Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Protein–protein interaction wikipedia , lookup
Drug design wikipedia , lookup
Magnesium transporter wikipedia , lookup
G protein–coupled receptor wikipedia , lookup
Ultrasensitivity wikipedia , lookup
Vesicular monoamine transporter wikipedia , lookup
Signal transduction wikipedia , lookup
Oxidative phosphorylation wikipedia , lookup
Ligand binding assay wikipedia , lookup
Metalloprotein wikipedia , lookup
Catalytic triad wikipedia , lookup
Two-hybrid screening wikipedia , lookup
Western blot wikipedia , lookup
SNARE (protein) wikipedia , lookup
NADH:ubiquinone oxidoreductase (H+-translocating) wikipedia , lookup
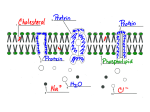
Membrane Transport Structure-function of the ADP/ATP carrier Martin Klingenberg Institute of Physical Biochemistry, University of Munich, Goethestrasse 33, 8000 Munich 2, Federal Republic of Germany ‘I’he A1 )P/A’I’P carrier (AAC) is an important member of the family of mitochondrial solute carriers. I hese transport prirnarily solute anions between the cytosol and the mitochondrial matrix. Although the proteins are of similar size, they can handle a very wide range of solute sizes, from the slidlest ones such as 0 1 I or I I in the uncoupling protein of brown fat adipose tissue to about the largest solutes involved in well defined transport, such ;IS A1 11’ and ATP. Our ideas of the structure of the mitochondrial carriers are derived from the priinary structures 11 I and from probing the carriers with membrane imperrneant or permeant agents [2J with substrate analogues and from reactions with antibodies [ 31. I:urthermore, ultracentrifugal studies on the isolated AAC and other proteins have shown that these proteins have a large hydrophobic surface which binds an extensive detergent micelle [41.Further crude structural information corms from circular dichroism studies, from the tumbling rates in the rnernbrane of spin-labelled carriers etc. [ 51. ‘I’he evaluation of the primary sequence for hydrophobic transmembrane helices is, in the case of the AAC carrier, quite equivocal because of the widely dispersed ionic residues. 1 Iere, the finding of internal repeat structures within the mitochondrial carriers permits a more detinite attribution of the hydrophobic regions. Internal homology analysis first tor the AAC and subsequently for all other mitochondrial carriers for which the structure is known showed that the whole sequence of around 300 residues is divided up into three similar repeats each of 100 residues IO-XI. Although the degree of sequence similarity between these repeats is very low, by appropriately inserting deletions and arranging these repeats, striking conservations o f certain residues were found. I n particular, some obviously critical glycine, proline and acidic residues were conserved. Additionally, by the overlay of the three repeats, the hydrophobic regions become more distinctly visible. In each repeat two hydrophobic stretches of about 20 residues can be attributed to transmembrane helices. The first helical region is more hydrophobic than the second one \vithin each repeat. T h e triplicate structure suggests r . ~ + an intraprotein symmetry based on three sirnilar building blocks. Each building block traverses the membrane twice. Two putative transmembrane helices are separated by hydrophilic stretches of 40-50 residues. In these central sections of each domain about 10- 15 polar residues are located. ‘I’he connections between the three repeat domains are considerably shorter consisting o f only about 12-1 5 residues. They are also quite hydrophilic and contain between three and six polar residues. In general, the threefold interdomain conservation of residues is inore pronounced in the central hydrophilic regions than between the helical stretches. The conserved localization of charged residues which often occur in clusters is quite striking. I n contrast, in the connecting regions the repetition of charged residues is less distinct. The membrane topography of the nucleotide carrier is deduced from different types of evidence. r . 