Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Epigenetics in stem-cell differentiation wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
Gene therapy of the human retina wikipedia , lookup
Polycomb Group Proteins and Cancer wikipedia , lookup
No-SCAR (Scarless Cas9 Assisted Recombineering) Genome Editing wikipedia , lookup
Site-specific recombinase technology wikipedia , lookup
Primary transcript wikipedia , lookup
Therapeutic gene modulation wikipedia , lookup
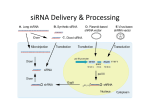
RNA silencing wikipedia , lookup
Vectors in gene therapy wikipedia , lookup
Silencing of Gene Expression in Cultured Cells Using Small Interfering RNAs UNIT 27.1 Kumi Sakurai,1 Pritsana Chomchan,1 and John J. Rossi1 1 Beckman Research Institute of City of Hope, Duarte, California ABSTRACT The discovery of RNA interference (RNAi) and related small RNA–mediated regulatory pathways has significantly altered the understanding of gene regulation in eukaryotic cells. In the RNAi pathway, small interfering RNAs (siRNAs) ∼21 to 23 nucleotides in length serve as the regulatory molecules that guide and induce sequence-specific gene silencing. The use of siRNA-mediated silencing as a tool for investigating gene function is well established in cultured mammalian cells. This unit provides basic approaches to explore the field of RNAi, and hopes to address the importance of optimizing transfection conditions after empirical determinations in order to understand various degrees of C 2010 by John Wiley silencing efficiency. Curr. Protoc. Cell Biol. 47:27.1.1-27.1.28. & Sons, Inc. Keywords: RNAi r small silencing RNAs r gene silencing r transfection r mammalian cell culture INTRODUCTION Since the first demonstration of RNA interference in cultured mammalian cells in 2001 (Elbashir et al., 2001a), small interfering RNA (siRNA)–mediated sequence-specific gene silencing has been widely used as a tool for investigating gene function, and has initiated a new wave of reverse genetics in mammalian systems. The siRNA-mediated silencing provides fast, target-specific down-regulation of gene expression. The extent to which one can successfully carry out the silencing for determination of gene function depends on the potency of siRNAs, transfection conditions, duration of analysis, and screening methods. This unit describes design of siRNAs, transfection of siRNAs into cultured cells, and testing and validation of gene knockdown. The silencing efficiency is based on degree of susceptibility of target transcripts to siRNAmediated RNA-induced silencing complex (RISC) activity. It is important to design multiple siRNAs and screen for siRNAs that are highly potent in silencing a given gene. There are several online siRNA design programs available, providing predicted siRNA sequences and opportunities to explore and compare the siRNA sequences generated by different design methods. Once several siRNA sequences with high prediction scores are obtained (Basic Protocol 1), one should carefully design and evaluate siRNA transfection conditions (Basic Protocol 2). Transfection of mammalian cells typically utilizes reagents that enhance the uptake of nucleic acids (e.g., plasmid DNA or siRNAs) by cultured cells. Transfection methods commonly used are: (1) cationic liposomes—a complex of the genetic materials and/or siRNAs with a cationic lipid—inducing fusion with the cell plasma membrane; (2) electroporation via destabilization of the cell plasma membrane; and (3) calcium phosphate/nucleic acid precipitate uptake. In this unit, we describe the use of liposomes to deliver siRNAs to cultured mammalian cells for silencing gene expression. RNA-Based Methods in Cell Biology Current Protocols in Cell Biology 27.1.1-27.1.28, June 2010 Published online June 2010 in Wiley Interscience (www.interscience.wiley.com). DOI: 10.1002/0471143030.cb2701s47 C 2010 John Wiley & Sons, Inc. Copyright 27.1.1 Supplement 47 As previously mentioned, it is desirable to test several siRNAs against a given gene and screen for efficacy. Target-site screening is tricky, but necessary to validate and observe outcomes that accurately reflect the effect of siRNAs. Currently, screening heavily relies on experimental observations such as monitoring phenotypic changes, detection of changes in reporter protein expression (e.g., fluorescence for GFP-target fusion proteins), RT-PCR (Support Protocol 1), or western blot analysis (Support Protocol 2). As fundamental processes for the validation of the siRNA-mediated silencing, we describe RNA isolation and protein collection following transfections. In addition, a dual-reporter system can be used for rapid and reliable screening for siRNA sequences (Support Protocol 3). The psiCHECK-2 vector system (Promega) incorporates expression of a fluorescent reporter protein (e.g., Renilla luciferase) and a control fluorescent protein (e.g., firefly luciferase) from a single vector, enabling one to evaluate the efficiency of siRNAs by comparing expression levels of reporter proteins. Support Protocols 4 and 5 are intended to supplement Basic Protocol 1 when a siRNA is obtained in single-stranded format. STRATEGIC PLANNING siRNA Design Algorithms The sequence of an siRNA can be by far the most important determinant of silencing efficiency. The identification and selection of highly potent siRNA sequences against a given gene prior to any experimental determinations can be overwhelming. To facilitate the designing process, there are several databases that archive siRNA sequences tested in mammalian RNAi experiments (Chalk et al., 2005; Shah et al., 2007; Ren et al., 2009). Validated siRNAs can also be found in commercial resources such as the Stealth RNAi siRNAs from Invitrogen, the Silencer validated siRNA from Ambion, and HP validated siRNAs from Qiagen. These vendors and Dharmacon also provide predesigned siRNAs and custom design services, making siRNA designing easier and providing a good starting point. However, many, if not most, siRNA-mediated RNAi assays require experimental validation for optimization of conditions to meet specific aims. NOTE: Throughout the protocols, RNA-handling techniques (APPENDIX 2A) should be practiced and aseptic cell culture conditions should be maintained to avoid accidental introduction of nucleases, cross-contamination of samples, and potential loss of cell culture. BASIC PROTOCOL 1 Silencing of Gene Expression in Cultured Cells DESIGN OF 21-NUCLEOTIDE-LONG siRNA (21-MER) If no matches are found for a given gene of interest in the literature and available siRNA databases, siRNAs can be designed using Web-based siRNA design programs. There are several siRNA selection algorithms to help predict siRNA sequences for effective silencing. Users need to provide the target messenger RNA sequence and are able to create some user-defined design criteria such as thermodynamic stability and AU/GC base pair contents that will influence the overall efficiency of predicted siRNAs. Each algorithm has a unique emphasis on design features, but fundamentally most of the algorithms provide siRNA sequences with scores or ranks for users to further analyze for specificity and select candidate siRNAs that meet their needs. Some algorithms are specialized in determining off-target effects of predicted siRNA sequences (Chalk and Sonnhammer, 2008; Gong et al., 2008) to reduce unwanted side effects. Once selected, siRNAs can be synthesized in-house or obtained from several siRNA-licensed vendors such as Ambion, Dharmacon, and Qiagen. While many of the siRNA design algorithms provide siRNAs in the form of 19 base pairs with 2 nucleotide overhangs at the 3 end on each strand, siRNAs can be generated by processing longer double-stranded RNAs (dsRNAs) by the RNase III Dicer enzyme. 27.1.2 Supplement 47 Current Protocols in Cell Biology One may find the use of such dsRNAs for target gene silencing advantageous, since transfecting 25- to 27-nt siRNAs showed improved silencing efficiency (Rose et al., 2005). Hannon and colleagues also reported similar observations with short hairpin RNAs with 29-bp stems (Siolas et al., 2005). These observations together with others suggest that the Dicer processing of its substrates results in better programming of RISC (Elbashir et al., 2001c; Liu et al., 2003; Gregory et al., 2005). While the 21-nt siRNAs often show a wide range of potency (Amarzguioui et al., 2003; Reynolds et al., 2004; Ui-Tei et al., 2004), transforming a 21-nt siRNA to a 27-nt Dicer substrate siRNA may improve the silencing efficiency. Furthermore, the structural modification that mimic secondary structure of existing Dicer substrates (e.g., pre-microRNAs) will introduce the defined polarity of Dicer processing, leading to preferential incorporation of the desired guide strand (Rose et al., 2005). Alternatively, D-siRNA sequences can be selected using an algorithm that provides fully automated D-siRNA designs using the mRNA target sequence (RNAi Design; Owczarzy et al., 2008; http://www.idtdna.com/Scitools/Applications/RNAi/RNAi.aspx). Materials Gene of interest Web-based siRNA design program (e.g., Table 27.1.1) Web-based specificity program (e.g., SpecificityServer; Table 27.1.1) Design siRNA 1. Obtain the most updated mRNA sequence of the gene of interest from the UCSC Genome Browser (http://genome.ucsc.edu/cgi-bin/hgGateway). 2. Use an online siRNA design program to enter the mRNA sequence. To increase the chances of designing highly potent siRNAs, include the 5 and 3 untranslated regions (UTRs) in the inquiry as well. Follow the Web site instructions to further define the siRNAs. Examples of Web-based siRNA design tools are listed in Table 27.1.1. 