors that influence viral neurotropism I. Viral cell attachment proteins and target cell receptors. TINS. 10, 455-460. Tyler, K.L. (1987b). Host and viral factors that influence viral neurotropism II. Viral genes, host genes, site of entry and route of spread of virus. TINS. 10, 492-497. Tyler, K.L., McPhee, D.A. and Fields, B.N. (1986). Distinct pathways of viral spread in the host determined by reovirus S1 gene segment. Science. 233, 770-774. Tyler, K.L., Virgin IVth, H.W., R., B.-D. and Feilds, B.N. (1989). Antibody inhibits defined stages in the pathogenesis of reovirus serotype 3 infection of the central nervous system. J. Exp. Med. 170, 887-900. Wahl, G.M., Lewis, K.A., Ruiz, J.C., Rothenberg, B., Zhao, J. and Evans, G.A. (1987). Cosmid vector for rapid genomic walking, restriction mapping, and gene transfer. Proc. Natl. Acad. Sci. USA. 84, 2160-2164. Webster, R.G. and Rott, R. (1987). Influenza virus pathogenicity: The pivotal role of hemagglutinin. Cell. 50, 665-666. 173 Wenner, H.A. and Kamitsuka, P. (1956). Further observations on the widesprea distribution of poliomyelitis virus in body tissues following intramuscular inoculation of cynomolgus monkeys. Virology. 2, 83-95. Wenner, H.A. and Kamitsuka, P. (1957). Primary sites of virus multiplication following intramuscular inoculation of poliomyelitis virus in cynomolgous monkeys. Virol. 3, 420443. Wenner, H.A. and Rabe, E.F. (1951). The recovery of virus from regional lymph nodes of fetal human cases of poliomyelitis. Am. J. Med. Sci. 222, 292-299. Westrop, G.D., Evans, D.M.A., Minor, P.D., Magrath, D., Schild, G.C. and Almond, J.W. (1987). Investigation of the molecular basis of attenuation in the Sabin type 3 vaccine using novel recombinant polioviruses constructed from infectious cDNA. In The Molecular Biology of the Positive Strand RNA Viruses, D.J. Rowlands, M.A. Mayo and B.W.J. Mahy, ed). (Alan R. Liss, Inc., New York), 53-60. Westrop, G.D., Wareham, K.A., Evans, D.M.A., Dunn, G., Minor, P.D., Magrath, D.I., Taffs, F., Marsden, S., Skinner, M.A., Schild, G.C. and Almond, J.W. (1989). Genetic basis of attenuation of the Sabin type 3 oral poliovirus vaccine. J. Virol. 63, 1338-1344. Wiegers, K., Uhlig, H. and Dernick, R. (1989). NAg1B of poliovirus type 1: A discontinuous epitope formed by two loops of VP1 comprising residues 96-104 and 141152. Virol. 170, 583-586. Williams, A.F. and Barclay, A.N. (1988). The immunoglobulin superfamily--Domains for cell surface recognition. Ann. Rev. Immunol. 6, 381-405. 174 Willingmann, P., Barnert, H., Zeichhardt, H. and Habermehl, K.O. (1989). Recovery of structurally intact and infectious poliovirus type 1 from HeLa cells during receptormediated endocytosis. Virol. 168, 417-420. Wimmer, E., Kuhn, R., Pincus, S., Yang, C.-F., Toyoda, H., Nicklin, M. and Takeda, N. (1987). Molecular events leading to picornavirus genome replication. In Virus Replication and genome Interaction: Seventh John Innes Symposium., J. Davies, ed). (The Company of Biologists LTD, Cambridge, England.), 251-276. Wolf, J.C., Rubin, D.H., Finberg, R., Kauffman, R.S., Sharpe, A.H., Trier, J.S. and Fields, B.N. (1981). Intestinal M cells: A pathway for entry of reovirus into the host. Science. 2, 471-472. Wyatt, H.V. (1990). Incubation of poliomyelitis as calculated from the time of entry into the central nervous system via the peripheral nerve pathways. Rev. Inf. Dis. 12, 547-556. Yeates, T.O., Jacobson, D.H., Martin, A., Wychowski, C., Girard, M., Filman, D.J. and Hogle, J.M. (1991). Three-dimensional structure of a mouse-adapted type2/type 1 poliovirus chimera. EMBO J. 10, 2331-2341. Yeh, I.T., O'Connor, D.M. and Kurman, R.J. (1990). Intermediate trophobalst: further immunocytochemical characterization. Mod-Pathol. 3, 282-287. Zack, J.A., Arrigo, S.J., Weitsman, S.R., Go, A.S., Haislip, A. and Chen, I.S.Y. (1990). HIV-1 entry into quiescent primary lymphocytes: Molecular analysis reveals a labile, latent viral structure. Cell. 61, 213-222. 175 Zeichhardt, H., Wetz, K., Willingmann, P. and Habermehl, K.-O. (1985). Entry of poliovirus type 1 and mouse Elberfeld (ME) virus into Hep2 cells: receptor-mediated endocytosis and endosomal or lysosomal uncoating. J. Gen. Virol. 66, 483-492. 176