1 he definition of putative trans-helical segments is strengthened first, by the repetition in the three domains, second, by probing certain residues with membrane impernieant 121 or permeant agents, and third, by using antibody probes in particular t o the N-terminus [ 3). Proteases were not useful because no clearly definite cleavage sites could be discerned. ‘I’he location of the C- or N-terminus is still uncertain as the terminal segments are not accessible t o specific probes or proteases. For the A1 )l’/Arl’l’ carrier the N-terminus was shown to protrude t o the cytosolic side by use of peptide-directed aritibodies [ 3 ] .It remains controversial whether there is an even or uneven nurnber of transmembrane segments. No direct evidence for the location of the Cterminus of the AAC has been produced so far. I Iowever, in the similar uncoupling protein o f brown adipose tissue iiii extended hydrophilic Cterminus was clearly shown t o be located on the cytosolic side. both by proteolytic digestion and by specific fluorescent SI I-probes [9]. These findings strongly suggested that the C-terminus in A A C is also on the cytosolic side. T h e location of both the C- and N-terminus o n the cytosolic side would also concur with the even-numbered transmembrane helices in the three segments. Probing \vith n~cmbr;inr-imperme;int rcagelits, such ;is ~,!.ridox;ilpliosptiate, showed that I992 547 Biochemical Society Transactions 548 visualized that a translocation channel exists within each subunit formed by the three repeat domains along the pseudo threefold symmetry axis. Another model depends on the dirneric structure of these carriers. T h e carrier homodirners have been shown to have only one binding site for specific inhibitors or substrates. This suggests that there is one central channel formed by the two dimers along the twofold symmetry axis [ 1 1 I. In this case the channel would be surrounded by the six domains of the two subunits. This model would be inore accorninodating to translocating very large solutes. such as A1 11’ and A‘I’P. An essential part of the single-binding centregated pore model of carrier action are the gates on both sides of the translocation channel (reviewed in [12]). T h e model stipulates that either the inner or outer gate is closed in the external or internal carrier state. Further conceptual and experimental developments of the gated pore model address the problem of how the catalytic energy for the transport is generated [ 13, 141. ‘1’0 put it in simpler terms: how is the activation barrier for the solute translocation across the membrane decreased by the solute-protein interaction? In an attempt t o answer this question, we conceived the ‘induced tit transition state’ mechanism in which we postulate that during the translocation a transition state occurs in which the binding centre has a conformation that exposes a maximum interaction with the some lysine groups in the hydrophilic intradomains are also accessible from the cytosolic side [2]. This accessibility from the outside and inside was dependent on the functional state of the carrier. The accessibility from the cytosolic side of the central regions seemed at first to contradict their localization on the matrix site according to the six-helical folding models. Ilowever, both types of data may be reconciled by assuming that a loop of each of these central portions is lining the hydrophilic translocation path Mithin the carrier molecule [ I ] . The lysines in these loops are proposed to be accessible through the hydrophilic transport channel depending on whether the external o r internal gate is open or closed (1:ig. 1). 