3. Analyze the target sequence of the candidate siRNAs for specificity using available Web servers (e.g., SpecificityServer). Utilize several online siRNA design tools to compare and select up to five candidate siRNAs with high prediction scores. Design Dicer substrate siRNA (D-siRNA) 4. Obtain a 21-nt siRNA sequence using the Web-based siRNA design tools. An example of a sequence is shown in step 5. Table 27.1.1 List of Web-Based siRNA Design Tools Tools URLs siRNA Target Finder http://www.ambion.com/techlib/misc/siRNA finder.html/ BLOCK-IT RNAi Designer http://rnaidesigner.invitrogen.com/rnaiexpress/ GeneAssist siRNA Workflow Builder http://www5.appliedbiosystems.com/tools/sirna/ siDESIGN Center http://www.dharmacon.com/DesignCenter/ DesignCenterPage.aspx siRNA Target Designer http://www.promega.com/siRNADesigner/ SpecificityServer http://informatics-eskitis.griffith.edu.au/ SpecificityServer/ RNA-Based Methods in Cell Biology 27.1.3 Current Protocols in Cell Biology Supplement 47 5. Extend the sequence of the sense strand to 25 nt and antisense strand to 27 nt, making one end of the duplex blunt-ended with 2 deoxy 3 terminal end to block the Dicer entry. An example of a target sequence and siRNA are shown below. Target 5 -AAGCUGACCCUGAAGUUCAUCUGCACCACCGGC-3 siRNA Sense: ACCCUGAAGUUCAUCUGCACC Antisense: ACUGGGACUUCAAGUAGACGU D-siRNA Sense: ACCCUGAAGUUCAUCUGCACCACCG Antisense: ACUGGGACUUCAAGUAGACGUGGUGGC 6. Exchange the last two ribonucleotides of the sense strand to corresponding deoxyribonucleotides. For instance, change the ribonucleotides to dCdG (shown in lowercase below) if the last two sequences are CG. Sense: ACCCUGAAGUUCAUCUGCACCACcg Antisense: ACUGGGACUUCAAGUAGACGUGGUGGC 7. Once selected, synthesize siRNAs in-house or obtain from several siRNA-licensed vendors such as Ambion, Dharmacon, and Qiagen. When an siRNA is obtained in single-stranded format, the sense and antisense strands need to be annealed, and the integrity and quality of annealed double-stranded RNA need to be determined. These procedures are described in Support Protocols 4 and 5. BASIC PROTOCOL 2 DETERMINING OPTIMAL TRANSFECTION CONDITIONS In order to set up experiments with the designed siRNAs, several experimental conditions need to be optimized. First, it is important to determine the optimal cell density at the time of transfection. Transfection efficiency varies based on the cell density and concentrations of siRNAs, in addition to variations in transfection reagents used in the reaction. Second, optimal siRNA-lipid concentrations should be determined while screening for potent siRNAs to verify the most effective siRNA duplexes and the lowest concentration that results in efficient silencing of target gene without toxicity. Finally, it is recommended to empirically determine which reagent works best for your cell line. A variety of proprietary transfection reagents are available, and the efficiency of siRNA delivery to a particular cell line will slightly differ among the reagents. In this protocol, we describe a transfection procedure using Lipofectamine 2000 reagent (Invitrogen), which showed high transfection efficiency with commonly used mammalian cell lines (e.g., HEK293, HeLa, and HCT116 cells). Procedures described here are for adherent cells grown in monolayer. For suspension cells, the same procedures will apply, except that detaching cells from the surface of culture dishes is not necessary. Otherwise, deviations in the procedures will be noted accordingly. Silencing of Gene Expression in Cultured Cells For successful gene silencing by transfecting mammalian cells with siRNAs, it is strongly recommended to determine the optimal cell density of your cell line, and optimal siRNA concentrations, before your experiments. To find an optimal cell density, perform transfections with at least two different levels of confluency. In accordance with the recommended 27.1.4 Supplement 47 Current Protocols in Cell Biology Table 27.1.2 Recommended Percent Confluence for Transfection Cell types Source Recommended % confluencea,b HEK293 Human embryonic kidney 90% HeLa Human cervical carcinoma 70%-80% HCT116 Human colon carcinoma 50% A549 Human lung carcinoma 60% D3 Mouse embryonic stem cells Transfect cells at the time of plating 3T3L1 Mouse fibroblast 60%-70% a The listed cell densities are for 16 to 24 hr incubation. Adjust the seeding cell density so that cells reach confluency at the end of incubation if a longer interval between transfection and harvesting cells is necessary. b Information also found at Invitrogen Web site http://www.invitrogen.com. Table 27.1.3 Reagent Mixes for Transfection with Six Different Concentrations of siRNAs (12-well plates) siRNA final conc. (nM)a Stuffer DNA plasmid (ng)c Total nucleic acid amount (μg) 0 (μg) 1100 1.1 1.0 1 pmol (13.37 ng) 1087 1.1 5.0 5 pmol (66.85 ng) 1033 1.1 25 25 pmol (334.3 ng) 765.7 1.1 50 50 pmol (668.5 ng) 431.5 1.1 75 75 pmol (1003 ng) 97.21 1.1 0 siRNA per well (ng)b a siRNA concentrations were calculated for the final volume of 1 ml per well of a 12-well plate. b siRNA (ng) was calculated with a dsRNA, mol. wt. = 1.34 × 104 g/mol. c Any cloning vectors not containing eukaryotic promoters (e.g., pBluescript, pCR2.1, etc) can be used to adjust the total amount of nucleic acid. percent confluence of adherent cells shown in Table 27.1.2, seed the cells in multiple-well plates at a minimum of two different densities 1 day prior to transfection. In this protocol, 75% seeding density is used to plate four 12-well plates to perform a transfection with two siRNAs, an irrelevant siRNA as a negative control, and a Cy3labeled siRNA as a positive control, at six different concentrations for an assay period of 24 hr. It is strongly recommended to perform another round of transfection with adjusted cell density to optimize the transfection conditions for your experiment. Cells with shorter doubling times should be seeded at a lower density so that cells reach confluency at the end of the assay period (e.g. HCT116, ∼16 hr; HEK293, ∼22 to 24 hr; HeLa, ∼ 24 hr). Within a well of a 12-well plate, you should be able to collect sufficient amounts of total RNA and proteins for your silencing analyses (Support Protocols 1 to 3). If the target gene expression is relatively low, consider plating in 6-well plates instead. When you are analyzing gene function by knocking down gene expression with siRNA transfection, it is strongly recommended to verify the highly potent siRNA duplexes among siRNAs that you designed, and to identify the lowest concentration that results in efficient silencing of the target gene and is least toxic to cell culture. Procedures described below refer to transfection of cells seeded on four 12-well plates to determine optimal cell density for transfection with two siRNAs against the same target transcript. When more than two siRNAs are designed, adjust the number of plates to have enough wells for experimental determinations. To find optimal siRNA concentrations, one should test at least six different concentrations (e.g., 0, 1, 5, 25, 50, and 75 nM) in duplicate for RNA-Based Methods in Cell Biology 27.1.5 Current Protocols in Cell Biology Supplement 47 step 1: preparation of siRNA/DNA mix tube: 2 1 3 5 4 6 siRNA (nM): 0 1.0 5.0 25 50 75 stuffer DNA (μg): 2.42 2.39 2.27 1.69 0.95 0.21 total vol (μl): 220 220 220 220 220 220 step 2: preparation of LipofectamineTM 2000 solution 5 min incubation 220 μl per tube step 3: liposome formation tubes from step 1: 1 2 3 4 5 6 20 min incubation step 4: transfection 200 μl per tube tube 1 2 2 3 1 3 5 5 6 4 4 6 Figure 27.1.1 Transfection of mammalian cells with siRNAs. Stepwise description of transfection of mammalian cells seeded onto a 12-well plate a day prior to transfection is shown above. After choosing five different concentrations of siRNAs to transfect the cells, mix siRNA and stuffer DNA in the Opti-MEM (or a serum-free medium) to bring the volume to 220 μl (step 1). Next, prepare the proper amount of Lipofectamine 2000 solution (two tubes of 720 μl for six tubes of siRNA/DNA mixes, step 2). After 5 min incubation, add 220 μl of Lipofectamine 2000 solution to each siRNA/DNA mix (step 3). Incubate for 20 min to let liposome formation, then add 200 μl of the liposome solution to transfect the cells (step 4). Transfection reagent preparations should be done in a tissue culture hood. each siRNA as a starting point. Also, the use of a fluorescent dye–labeled siRNA as a transfection control (e.g., FITC, Cy3) will help visualize the transfection efficiency at different concentrations of an siRNA. See below for reagents contained in each transfection reaction (Table 27.1.3). A brief summary of the procedure is also shown in Figure 27.1.1. Silencing of Gene Expression in Cultured Cells NOTE: It is desirable to use early-passage-number cells if available. The effects vary among cell lines; however, passage number of the cells at the time of transfection may have impact on the efficiency of liposome uptake and may result in cellular toxicity. 27.1.6 Supplement 47 Current Protocols in Cell Biology NOTE: In the case of transfection with suspension cells, adjust the cell numbers accordingly. NOTE: Transfection reagent preparations should be done in a tissue culture hood. NOTE: The use of stuffer DNA plasmid will adjust the final amounts of nucleic acid to be consistent throughout the experiment; hence, maintaining the volume of Lipofectamine 2000 used in the reaction. We have often observed inconsistency in knockdown levels of target genes that result from varying the volume of cationic lipid solution for delivery of different concentrations of an siRNA. Hence, maintaining the volume of Lipofectamine 2000 throughout the experiment will not only prevent variation in transfection efficiencies among the six siRNA concentrations, but also will rectify cellular toxicity, which may be caused by the different amounts of cationic lipid used in the reaction. Any cloning vectors not containing eukaryotic promoters, such as pBluescript and pCR2.1, can be used as a stuffer DNA plasmid to adjust the total amount of nucleic acid. Materials Mammalian cell line cultured in the appropriate growth medium in 10-cm2 dishes Calcium- and magnesium-free Dulbecco’s phosphate-buffered saline (CMF-DPBS; Cellgro, cat. no. 21-031 CV, or see APPENDIX 2A), 37◦ C 1× trypsin/EDTA solution (e.g., Invitrogen; also see UNIT 1.1), 37◦ C Complete DMEM with 10% (v/v) FBS (see recipe), 37◦ C Complete DMEM with 10% (v/v) FBS (see recipe), without antibiotics, 37◦ C Trypan blue staining solution (UNIT 1.1) 10 μM siRNA working solution (Basic Protocol 1) 10 μM irrelevant siRNA as a negative control 10 μM fluorescent dye (e.g., Cy3 or FITC)–labeled siRNA as a transfection control 250 ng/μl stuffer DNA plasmid (e.g., pBluescript, pCR2.1; Clontech, Invitrogen) Opti-MEM I (a reduced-serum medium from Invitrogen), or serum-free growth medium Lipofectamine 2000 (Invitrogen) Microscope Hemacytometer with coverslip (Figure 1.1.1) 12-well tissue culture plates Standard microscope and UV lamp or fluorescence microscope Additional reagents and equipment for basic cell culture techniques including trypsinization and counting viable cells on a hemacytometer by trypan blue exclusion (UNIT 1.1) NOTE: All solutions and equipment coming into contact with living cells must be sterile, and aseptic technique should be used accordingly. NOTE: All cell culture incubations should be carried out in a 37◦ C, 5% CO2 humidified incubator. Seed cultured cells 1 day prior to transfection 1. For adherent cells grown at subconfluency on 10-cm2 dishes, rinse with 5 ml 1 CFM-DPBS and add 2 ml of 1× trypsin/EDTA solution per dish to detach cells from surface. This will be enough to cover the monolayer culture. In order to seed four 12-well plates at 75% cell confluency, prepare two 10-cm2 dishes of cells at ∼80% confluency. UNIT 1.1 includes protocols for basic cell culture techniques including trypsinization. RNA-Based Methods in Cell Biology 27.1.7 Current Protocols in Cell Biology Supplement 47 2. After 2 min incubation at 37◦ C, gently tap bottom of dish to dislodge cells. Also check culture under a microscope to see that cells are detached from the surface. The cells should look rounded up. For cells grown in suspension, skip steps 1 and 2. First centrifuge the cell culture 5 min at 200× g, room temperature, aspirate medium, and follow steps 3 to 9. 3. Gently resuspend cells by adding ∼5 ml of complete DMEM/10% FBS and pipetting up and down. The addition of the serum-containing medium will inhibit further trypsin digestion. 4. Transfer the cell suspension to a sterile 50-ml centrifuge tube, and centrifuge for 3 min at 200 × g room temperature. 5. Aspirate medium and resuspend the cells with 10 ml complete DMEM/10% FBS without antibiotics, so that maximum cell count will be ∼50 cells per 1 mm2 . If cells on a 10-cm dish are below 80% confluency, lower the volume of medium to adjust the maximum cell count to ∼50 cells per 1 mm2 . 6. Add 10 μl of trypan blue staining solution and a 10-μl aliquot of cell suspension to a 1.5-ml microcentrifuge tube. Mix thoroughly and dispense 10 μl of the mix gently to edge of hemacytometer counting chamber by placing the tip of pipet under the coverslip. UNIT 1.1 includes a protocol for viable cell counting using trypan blue and a hemacy- tometer. 7. Count cells under a microscope and calculate cell numbers as follows (also see UNIT 1.1): Cell numbers (cells/ml) = average count per square × 2 × 104 Total cells = cell numbers (cells/ml) × volume of medium used to dilute cells where 2 is the dilution factor, and 104 is the volume correction factor. 8. Determine cell viability by counting total number of viable (unstained) cells. % viable cells = [number of unstained cells/total number of cells] × 100 To plate cells at 75% seeding density on four 12-well plates, you will need a total of ∼1.5 × 107 viable cells. 9. Plate cells at a seeding density of 3.0 × 105 cells (initial 75% confluence) in 0.8 ml medium into wells of four 12-well plates. Make sure to distribute cells uniformly across the well to ensure even uptake of liposomes. On the day of transfection (i.e., the next day), cells with slow doubling time will reach ∼85% to 90% confluency. After the transfection efficiency is determined, scale up or down the seeding density and perform another round of transfection. For different cell lines, the numbers of cells at seeding should be adjusted so that they will reach confluency at harvesting. For instance, seed 4.0 × 105 HEK293 cells to obtain 90% confluency for a 24-hr assay period. Also refer to Table 27.1.2 for the recommended % confluency at transfection for different cell lines so that the optimal seeding density can be empirically determined. 10. Incubate the cells at 37◦ C, 5% CO2 for subsequent transfection. Silencing of Gene Expression in Cultured Cells Do not add antibiotics to the medium during transfection. If complete DMEM/10% FBS is used to seed cells, exchange the medium to complete DMEM/10% FBS without antibiotics once the cells are attached to wells, or at the latest 4 to 6 hr prior to transfection. 27.1.8 Supplement 47 Current Protocols in Cell Biology Prepare siRNA/DNA mixes 11. Prepare 40 μl of 1 μM siRNA by diluting 4 μl of the 10 μM siRNA working solution in 36 μl of Opti-MEM I (or serum-free medium) so that there are two different concentrations for a given siRNA. Using the same technique, prepare 1 μM solutions for three other siRNAs—the second siRNA to be tested, one Cy3-labeled siRNA, and one irrelevant control siRNA. It is recommended that you use serum-free medium to dilute siRNAs, to prevent potential reduction in transfection efficiency. It is not necessary to perform annealing (Support Protocol 4 and 5) if the siRNA is obtained in the double-stranded form. If the siRNA is purchased based on the match in the literature and available siRNA databases, it will be still ideal to determine the transfection efficiency (Support Protocols 1 to 3) to evaluate technical skills/conditions. One can interpret an irrelevant siRNA control as an siRNA whose sequence cannot be found in the system that the investigator tests with. An irrelevant/control siRNA can be purchased from several siRNA-licensed vendors such as Ambion, Dharmacon, and Qiagen. The sequences of such control siRNAs may not be available, for proprietary reasons. For the fluorescently labeled siRNA, in an ideal experimental setting, one should obtain the same siRNA that was designed in Basic Protocol 1, but with a fluorescent label. This can be done by custom ordering from the vendor. However, synthesizing the fluorescently labeled siRNA for each siRNA designed in Basic Protocol 1 is not cost-effective. Instead, one can purchase a fluorescently labeled control siRNA from vendors such as Invitrogen. 12. Prepare six 1.7-μl microcentrifuge tubes with proper labeling (0, 1, 5, 25, 50, 75 nM) for each siRNA, add each reagent as described in Table 27.1.3, and bring the final volume to 100 μl per tube with Opti-MEM I. Mix gently. It is strongly recommended to perform transfection in duplicate. From this point forward, volumes of reagents are calculated for duplicate reactions and shown on the right-hand side of Table 27.1.4 and Figure 27.1.1. Table 27.1.4 Reagent Mixes for 0, 1, 5, 25, 50, and 75 nM of siRNA (12-well plates) Per well For 2.2x siRNA per well siRNA per wellb siRNA final conc. (nM)a 1 μM (μl) 1 0 – – 1100 2 1.0 2.0 – 3 5.0 10.0 4 25 5 6 Tubes 10 μM Stuffer DNA Total volume (μl) plasmid (ng)c per well (μl) 1 μM (μl) 10 μM (μl) Stuffer DNA plasmid (ng) Total volume per well (μl)d 100 – – 2420 220 1087 100 4.4 – 2391 220 – 1033 100 22.0 – 2273 220 – 5.0 765.7 100 – 11.0 1685 220 50 – 10.0 431.5 100 – 22.0 949.3 220 75 – 15.0 97.2 100 – 33.0 213.8 220 a siRNA concentrations were calculated for the total volume of 1 ml per well of a 12-well plate. b siRNA (ng) was calculated with a dsRNA, mol. wt. = 1.34 × 104 g/mol. c Any cloning vectors not containing eukaryotic promoters (e.g., pBluescript, pCR2.1, etc) can be used to adjust the total amount of nucleic acid. d The 220 μl of siRNA/DNA mixes contains a 0.2× volume excess to the actual volume for a well. (220 μl = 100 μl × 1.2 × 2 wells). RNA-Based Methods in Cell Biology 27.1.9 Current Protocols in Cell Biology Supplement 47 75 nm 50 nm 25 nm 5 nm 1 nm Figure 27.1.2 Detection of Cy3-labeled siRNA uptake by HEK293 cells. After 24 hr incubation, the transfection efficiency of 0, 1, 5, 25, 50, and 75 nM Cy3-labeled siRNAs were observed under a microscope with a UV lamp. Excitation wavelength of Cy3 is ∼550 nm and emission wavelength is ∼570 nm. Representative results are shown. For the color version of this figure go to http://www.currentprotocols.com/protocol/cb2701. Silencing of Gene Expression in Cultured Cells 27.1.10 Supplement 47 Current Protocols in Cell Biology Prepare Lipofectamine 2000 solution 13. Mix Lipofectamine 2000 gently and microcentrifuge briefly to bring the solution to the bottom of the tube. 14. Dilute 2.0 μl Lipofectamine in 100 μl Opti-MEM I per well. For six different concentrations of a siRNA (for 12 wells), prepare 1440 μl of Lipofectamine 2000 solution in two tubes (=14.4 μl Lipofectamine 2000 in 720 μl Opti-MEM I per tube). Prepare three more sets for other siRNA/DNA mixes (total of eight tubes) (step 2, Fig. 27.1.1). The 1440 μl of Lipofectamine 2000 solution contains a 0.091× volume excess of the volume for six tubes of the siRNA/DNA mixes (tubes 1 to 6 in the table; 1440 μl =220 μl × 1.091 × 6 siRNA/DNA mixes). 15. Mix gently and incubate for 5 min at room temperature. Form liposomes 16. Prepare DNA-siRNA-lipid complex (liposome) by adding 220 μl of the Lipofectamine 2000 solution to 220 μl of the siRNA/DNA mix. Mix gently and incubate for 20 min at room temperature for liposome formation (step 3, Fig. 27.1.1). Transfect cells 17. Add 200 μl of the liposome solution dropwise to each well containing cells and 0.8 ml medium (step 4, Fig. 27.1.1). Mix gently by rocking the plate back and forth, left to right several times. 