1;urther support of this model is derived from studies on the localization of the nucleotide- and inhibitor-binding sites. With 2-azido- or X-azidoATP the central region of the second domain was labelled 110, 10aJ.T h e nucleotides are incorporated to the same position in intact mitochondria as in the isolated protein. This means that they probably gain access from the cytosolic side to the same binding site as from the membrane side. In fact, according to the single-binding centre-gated pore model the nucleotide-binding region should be accessible from both the outside and inside. The three-dimensional folding model is critically dependent on assumptions about the localization o f the translocation channel. Thus, it can be Fig. I The proposed charge distribution in the folding of the ADP/ATP carrier (AAC-2) from yeast The three-repeat-domain structure i s stressed A hydrophobic loop extends into the membrane region from the intradomain central matrix section containing some polar residues These loops are visualized to line the translocation channel The loop of the second domain contributes t o the nucleotide-binding site 2s .. 38 Volume 20 48 10 40 40 Membrane Transport Fig. 2 Fig. 3 The role of the gates in the induced-fit mechanism of the single-binding centre-gated pore model The charge-oscillating model of gating in the singlebinding centre-gated pore mechanism In the transition state the binding centre i s best fitted t o the substiate The released binding energy results in a conformational-labile transition state, in which both gates around the binding centre are open t o permit easy substrate transfer t o either side of the membrane For details see text. A - , anionic subrtrate. 549 8 / Closed \ ~ (Half) open solutes (Fig. 2). Whereas in the external and internal states the binding centre is postulated not t o display substrate-like conformations, these are induced by the solute-protein interaction in the transition state. This ‘induced fit’ is the key element of transport catalysis in carriers. In contrast, in enzymes an induced fit is catalytically counterproductive and may only enhance specificity. Recause the gating is an essential part of the conforrnational changes during the translocation process, we have to ask how the gates are configured in the transition state. An approximately symmetrical state is probable. Originally we assumed closed gates around the substrate. I lowever, for considerations of catalytic efficiency we now favour that both gates are open (Fig. 2). ‘I’he gating and its control is suggested to involve alternating formation and breaking of ionic pairs. The control conies from the binding of the anionic substrate t o one or more positive charges in the binding centre. This case is illustrated in Fig. .? as what we call the ‘charge oscillating model’. Negative residues on both sides of the binding centre are postulated to be rnobile and can pair either with the positive residue of the binding centre or, by moving outwards, with the positive residues of the gates. Without a solute the central positive charge is free and can pair with either the internal or external mobile negative charge. Hy neutralizing one mobile negative charge it releases one positive charge at a gate whic*h can now form an ion bond across the channel ; i d then close the gate. At the same time, the opposite gate is open because its positive residue is neutralized by the mobile negative charge. On binding of the anionic solute the central positive residue is sequestered and the released mobile negative charge moves towards the outside and pairs with the positive charge of the gate. Thus, binding of a substrate is propagated to the gates by the two mobile negative residues such that both gates are open in the transition state. If both gates were closed. too much binding energy might be consumed and the energy level of the transition state would form a trap for the substrate from which i t would dissociate too slowly. The open-gated transition state induces t o the carrier a highly labile state. It can flip either into the external or internal state from which the substrate is readily released. ‘I’he model requires the proper arrangement of charges along the translocation channel. As seen in 1:ig. 1, a sufficient number of charges is available, particularly in the three central domain regions which are thought to loop into the translocation channel. It would be most intriguing to investigate by site-directed rnutagenesis which of these charges interrupt the gating process. This is n o w in progress for the AAC-2 of Saccharomyces cereokiae. 1. Klingenberg, bl. ( 10x0) Arch. Hiochem. I3iophys. 270, 1-14 2. Hogner. W., Acluila. i 1. & Klingenberg. M. (10x0) E u r . J. I3iochcm. 