18. Incubate the cells at 37◦ C, 5% CO2 until cells are ready for harvesting. Generally the assay period is 24 to 48 hr. 19. At the end of incubation, observe transfection efficiency with the Cy3-labeled siRNA under a microscope with a UV lamp or fluorescence microscope. The excitation wavelength of Cy3 is ∼550 nm and the emission wavelength is ∼570 nm. Representative results are shown in Figure 27.1.2. Under some experimental conditions (e.g., multiple cycles of transfection for a prolonged assay period), growth medium may be replaced after 4 to 6 hr of incubation without loss of transfection activity; this helps to reduce cellular toxicity. After 24 hr transfection, one can perform one of following procedures to estimate/measure transfection efficiency (should include a positive control for Cy3 fluorescence detection); approaches are listed in order from estimations to accurate measurements: Visual observation: Observe Cy3 fluorescence cells under a microscope with a UV lamp/fluorescence microscope and compare and estimate a ratio Cy3 fluorescence cells to total cells. Counting cell numbers: In a visual field under the microscope, count Cy3 fluorescence cell numbers and total cell numbers, and take a ratio. Transfection efficiency = Cy3 fluorescing cells/total cells. FACS analysis: Count cell numbers of negative control cells, positive control cells, and transfected cells, and take a ratio. Transfection efficiency = Cy3 fluorescing cells/total cells). 20. Once you optimize the transfection conditions, perform your experiment with the most potent siRNAs at an optimal cell density and siRNA concentration and using Lipofectamine 2000 at least three times for validating the reproducibility. Once the potent siRNAs are evaluated on 12-well plates, we run scaled-up experiments on 6-well plates to obtain sufficient samples for subsequent analyses. 21. Assess the results of transfection by PCR (Support Protocol 1), immunoblotting (Support Protocol 2), or a dual-reporter system (Support Protocol 3). RNA-Based Methods in Cell Biology 27.1.11 Current Protocols in Cell Biology Supplement 47 SUPPORT PROTOCOL 1 ASSESSING siRNA TRANSFECTION EFFICIENCY BY RT-PCR The importance of detection of target transcript levels after siRNA treatment is often overlooked. Potentially, the target gene might express a protein with a long half-life; therefore, the effect of the siRNA treatment may not be apparent by immunoblot analyses (described in Support Protocol 2). Thus, following a short period of time post-transfection, it is recommended to validate the effect of designed siRNAs at the transcript level. The efficiency of down-regulation of target gene expression by siRNAs can be determined by real-time quantitative RT-PCR (qRT-PCR) or end-point RT-PCR reactions. Here, we describe the isolation of total RNA from siRNA-treated cells, and cDNA preparation, in detail. Reagents available for RNA extraction are RNA STAT-60 (TEL-TEST, INC.) or TRIzol (Invitrogen), and procedures for both reagents are very similar. In this protocol, we will describe the use of RNA STAT-60 in total RNA isolation. Materials siRNA-treated cells on 12-well plates (Basic Protocol 2) RNA STAT-60 (Tel-Test, Inc., http://www.tel-test.com/) at 4◦ C Chloroform 75% ethanol made with nuclease-free or diethylpyrocarbonate (DEPC)-treated H2 O (see APPENDIX 2A fro DEPC treatment) Nuclease-free or DEPC-treated water (APPENDIX 2A) 2 U/μl DNase I (RNase-free) and 10× DNase buffer (Ambion) 1 U/μl RNase inhibitor (RNasin, Promega) Random hexamer primers [or oligo(dT)12-18 ] 10 mM dNTP mix(10 mM dATP, dCTP, dGTP, and dTTP) 5× first-strand buffer (Invitrogen) 0.1 M DTT MMLV reverse transcriptase (Invitrogen) Forward and reverse PCR primers iQ SYBR Green Supermix (for real time qPCR; BioRad) Refrigerated centrifuge NanoDrop 1000 (Thermo Fisher Scientific) or UV/Vis spectrophotometer PCR tubes 65◦ and 80◦ C water baths or heating block Additional reagents and equipment for spectrophotometric determination of RNA concentration (APPENDIX 3D), standard PCR (Kramer and Coen, 2001), and real-time qPCR (Bookout et al., 2006) Collect total RNA from siRNA treated cell culture 1. At the end of siRNA treatment (Basic Protocol 2), aspirate medium from the cell culture. Do not wash cells with CMF-DPBS, since this may result in RNA degradation. For cells grown in suspension, centrifuge the suspension and collect the cell pellet. 2. Lyse the cells by directly adding RNA STAT-60 (1 ml/2.5 to 5 × 106 cells; e.g., 300 μl of RNA STAT-60 per well on a 12-well plate) and pipetting up and down with a 200-μl pipet tip until the viscosity disappears. If the viscosity persists, add 50-μl increments of RNA STAT-60 until the lysate becomes transparent. It is critical to lyse cells thoroughly to extract RNA of high quality. 3. Transfer the lysate to a 1.7-ml microcentrifuge tube. Silencing of Gene Expression in Cultured Cells In the case of collecting lysates from a 10-cm dish, use a 2.0-ml microcentrifuge tube or aliquot the lysate into multiple tubes. However, aliquotting into multiple tubes may cause RNA extraction efficiency to vary among tubes. 27.1.12 Supplement 47 Current Protocols in Cell Biology Extract RNA 4. Let the lysate stand for 5 min at room temperature to let nucleoprotein complexes dissociate. 5. Add 0.2 vol of chloroform to the RNA STAT-60 (i.e., 0.2 ml of chloroform to the 1 ml of RNA STAT-60 solution). 6. Cap tightly and vortex for 15 sec. Let it stand for 2 to 3 min at room temperature. 7. Centrifuge 15 min at 12,000 ×g, 4◦ C. 8. Transfer the upper aqueous phase to a 1.7-ml microcentrifuge tube. The volume of the aqueous phase will be ∼0.6 vol of RNA STAT-60 used for cell lysis. DNA and proteins remain in the interphase and organic phase. While you are pipetting out the aqueous phase, be careful not to disturb the white debris. Precipitate RNA 9. Add 0.5 volume of isopropanol (with respect to the volume of RNA STAT-60 used, i.e., 150 μl of isopropanol when 300 μl of RNA STAT-60 is used). Mix by pipetting up and down and let stand 5 min at room temperature. 10. Centrifuge 10 min at 12,000 × g, 4◦ C. You will see RNA precipitated as a white pellet at the bottom of the tube. 11. Discard supernatant, and add 1 vol of 75% ethanol made with DEPC-treated water to wash the RNA pellet. Mix by inverting the tube a couple of times. 12. Centrifuge 5 min at 7500 × g, 4◦ C. 13. Carefully discard supernatant and air-dry the RNA pellet. It is important to avoid drying the pellet completely, since that will reduce its solubility. 14. Dissolve the RNA pellet in nuclease-free water (or DEPC-treated water) so that the concentration of total RNA will be around 1 to 2 μg/μl. Generally, you will obtain about 5 to 10 μg of total RNA from 0.4 × 106 cells. Adding 15 μl of nuclease-free water to the RNA pellet will be enough to dissolve the RNA pellet. 15. Measure the concentration and check the quality of the total RNA using a NanoDrop assay or a spectrophotometer with UV lamp. APPENDIX 3D includes protocols for spectrophotometric determination of RNA con- centration. The quality of the RNA can be estimated by measuring absorbance at 260 nm and 280 nm and take a ratio 260/280 for purity. In general, a ratio ≥1.8 is considered to be good quality for real-time PCR analysis (with 2.0 being the highest). Treat with DNase I 16. For a 15-μl reaction, mix the following reagents in a PCR tube at room temperature (15 μl total volume). Prepare two tubes per sample so that one tube will be used for reverse transcription (RT) negative control. 4.0 μl 0.5 μg/μl total RNA 1.5 μl 10× DNase buffer 1.0 μl 2 U/μl RNase-free DNase I 0.5 μl 1 U/μl RNase inhibitor 8.0 μl RNase-free water (or DEPC-treated water). RNA-Based Methods in Cell Biology 27.1.13 Current Protocols in Cell Biology Supplement 47 17. Mix, then microcentrifuge briefly to bring the mixture to the bottom of the tube. Incubate the reaction mix at 37◦ C for 60 min. 18. Heat-inactivate the DNase I by incubating the reaction mix at 80◦ C for 10 min. Immediately place the tube on ice and leave it for 3 min. 19. Centrifuge the tube briefly to bring the solution to the bottom of the tube. 20. Store the reaction mix at −20◦ C if necessary. Normalized relative Gene A/HRPT expression (%) A 120 100 80 60 40 20 0 si A si IR si B 10 nM siRNAs si A si B si IR A antibody tubulin Normalized relative protein level of Gene A/tubulin(%) B 120 100 80 60 40 20 0 si A si B si IR 10 nM siRNAs Figure 27.1.3 Effects of siRNA target gene down-regulation. (A) Determination of siRNA silencing efficiency by real-time qRT-PCR. Silencing of Gene A by si A or si B (10 nM) was determined with qRT-PCR. An irrelevant siRNA, si IR, was used as a negative control and used to normalize the Gene A expression level. The expression levels of the gene A was also analyzed relative to HRPT expression level set as 100%. The effects of siRNAs were determined in three independent transfections, and qRT-PCR reactions were performed in duplicate. (B) Determination of siRNA silencing efficiency by immunoblot analyses. Silencing of a gene A by si A or si B (10 nM) was determined at protein level. Quantification of protein expression levels are shown. An irrelevant siRNA, si IR, was used as a negative control and used to normalize the Gene A expression level. The expression levels of the Gene A was also analyzed relative to tubulin expression level set as 100%. The effects of siRNAs were determined in three independent transfections, and immunoblot analyses were performed independently. Silencing of Gene Expression in Cultured Cells 27.1.14 Supplement 47 Current Protocols in Cell Biology Synthesize cDNA 21. Add following reagents to the 15-μl DNase I reaction mix in PCR tubes (18 μl total volume): 2.0 μl 50 ng/μl random hexamers 1.0 μl 10 mM dNTP mix. 22. Incubate the reaction mix at 65◦ C for 5 min. In the meantime, prepare the solution described in step 23. After 5 min incubation, immediately place tubes on ice. 23. Add the following reagents to the 18-μl reaction mix (27 μl total volume). For RT negative control, add 1.0 μl of DEPC-treated water in place of the MMLV-RT. 5.0 μl 5× first-strand buffer 2.5 μl 0.1 M DTT 0.5 μl 1 U/μl RNase inhibitor 1.0 μl MMLV-RT. 24. Incubate the 27-μl cDNA synthesis mix at 27◦ C for 10 min and 37◦ C for 50 min. 25. Heat-inactivate MMLV-RT by incubating the synthesis mix at 70◦ C for 15 min. Immediately place on ice and add 173 μl DEPC-treated water to bring the final volume to 200 μl. Perform PCR 26. Perform either end-point PCR (standard PCR, e.g., Kramer and Coen, 2001, with determination of reaction product on a gel) or real-time qPCR (Bookout et al., 2006) to enable one to determine the gene silencing levels after siRNA treatment. In a PCR reaction, use 10 μl of synthesized cDNA to assess target RNA levels. Representative qRT-PCR results are shown in Figure 27.1.3A. ASSESSING siRNA TRANSFECTION EFFICIENCY BY IMMUNOBLOTTING Protein levels of target genes after siRNA treatments are often determined to validate the effects of designed siRNAs. To fully describe the effect of siRNA on protein levels, it is important to know the half-life of the target protein. With a protein that has a long half-life, you may adjust the length of incubation with siRNAs and/or numbers of transfections during the assay period or perform multiple cycles of transfection for a long-term assay to see the effects of siRNAs at the protein level. Here, we describe cell lysis for collecting total proteins for further analysis. Refer to UNIT 6.2 for immunoblotting procedures. Prepare total protein from siRNA-treated cells for determination of the effects of transfection experiments, and mock-treated cells as a control for protein detection. SUPPORT PROTOCOL 2 Materials siRNA-treated cell culture in 12-well plate (Basic Protocol 2) Mock-treated cell culture Calcium- and magnesium-free Dulbecco’s phosphate-buffered saline (CMF-DPBS; Cellgro, cat. no. 21-031 CV, or see APPENDIX 2A), 4◦ C RIPA buffer (see recipe) with freshly added 1× protease inhibitor cocktail (Roche), ice cold End-over-end rotator (e.g., Labquake from Thermo Scientific) Additional reagents and equipment for Bradford protein assay (APPENDIX 3H) and immunoblotting (UNIT 6.2) RNA-Based Methods in Cell Biology 27.1.15 Current Protocols in Cell Biology Supplement 47 Lyse cells 1. At the end of siRNA treatment (Basic Protocol 2), aspirate medium from the cell culture. Wash cells once with 0.5 ml ice-cold CMF-DPBS. Keep the plate on ice. Also perform this and all subsequent steps on a mock-transfected culture as a control. For cells grown in suspension, collect the cell suspension, centrifuge 5 min at 200 × g, 4◦ C, and collect cell pellet. Wash the pellet with 1 ml ice-cold CMF-DPBS, and centrifuge again at 4◦ C. 2. Lyse the cells by directly adding ice-cold RIPA buffer with freshly added 1× protease inhibitor cocktail (1 ml per 2–5 × 106 cells, e.g., 300 μl per well on a 12-well plate). Let it sit ∼ 2 min on ice so that the cells detach from the surface. 3. Collect cells with a pipettor and 1000-μl pipet tip by tilting the plate on ice, and transfer to a cold 1.7-ml microcentrifuge tube. It is critical to keep cells on ice to prevent protein degradation. If necessary, use a scraper to collect cells. Adding more RIPA buffer may lower total protein concentrations. 4. Incubate the cells for 15 min at 4◦ C with gentle agitation on an end-over-end rotator. 5. Microcentrifuge 15 min at 14,000 rpm, 4◦ C, and transfer supernatant to a new, cold 1.7-ml tube. 6. Measure total protein concentration with Bradford assay (APPENDIX 3H), and aliquot the lysate into several tubes at ∼50 or 100 μl/tube to avoid multiple freeze-thaw cycles. Store the aliquots at −80◦ C. 7. Perform immunoblotting (UNIT 6.2). Representative results are shown in Figure 27.1.3B. SUPPORT PROTOCOL 3 ASSESSING siRNA TRANSFECTION EFFICIENCY BY A DUAL-REPORTER ASSAY SYSTEM Prevalidation of siRNA potency and optimization of transfection efficiency can be performed by developing reporter genes fused to a target gene. For instance, green fluorescent protein (GFP) fluorescence in cell culture can be quantified by flow cytometry, or a FLAG epitope tag can be used to detect the change in protein expression levels by immunoblot analysis. However, the former method requires that a FACS facility be readily available, and the latter tends to be time consuming. Conversely, a dualluciferase reporter assay system by Promega accelerates the identification of potent siRNAs among designed siRNAs in 24 to 48 hr by detecting changes in expression of a reporter gene. With this system, the transfection of the dual-luciferase reporter vectors (psiCHECK vectors, Promega), carrying the target sequence, into mammalian cell lines leads to expression of the target sequence fused to the reporter gene, which is translated into the functional Renilla luciferase (http://www.promega.com). The catalytic activity of Renilla luciferase measured by bioluminescent reactions is used to determine how effective siRNAs initiate RNAi by observing reductions in the enzymatic activity. Silencing of Gene Expression in Cultured Cells Simultaneous transfection of two plasmids, one expressing a target gene and another expressing a control protein, can provide evaluation of siRNA potency by normalizing expression levels of the reporter protein to the control protein at time of transfection. However, results may vary from transfection to transfection due to inconsistent transfection efficiencies between the reporter and control plasmids. The psiCHECK-2 (Fig. 27.1.4, Promega) contains an additional internal control reporter gene that improves the reproducibility and that enables evaluating well-to-well variation of transfection (http://www.promega.com). The Renilla luciferase is expressed from the vector 27.1.16 Supplement 47 Current Protocols in Cell Biology SV40prom intron1 T7 Amp R hRluc psiCheck2-GFP-sense 6524 bp SV40late polyA I-Rluc-3F primer S1 siRNA site GFPfragment, sense synth poly A HSV-TK prom hFluc Figure 27.1.4 A reporter plasmid psiCHECK-2 (Promega) containing a target site GFP S1. A reporter plasmid psiCHECK2-GFP-sense was prepared by cloning a PCR fragment that contains the target sequence complementary to siRNAs into the multiple cloning region of the psiCHECK-2 (S1 siRNA site). The psiCHECK-2 contains an additional internal control reporter gene, firefly luciferase (hluc+), which is used to normalize the Renilla luciferase activity. Depending on experimental settings, the siRNAs can be either cotransfected or transfected sequentially. Small interfering RNA silencing efficiency was evaluated by measuring the activities of firefly and Renilla luciferases sequentially from a single sample using a luminometer (http://www.promega.com). containing the target gene sequence cloned into the multiple cloning site located in the 3 untranslated region (UTR) of a humanized Renilla luciferase (hRluc) reporter gene (http://www.promega.com). The firefly luciferase (hluc+) expressed from the same vector is used to normalize the Renilla luciferase activity, serving as the baseline response (http://www.promega.com). Depending on experimental settings, the siRNAs can be either cotransfected or transfected sequentially. In this protocol, cotransfection of siRNAs and the psiCHECK-2 vector is described in detail. Prepare a reporter plasmid by cloning a PCR fragment that contains the target sequence complementary to siRNAs into the multiple cloning region of the psiCHECK-2 (S1 siRNA site, Fig. 27.1.4). The efficiency of siRNA silencing is evaluated by measuring the activities of firefly and Renilla luciferases sequentially from a single sample using a manual luminometer or a luminometer with one or two reagent injectors. Promega provides the Dual-Luciferase Reporter Assay system, containing reagents necessary for cell lysis and measuring Renilla and firefly luciferase activities. Refer to the manufacturer’s protocol for more information (http://www.promega.com). In this section, we describe cotransfection of mammalian cell culture with psiCHECK-2 reporter plasmid containing a target sequence and siRNAs for validation of the most effective siRNA duplexes among siRNAs, and the lowest concentration that results in efficient silencing. To find optimal siRNA concentrations, one should test at least six different concentrations (e.g., 0, 0.05, 0.5, 1.0, 20, and 45 nM) for each siRNA with a constant amount of the reporter plasmid as a starting point. The procedure described here refers to transfection of cells seeded in 48-well plates at two different levels of confluence (one at 75% and another at 90% confluency) with the reporter plasmid in addition to the siRNAs. See below for the summary of compositions for transfection reaction mixes shown in Table 27.1.5. Transfection procedures deviating from the previous section (with 12-well plates) are emphasized in this section (see the flow chart in Fig. 27.1.5). Once the conditions are optimized, you may perform transfection in a different format (e.g., 6- or 12-well plates) to accommodate your subsequent RNA-Based Methods in Cell Biology 27.1.17 Current Protocols in Cell Biology Supplement 47 Table 27.1.5 Transfection Reaction Mixes for Six Different siRNA Concentrations (48-Well Plates) siRNA final siRNA per well conc. (nM)a (ng)b psiCHECK reporter plasmid (ng) Stuffer DNA plasmidc Total nucleic acid amount (ng) 0 0 (0 ng) 40 120.00 160 0.05 10 fmol (0.134 ng) 40 119.87 160 0.5 100 fmol (1.34 ng) 40 118.66 160 1.0 200 fmol (2.67 ng) 40 117.30 160 20 4 pmol (53.5 ng) 40 66.50 160 45 9 pmol (120 ng) 40 0 160 a siRNA concentrations were calculated for the total volume of 200 μl per well of a 48-well plate. b siRNA (ng) was calculated with a dsRNA, mol. wt. = 1.34 × 104 g/mol. c Any cloning vectors not containing eukaryotic promoters (e.g., pBluescript, pCR2.1, etc) can be used to adjust the total amount of nucleic acid. experimental procedures (e.g., when collecting total proteins for immunoblot analyses or isolating total RNA for qRT-PCR, seed cells in 6- or 12-well plates while keeping the optimal percent confluency and optimal siRNA concentrations consistent at the time of transfection). NOTE: Transfection reagent preparations should be carried out in a tissue culture hood. Materials 10 μM siRNA working solution (Basic Protocol 1) 10 μM irrelevant siRNA as negative control psiCHECK-2 vector with target cloned into the MCS (psiYTC) Stuffer DNA plasmid (see Basic Protocol 2) Opti-MEM I (a reduced-serum medium from Invitrogen), or serum-free growth medium Lipofectamine 2000 (Invitrogen) Mammalian cells seeded in two 48-well plates Complete medium with 10% FBS (e.g., complete DMEM/10% FBS; see recipe), without antibiotics, 37◦ C Dual-Luciferase Reporter Assay System (Promega) Orbital shaker Luminometer (e.g., Veritas Microplate Luminometer) NOTE: All solutions and equipment coming into contact with living cells must be sterile, and aseptic technique should be used accordingly. NOTE: All cell culture incubations should be carried out in a 37◦ C, 5% CO2 humidified incubator. Prepare DNA master mixes 1. Prepare six tubes properly labeled (0, 0.05, 0.5, 1.0, 20, 45 nM) for each corresponding siRNA mix, add each reagent as described in Table 27.1.6 and step 1 in Fig. 27.1.5, and bring the final volume to 16 μl per well with Opti-MEM I. Mix gently. Silencing of Gene Expression in Cultured Cells It is strongly recommended to perform transfections in duplicate. From this point forward, volumes of reagents are calculated for duplicate transfections. 27.1.18 Supplement 47 Current Protocols in Cell Biology step 1: preparation of DNA mix tube: D1 D2 D3 D4 D5 D6 psiYTC (ng): 576 576 576 576 576 576 stuffer DNA (ng):1728 1726 1709 1689 957.6 0 total vol (μl): 230.4 230.4 230.4 230.4 230.4 230.4 step 3: preparation of siRNA/DNA mix 70.4 μl of D6 per tube step 2: preparation of siRNA mixes SiRNA-A 1 2 3 4 5 6 2 3 4 5 6 tube: 1 siRNA-B 1 2 3 4 5 6 siRNA-C 1 2 3 4 5 6 siRNA-C 1 2 3 4 5 6 0.05 0.5 1.0 20 45 siRNA (nM): 0 total vol (μl):17.6 17.6 17.6 17.6 17.6 17.6 step 4: preparation of LipofectamineTM 2000 solution 5 min incubation 88 μl per tube SiRNA-A 1 2 3 4 5 step 5: liposome fomation siRNA-B 1 2 3 6 4 20 min incubation step 6: transfection si-RNA siRNA-B siRNA-C seeding density: 2 1 1 2 75% 4 5 4 4 3 3 40 μl per well 3 2 1 6 5 6 3 2 1 1 3 2 1 6 5 5 6 2 3 5 4 4 5 4 5 6 6 6 si-RNA siRNA-B siRNA-C 90% Figure 27.1.5 Transfection of mammalian cells with psiCHECK2-GFP-sense and siRNAs (48-well plates). Stepwise procedures of transfection of mammalian cells seeded at two different cell densities (75% and 90%) a day prior to transfection are shown. After choosing five different concentrations of siRNAs (0 to 45 nM) to transfect the cells, prepare DNA master mixes and siRNA mixes separately in the Opti-MEM (or a serum-free medium) as indicated (steps 1 and 2). Next, add 70.4 μl of D1 to tubes containing 0 nM of siRNAs A, B, or C (tubes A1, B1, and C1). Repeat with D2 to D6 to prepare siRNA/DNA mixes (step 3). Prepare proper amount of Lipofectamine 2000 solution (three tubes of 576 μl for six tubes of siRNA/DNA mixes, step 4). After 5 min incubation, add 88 μl of Lipofectamine 2000 solution to each siRNA/DNA mix (step 5). Incubate for 20 min to allow liposome formation, then add 40 μl of the liposome solution to two wells on the 75% seeding density plate and two wells on the 90% seeding density plate (step 6). Transfection reagent preparations should be done in a tissue culture hood. RNA-Based Methods in Cell Biology 27.1.19 Current Protocols in Cell Biology Supplement 47 Table 27.1.6 DNA Master Mixes for 0, 0.05, 0.5, 1.0, 20, and 45 nM siRNAs for Dual Reporter Assays Per well For 14.4× For siRNA final conc. (nM)a psiYTC reporter plasmid (ng)b Stuffer DNA plasmid (ng)c Total volume per well (μl) psiYTC reporter plasmid (ng)b Stuffer DNA plasmid (ng)c Total volume per well (μl) D1 0 40 120.00 16 576 1728 230.4 D2 0.05 40 119.87 16 576 1726 230.4 D3 0.5 40 118.66 16 576 1709 230.4 D4 1.0 40 117.30 16 576 1689 230.4 D5 20 40 66.50 16 576 957.6 230.4 D6 45 40 0 16 576 0 230.4 Tube a siRNA concentrations were calculated for the total volume of 200 μl per well of a 48-well plate. b The optimal amount of psiCHECK-2 vector with your target site (psiYTC) should also be experimentally determined. Here an example of 40 ng of psiYTC is used. c Any cloning vectors not containing eukaryotic promoters (e.g., pBluescript, pCR2.1, etc) can be used to adjust the total amount of nucleic acid. Table 27.1.7 siRNA Mixes for Six Different siRNA Concentrations Per well For 14.4× For siRNA final conc. (nM)a psiYTC reporter plasmid (ng)b Stuffer DNA plasmid (ng)c Total volume per well (μl) psiYTC reporter plasmid (ng)b Stuffer DNA plasmid (ng)c Total volume per well (μl) D1 0 40 120.00 16 576 1728 230.4 D2 0.05 40 119.87 16 576 1726 230.4 D3 0.5 40 118.66 16 576 1709 230.4 D4 1.0 40 117.30 16 576 1689 230.4 D5 20 40 66.50 16 576 957.6 230.4 D6 45 40 0 16 576 0 230.4 Tube a siRNA concentrations were calculated for the total volume of 200 μl per well of a 48-well plate. 2. For three siRNAs—two test siRNAs and an irrelevant siRNA—and two 48-well plates with different cell densities, prepare 230.4 μl (14.4× volume) of DNA master mix per tube. The 230.4 μl of DNA master mix per tube contains 0.091× volume excess of the volume of the DNA mix added per siRNA mix tube (230.4 μl = 17.6 μl × 1.091 × 4 wells ×3 siRNA concentrations). Prepare siRNA mixes 3. Prepare 20 μl of 0.1 and 1 μM siRNA by diluting 2 μl of the 10 μM siRNA working solution in 18 μl of Opti-MEM I and 2 μl of the 1 μM siRNA solution in 18 μl of Opti-MEM I so that you have three different concentrations for each siRNA. Silencing of Gene Expression in Cultured Cells 4. Prepare three sets of six tubes properly labeled (0, 0.05, 0.5, 1.0, 20, 45 nM) for a set per siRNA, add reagents to each tube as described in Table 27.1.7 and step 2 in 27.1.20 Supplement 47 Current Protocols in Cell Biology Fig. 27.1.5, and bring the final volume with Opti-MEM I to 17.6 μl per well. Mix gently. To perform transfections in duplicate, and since the cells are seeded in two 48-well plates with different cell densities, prepare 17.6 μl (4.4 × volume) of siRNA mix for 4 wells. The 17.6 μl of siRNA/DNA mix per tube contains 0.1× volume excess of the actual volume of siRNA mix used for a well (17.6 μl = 4.0 μl × 1.1 × 4 wells). Prepare siRNA/DNA mixes 5. Add 70.4 μl of DNA master mixes to corresponding siRNA mixes [i.e., add 70.4 μl of the DNA master mix for 45 nM siRNA (tube D6) to tubes containing 17.6 μl of 45 nM siRNA-A, siRNA-B, and irrelevant siRNA (siRNA-C) so that the total volume now is 88 μl per tube]. Add other DNA master mixes for 0, 0.05, 0.5, 1, and 20 nM to corresponding siRNA mix tubes (step 3 in Fig. 27.1.5). Prepare Lipofectamine 2000 solution 6. Mix Lipofectamine 2000 gently and centrifuge briefly to bring the solution to the bottom. 7. Dilute 0.5 μl in 20 μl Opti-MEM I per well. For six different concentrations of an siRNA at four wells per concentration, prepare 576 μl of Lipofectamine 2000 solution (=14.4 μl Lipofectamine 2000 in 576 μl Opti-MEM I) for 24 wells (=6 siRNA concentrations × duplicate × 2 plates). Prepare two more tubes of the Lipofectamine 2000 solution for other siRNAs as well (total of three tubes; step 4, Fig. 27.1.5). The 576 μl of Lipofectamine 2000 solution contains 0.091× volume excess of the volume of the Lipofectamine 2000 solution to be added to the siRNA/DNA mix per tube (576 μl = 88 μl × 1.091 × 6 siRNA concentrations). 8. Mix gently and incubate for 5 min at room temperature. Form liposomes 9. Prepare DNA/siRNA/lipid complex (liposome) by adding 88 μl of the Lipofectamine 2000 solution to 88 μl of the siRNA/DNA mix. Mix gently and incubate for 20 min at room temperature for liposome formation (step 5, Fig. 27.1.5). Transfect cells 10. Add 40 μl of the liposome solution drop-wise to each well containing cells and 160 μl medium (step 6, Fig. 27.1.5A). Mix gently by rocking the plate back and forth, and left to right several times. 11. Incubate the cells at 37◦ C, 5% CO2 until cells are ready for harvesting. Generally the cells are harvested at 24 to 48 hr. Lyse cells 12. Prepare ∼8 ml of 1× passive lysis buffer from 5× passive lysis buffer stock, which is part of the Dual-Luciferase Reporter Assay System (Promega) for 96 wells (=75 μl/well × 48 wells × 2 plates + extra). 13. Add 75 μl per well and shake the culture plates on an orbital shaker for 15 min at room temperature. The rocking motion will ensure covering the cells evenly and completely with the 1× passive lysis buffer. 14. Immediately proceed to dual-luciferase assays (using kit from Promega and luminometer) or store samples at −80◦ C. RNA-Based Methods in Cell Biology 27.1.21 Current Protocols in Cell Biology Supplement 47 1.2 Normalized FLuc/RLuc 1.0 0.8 0.6 0.4 0.2 0.0 0 nM 0.05 nM 0.5 nM siRNA-A 1.0 nM 20 nM 45 nM siRNA-B Figure 27.1.6 Determination of transfection efficiency by the dual-luciferase assays. The efficiency of siRNA silencing was evaluated by measuring the activities of firefly and Renilla luciferases. The relative luminescence units were normalized to the negative control (0 nM) set as 100%. Results showed concentration dependent silencing efficiency of the two siRNAs A and B. Once an optimal concentration of a siRNA is determined, perform your experiment with the highly potent siRNA(s) at optimal cell density and concentrations at least three times to assess reproducibility. 15. Once you optimize the transfection conditions, perform your experiment with the most potent siRNAs at optimal cell density and concentrations at least three times to assess reproducibility. Representative results are shown in Figure 27.1.6. SUPPORT PROTOCOL 4 ANNEALING SINGLE-STRANDED OLIGOS FOR A DOUBLE-STRANDED RNA Depending upon the purpose of the experiments (e.g., siRNA structural modifications, chemical modifications on siRNA strands, etc), you may obtain siRNAs as singlestranded RNA oligos or duplex siRNAs. When single-stranded oligos are obtained, you should anneal equal amounts of each single-stranded oligo to generate a double-stranded RNA. NOTE: Gloves and plastic ware (no glass) should be used to avoid RNA degradation and cross-contamination of samples. Materials Lyophilized single-stranded sense and antisense oligos (Basic Protocol 1) RNase-free (e.g., DEPC-treated) H2 O or TE buffer (APPENDIX 2A) 10× annealing buffer (see recipe) Nuclease-free (e.g., DEPC-treated) H2 O 95◦ C heat block NanoDrop 1000 (Thermo Fisher Scientific) or UV/Vis spectrophotometer Silencing of Gene Expression in Cultured Cells Additional reagents and equipment for spectrophotometric determination of RNA concentration (APPENDIX 3D) 27.1.22 Supplement 47 Current Protocols in Cell Biology Prepare for annealing 1. Dissolve the single-stranded RNA oligos in nuclease-free water or TE buffer to a final concentration of 100 μM. Make sure the oligos are completely dissolved. Place them on ice. It is recommended to measure actual concentrations of single-stranded oligos after dissolving by measuring A260 with a NanoDrop or a spectrophotometer with UV lamp (APPENDIX 3D) and assess the purity of your RNA oligos before use. Anneal the oligos 2. For a 50-μl total volume reaction, mix the following reagents in a 1.7-ml sterile microcentrifuge tube at room temperature to generate a 25 μM dsRNA solution. 12.5 μl 100 μM sense RNA oligo 12.5 μl 100 μM antisense RNA oligo 5 μl 10× annealing buffer Nuclease-free H2 O for 50 μl. 3. Incubate the reaction mix at 95◦ C for 4 min. 4. Microcentrifuge the tube briefly to bring the solution to the tube bottom, and remove the heat block from the heat source and set on your lab benchtop. Protect the surface of the bench top from heat by placing an insulator between the heat block and the bench top. 5. Place the tube back in the heat block to slowly cool to room temperature by allowing the heat block to reach ambient temperature. This will gradually anneal the single-stranded oligos to dsRNA. It will take about 1.5 to 2 hr to reach room temperature. 6. Briefly mix and centrifuge the solution and place on ice. 7. Dilute the dsRNA to 10 μM with RNase-free water and use as a working solution. Store the remaining sample up to 6 months at −20◦ C or 2 to 3 years at −80◦ C as the master stock. Double-stranded siRNA is diluted in RNase-free water for short-term storage or 1× RNase-free siRNA buffer (100 mM potassium acetate, 30 mM HEPES, pH 7.5) for longterm storage. The siRNA buffer can also be purchased from various vendors. It is strongly recommended to verify the quality of the annealed dsRNA using polyacrylamide gel electrophoresis (see Support Protocol 5). CHECKING THE INTEGRITY OF dsRNAs Before proceeding to transfection, it is strongly recommended to verify the integrity of the dsRNA annealed above using non-denaturing polyacrylamide gel electrophoresis. The presence of free single-stranded oligos will not only reduce the amount of siRNAs used in the transfection but also, more significantly, it will affect the silencing efficiency. SUPPORT PROTOCOL 5 Materials 10× TBE buffer (APPENDIX 2A) 40% (w/v) 19:1 acrylamide:bisacrylamide (AC:BC) solution TEMED 10% (w/v) ammonium persulfate (APS) solution Annealed dsRNAs (Support Protocol 4) 4× native gel loading dye (see recipe) 10 mg/ml ethidium bromide solution RNA-Based Methods in Cell Biology 27.1.23 Current Protocols in Cell Biology Supplement 47 14 × 16–cm gel electrophoresis apparatus Power supply Flat gel loading tips UV lamp NOTE: When a minigel is used, adjust the volume and time of gel running. Prepare gel apparatus 1. Assemble a glass plate sandwich on a clean surface. Make sure the plates are properly aligned with spacers to avoid leakage. 2. Prepare 35 ml of 8% polyacrylamide nondenaturing solution by mixing the following: 24.5 ml Milli-Q water 3.5 ml 10× TBE buffer 7 ml 40% (w/v) AC:BC 15 μl TEMED. 3. Add 150 μl of 10% APS solution to the gel solution and mix quickly, but thoroughly, by swirling and avoid foaming. To avoid polymerization while casting the gel, make sure to prepare the solution at room temperature. 4. Working quickly, fill the gel mold (without the comb) to 1 mm below the top of the smaller piece of glass using a disposable transfer pipet. Hold the tip of the pipet against the large piece of glass and at the end of the glass so the solution fills along the spacer. Insert the comb to its fully seated position. Allow gel to polymerize 10 to 15 min. This will cause some of the solution to overflow. Add more if necessary. 5. Assemble the 8% nondenaturing polyacrylamide gel into the running apparatus and pre-run for ∼0.5 hr at 200 V. 6. Prepare dsRNA samples by diluting part of the 10 μM working solution to a final concentration of 1 μM. 7. In a 1.7-ml tube, mix 1 μl of 10 μM dsRNA with 2 μl 1× TBE buffer and 1 μl 4× native gel loading dye. In another tube, mix 1 μl of 1 μM dsRNA with 2 μl 1× TBE and 1 μl 4× native gel dye. Run and analyze the gel 8. Load samples at the bottom of the wells with flat gel loading tips. 9. Run the gel for 1.5 to 2 hr at 200 V. 10. Disassemble the gel and stain with 0.4 μg/ml ethidium bromide solution (prepared from 10 mg/ml stock) for 20 min. 11. Visualize dsRNA samples under a UV lamp. You should avoid leaving the gel in the stain solution too long because RNA samples will diffuse out from the gel and may no longer be detectable under the UV lamp. Silencing of Gene Expression in Cultured Cells You should see results similar to those shown in Figure 27.1.7 when you perform nondenaturing polyacrylamide gel analysis.When the annealing is successful, you should see a detectable higher-molecular-weight band, which represents annealed dsRNA, and no remaining single-stranded oligo, which is seen as a lower-molecular-weight band, not shown in the figure. 27.1.24 Supplement 47 Current Protocols in Cell Biology ol pm 10 1 pm ol dsRNA Figure 27.1.7 Verification of dsRNA integrity. The integrity of double-stranded RNAs was determined by gel electrophoresis. Either 1 pmol or 10 pmol of a 21-nt long dsRNA were loaded on a 8% non-denaturing polyacrylamide gel. Successful dsRNA annealing should result in a detectable higher-molecular-weight band and no remaining single-stranded oligo as a low-molecular-weight band, which was not detected in the gel. REAGENTS AND SOLUTIONS Use deionized, distilled water in all recipes and protocol steps. For common stock solutions, see APPENDIX 2A; for suppliers, see SUPPLIERS APPENDIX. Annealing buffer, 10× 100 mM Tris·Cl, pH 8.0 (APPENDIX 2A) 1.5 M NaCl Store up to 2 to 3 years at 4◦ C Complete DMEM/10% (v/v) FBS Dulbecco’s modified Eagle medium, high-glucose formulation supplemented with: 10% (v/v) FBS 2 mM L-glutamine 1 mM sodium pyruvate 100 U/ml penicillin 100 μg/ml streptomycin Store up to 3 months at 4◦ C For complete DMEM/10% FBS without antibiotics, omit adding penicillin and streptomycin. Native gel loading dye, 4× 0.1% (w/v) bromphenol blue 0.1% (w/v) xylene cyanol FF 0.1% (w/v) Orange G 1 mM EDTA 10 mM Tris·Cl, pH 7.5 (APPENDIX 2A) 40% (v/v) glycerol Store up to 2 to 3 years at 4◦ C (or long-term at −20◦ C) RNA-Based Methods in Cell Biology 27.1.25 Current Protocols in Cell Biology Supplement 47 RIPA buffer 150 mM NaCl 50 mM Tris·Cl, pH 8.0 (APPENDIX 2A) 1% (v/v) NP-40 0.25% (v/v) sodium deoxycholate 0.1% (w/v) SDS 1× protease inhibitor cocktail (Roche), added fresh Adjust pH to 8.0 Store up to 1 year at 4◦ C COMMENTARY Background Information Silencing of Gene Expression in Cultured Cells The mechanism of RNA interference (RNAi) was first described in detail in Caenorhabditis elegans by Andrew Fire and Craig C. Mello (Fire et al., 1998), who later shared the Nobel Prize in Physiology or Medicine in 2006. Soon after, approximately 21 nucleotide-long small interfering RNAs (siRNAs) were shown to mediate sequencespecific gene silencing mammalian cells and Drosophila (Zamore et al., 2000; Elbashir et al., 2001b). The selective and immediate effect of RNAi on the gene silencing within living cells made it a valuable tool for investigating gene function and initiated a new wave of reverse genetics. The RNAi pathway is found in many eukaryotes including plants and animals. It is a small RNA–mediated gene silencing process controlled by the RNA induced silencing complex (RISC) activity at the posttranscriptional level. In living cells, small RNAs have been shown to participate in: (1) regulating gene expression, (2) maintaining genome integrity, (3) controlling cell growth, differentiation, and cellular metabolism, and (4) defending from viral invasion. Biochemical analysis of the RNAi pathway led us to realize that introducing double-stranded RNAs (dsRNAs) with sequences complementary to target transcripts can elicit an RNAi response, demonstrating the versatility of the process in cells. RNAi can be described as having at least two well defined steps: the initiation step, where the ribonuclease (RNase) III enzyme Dicer processes dsRNAs into 21- to 22nucleotide (nt)–long duplexes (Bernstein et al., 2001), and the effector step, in which Argonaute 2 (Ago 2), a core endonuclease of the RISC, executes RNAi (Liu et al., 2004; Meister et al., 2004; Rivas et al., 2005). With exogenously introduced 21-nt siRNAs, the Dicer processing step is skipped and directly incorporated into RISC, where Ago2 carries out cleavage of the target transcript. Conversely, dsRNAs longer than 25 bp can be used to trigger RNAi response by undergoing Dicer processing. It is apparent that not only siRNA sequences that recruit RISC to target transcripts, but also incorporation of siRNAs into RISC, become critical determinants for the RNAi efficiency. The protocol presented here describes basic approaches that one can use as a starting point. It is important to optimize transfection conditions after empirical determinations. Critical Parameters siRNA sequences It is common to observe various degrees of silencing efficiency among designed siRNAs. This could simply be due to insufficient incorporation of siRNA into RISC, or it could be due to poor interaction between the siRNA and target transcript that might form secondary structure, making RISC inaccessible to the target sequence. To find target sequences that are readily accessible, it is advisable to design multiple siRNAs targeting different sites and assay the siRNAs at several concentrations. Some poorly designed siRNAs can still elicit RNAi when they are used in extremely high concentrations, a potential cause of cellular toxicity. In this protocol, 75 nM of siRNAs are included as an example to determine the gene-silencing efficiency. If designed siRNAs show some knockdown effects only at higher concentrations, you may consider redesigning siRNAs for the abovementioned reasons. Also there are siRNA design algorithms that consider the secondary structure of target transcripts (Ding et al., 2004; Yiu et al., 2005), e.g., siRNA Site selector (Heale et al., 2005; http://www1.infosci.coh.org/hpcdispatcher/). Although an siRNA is designed against a specific target mRNA site, it can cause silencing of unspecific genes, termed off-target effects. If a sequence match is found between 27.1.26 Supplement 47 Current Protocols in Cell Biology a guide strand and an mRNA in the 3 UTRs, the first 6 to 7 nucleotides of the guide strand can lead to translational inhibition via the microRNA-mediated pathway. Each strand of the siRNA has the potential to be assembled into RISC as a guide, so that it doubles the chance of creating off-target effects. If this is suspected, the psiCHECK-2 vector with the target sequence inserted in the antisense orientation can be used for the assessment. Design of Dicer substrate siRNAs (DsiRNAs; Rose et al., 2005) described in this unit emanated from ideas of (1) inducing Dicer processing to increase the possibility of assembly into RISC, and (2) reducing a likelihood of passenger-strand incorporation by introducing structural asymmetry. However, it is difficult to completely eliminate all the potential off-targets. There are chemical and structural modifications such as 2 O-methyl at a few positions that reduce the off-target effects. Transfection conditions In this protocol, the use of Lipofectamine 2000 has been described as a transfection reagent. Certain cell types may not achieve high levels of transfection efficiency with this reagent. If so, there are several other transfection reagents available from Invitrogen (e.g., Lipofectamine RNAiMax) and Mirus (e.g., TransIT-siQUEST reagent), to name a few. It is also important to adjust the seeding density for optimization of the transfection condition. Validations of siRNA silencing efficiency To validate the silencing efficiency of the designed siRNAs, it is critical to assess the level of the target mRNA, since there could be a lag between degradation of mRNA and actual reduction in protein level. If necessary, you may need to perform multiple cycles of transfection to observe effects of siRNAs at the protein level. It is also strongly recommended to perform validations on both mRNA and protein levels (Support Protocols 1 and 2, respectively). Anticipated Results transfection reagent preparation and transfection takes ∼1.5 hr. Time for maintaining cultured cells varies. For dual-luciferase reporter assays, allow ∼1.5 hr from cell lysis to bioluminescence measurements. For RT-PCR, it will take ∼1 hr for RNA extraction, ∼1 hr for DNase treatment, ∼1.5 hr for cDNA synthesis, and ∼2.5 hr for realtime PCR. For immunoblotting, time considerations are ∼1 hr for preparation of SDS-PAGE gel, ∼1.5 to 3 hr for electrophoresis, ∼2 hr for protein transfer, and ∼4 to 5 hr for blocking a PVDF membrane and antibody incubation. The total time required to identify a valid siRNA for a gene can be ∼ 2 weeks. Literature Cited Amarzguioui, M., Holen, T., Babaie, E., and Prydz, H. 2003. Tolerance for mutations and chemical modifications in a siRNA. Nucleic Acids Res. 31:589-595. Bernstein, E., Caudy, A.A., Hammond, S.M., and Hannon, G.J. 2001. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 409:363-366. Bookout, A.L., Cummins, C.L., Kramer, M.F. Pesola, J.M., and Mangelsdorf, D.J. 2006. High-throughput real-time quantitative reverse transcription PCR. Curr. Protoc. Mol. Biol. 73:15.8.1-15.8.28. Chalk, A.M. and Sonnhammer, E.L. 2008. siRNA specificity searching incorporating mismatch tolerance data. Bioinformatics 24:1316-1317. Chalk, A.M., Warfinge, R.E., Georgii-Hemming, P., and Sonnhammer, E.L. 2005. siRNAdb: A database of siRNA sequences. Nucleic Acids Res. 33:D131-134. Ding, Y., Chan, C.Y., and Lawrence, C.E. 2004. Sfold web server for statistical folding and rational design of nucleic acids. Nucleic Acids Res. 32:W135-141. Elbashir, S.M., Harborth, J., Lendeckel, W., Yalcin, A., Weber, K., and Tuschl, T. 2001a. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 411:494498. Elbashir, S.M., Lendeckel, W., and Tuschl, T. 2001b. RNA interference is mediated by 21and 22-nucleotide RNAs. Genes Dev. 15:188200. Well designed siRNA-mediated silencing of a given gene of interest can result in greater than 95%, if not 100%, reduction in the mRNA and protein levels. Elbashir, S.M., Martinez, J., Patkaniowska, A., Lendeckel, W., and Tuschl, T. 2001c. Functional anatomy of siRNAs for mediating efficient RNAi in Drosophila melanogaster embryo lysate. EMBO J. 20:6877-6888. Time Considerations Fire, A., Xu, S., Montgomery, M.K., Kostas, S.A., Driver, S.E., and Mello, C.C. 1998. Potent and specific genetic interference by doublestranded RNA in Caenorhabditis elegans. Nature 391:806-811. Synthesis of siRNA from ordering to receiving may take ∼1 week, while annealing and verification take ∼4.5 to 5 hr. Seeding cells in multiple-well plates takes ∼1 hr, and Gong, W., Ren, Y., Zhou, H., Wang, Y., Kang, S., and Li, T. 2008. siDRM: An effective and RNA-Based Methods in Cell Biology 27.1.27 Current Protocols in Cell Biology Supplement 47 generally applicable online siRNA design tool. Bioinformatics 24:2405-2406. binant human RISC. Nat. Struct. Molec. Biol. 12:340-349. Gregory, R.I., Chendrimada, T.P., Cooch, N., and Shiekhattar, R. 2005. Human RISC couples microRNA biogenesis and posttranscriptional gene silencing. Cell 123:631-640. Rose, S.D., Kim, D.H., Amarzguioui, M., Heidel, J.D., Collingwood, M.A., Davis, M.E., Rossi, J.J., and Behlke, M.A. 2005. Functional polarity is introduced by Dicer processing of short substrate RNAs. Nucleic Acids Res. 33:41404156. Heale, B.S., Soifer, H.S., Bowers, C., and Rossi, J.J. 2005. siRNA target site secondary structure predictions using local stable substructures. Nucleic Acids Res. 33:e30. Kramer, M.F. and Coen, D.M. 2001. Enzymatic amplification of DNA by PCR: Standard procedures and optimization. Curr. Protoc. Mol. Biol. 15.1.1-15.1.14. Liu, J., Carmell, M.A., Rivas, F.V., Marsden, C.G., Thomson, J.M., Song, J.J., Hammond, S.M., Joshua-Tor, L., and Hannon, G.J. 2004. Argonaute2 is the catalytic engine of mammalian RNAi. Science 305:1437-1441. Liu, Q., Rand, T.A., Kalidas, S., Du, F., Kim, H.E., Smith, D.P., and Wang, X. 2003. R2D2, a bridge between the initiation and effector steps of the Drosophila RNAi pathway. Science 301:19211925. Meister, G., Landthaler, M., Patkaniowska, A., Dorsett, Y., Teng, G., and Tuschl, T. 2004. Human Argonaute2 mediates RNA cleavage targeted by miRNAs and siRNAs. Molec. Cell 15:185-197. Owczarzy, R., Tataurov, A.V., Wu, Y., Manthey, J.A., McQuisten, K.A., Almabrazi, H.G., Pedersen, K.F., Lin, Y., Garretson, J., McEntaggart, N.O., Sailor, C.A., Dawson, R.B., and Peek, A.S. 2008. IDT SciTools: A suite for analysis and design of nucleic acid oligomers. Nucleic Acids Res. 36:W163-W169. Ren, Y., Gong, W., Zhou, H., Wang, Y., Xiao, F., and Li, T. 2009. siRecords: A database of mammalian RNAi experiments and efficacies. Nucleic Acids Res. 37:D146-D149. Reynolds, A., Leake, D., Boese, Q., Scaringe, S., Marshall, W.S., and Khvorova, A. 2004. Rational siRNA design for RNA interference. Nat. Biotechnol. 22:326-330. Rivas, F.V., Tolia, N.H., Song, J.J., Aragon, J.P., Liu, J., Hannon, G.J., and Joshua-Tor, L. 2005. Purified Argonaute2 and an siRNA form recom- Shah, J.K., Garner, H.R., White, M.A., Shames, D.S., and Minna, J.D. 2007. sIR: siRNA Information Resource, a web-based tool for siRNA sequence design and analysis and an open access siRNA database. BMC Bioinformatics 8:178. Siolas, D., Lerner, C., Burchard, J., Ge, W., Linsley, P.S., Paddison, P.J., Hannon, G.J., and Cleary, M.A. 2005. Synthetic shRNAs as potent RNAi triggers. Nat. Biotechnol. 23:227-231. Ui-Tei, K., Naito, Y., Takahashi, F., Haraguchi, T., Ohki-Hamazaki, H., Juni, A., Ueda, R., and Saigo, K. 2004. Guidelines for the selection of highly effective siRNA sequences for mammalian and chick RNA interference. Nucleic Acids Res. 32:936-948. Yiu, S.M., Wong, P.W., Lam, T.W., Mui, Y.C., Kung, H.F., Lin, M., and Cheung, Y.T. 2005. Filtering of ineffective siRNAs and improved siRNA design tool. Bioinformatics 21:144-151. Zamore, P.D., Tuschl, T., Sharp, P.A., and Bartel, D.P. 2000. RNAi: Double-stranded RNA directs the ATP-dependent cleavage of mRNA at 21 to 23 nucleotide intervals. Cell 101:25-33. Internet Resources http://genome.ucsc.edu/cgi-bin/hgGateway Source for mRNA sequences of genes. http://www.idtdna.com/Scitools/Applications/ RNAi/RNAi.aspx Site for assistance with RNAi design. http://www1.infosci.coh.org/hpcdispatcher/ Home of RNAi Site selector which considers secondary structure of target transcripts. http://www.invitrogen.com Cell-type specific transfection protocols for stealth RNAi and siRNA. http://www.promega.com/paguide/chap8.htm Protocols and applications guide. Silencing of Gene Expression in Cultured Cells 27.1.28 Supplement 47 Current Protocols in Cell Biology