161. 01 1-020 3 . Ihandolin, (;., Houlay, I:., I);ilbon, 1’. 8( Vigiuis, 1’. V. (10x9) Hiochemistry 28, 1003-1 100 4. I lackenberg, 11. & Klingciiberg, M. (10x0) Hioc~hemistry19, 548-555 5. I lorwath, I,. I., Munding, A,, I k y c r , K., Klingenberg, \I. 8( Marsh, I ) . (19XO) Hiochemistry 28, 407-41-1 I992 Biochemical Society Transactions 550 6. Sarastc. M. It Walker, J. 1.: (10x2) FEHS 1,ett. 144, 2 5 0-2 5 4 7 . Aqiiila, 11.. Idink, T. A. It Klingenberg. IL1. (1085) EMHO J. 4. 2.700-2.77f) 8. Acluila, 11.. ],ink. T. A. rU tilingenberg, hl. (1087) F m S I d t . 212, 1-0 0 . Klingcnberg, XI. It Appcl, M.(1080) Eur. J. Hiochcm. 180,123-131 10. Mayingw. I’.. M’inklcr. li, It Klingenberg, M. (1080) 1;EHS I,ctt. 244, 421-420 10a. Ihlbon. I)., Hrandolin, C.. Houlay. F. et al. (1088) Hiochemistry 27. .5 141- 5 140 11. Klingcnlwrg, hl. (1081) Nature ( I m ~ d o n ) 290. 440-45 4 12. Klingciibcrg. M. ( 1070) T h e Fnzymes of 13iologic;il Menilmrics; !Llemhranc 7’r;Irlsport (Akrrtonosi,A. N.. ed.). Vol. 3 . pp. 3x3-438, I’lcnum I’ublishing C‘orp, New York/l.ondon 1.3. Klingenbcrg, hl. (1085) Ann. N.Y. Ac-ad. Sci. 456, 270-288 14. Klingenbcrg, 14. (1001) A Stud!. of linzymcs; Mechanism of Enzyme Action (Kuby, S.A,, cd.).Vol. 11. pp. 307-388. CKC I’rcbss, 1ioc;i K;rton Received 28 April 1092 Studies of the structure and function of sarcoplasmic reticulum (CaZ -Mg2 )ATPase using immunological approaches + + J. Malcolm East, Ian Matthews, Richard E. A. Tunwell,* Ana M. Mata,t and Anthony G. Lee SERC Centre for Molecular Recognition, Department of Biochemistry, University of Southarnpton, Southarnpton SO9 3TU. U.K. Introduction Amino acid sequence data and site-directed mutagenesis studies have contributed greatly to our understanding of the uay in \bhich the P-type cation transporters operate I 1. 2 1. In particular, sitedirected rnutagenesis experiments carried out on the (<’a’ -Mg’ )-A‘l’Pase of sarcoplasmic reticulum (SK) have provided invaluable information about the location of the Ca”-binding sites 1.3 I, the nature of the nucleotide-binding domain [-I I and the function of the P-strand domain 15 I, as \\ell as highlighting critical proline residues in the transmembraneous domain 16 I. I kspite these recent advances, information about the molecular events which characterize membrane transport and high resolution structural information about such transporters is still lacking. The (<’a’ -Mg’+ )-ATI’ase of SR is one of the most studied of the P-type cation A‘I’Pases [ 7 1. It is available in large amounts and ;it high purity from skeletal muscle, making it ;in obvious candidate for study. The 1’-type ATPases also include a number of other physiologically important cation transporters such as the plasma membrane (Ka K )-A‘l’l’ase and the 1 I -A‘l’Pase from stom;ich. + + + + + Volume 20 + Members of this class of A‘l’l’ases have similar amino acid sequences and are thought to pump their respective ions by ;in analogous mech;inism [ 1 1. Thus. any information gained about the structure-function relationships of the (Ca’+-Mg”)ATPase mill provide important insights into the working of other members of this family of transporters. I he series of events which lead t o cation translocation has been elucidated in some detail for a number of these transporters and similar pathmays appear t o be folloued in each instance I X I. In the case of (Ca”-1Llg’+)-A‘l‘l’;tse this involves the sequential binding of 2C;i’+ and Nl’P t o the El form of the ATI’ase, which has two high-;iffinity calcium-binding sites exposed t o the cytopl;ismic environment, to give E 1 .X’;i’ .ArI’I’. 1;ollouing phosphorylation of the A‘l’l’use t o give 1: 1 I’X‘;i’+ the A‘I’I’ase undergoes a conform;itioiial cliange t o E21’.2Ca’+ . In the E2 conforination the <’a’+ -binding sites face the lumen and are of IOU affinity. A s a consequence the 2Ca’ dissociates from the Al’Pase and the Al’l’ase is depIios~~lioryl;itedto give E2 which can then rindergo ;I confi)rm;ition change back t o E l so that the cycle can continue. In contrast to our understanding of the reactions which make up the catalytic cycle of the AW’ase, relatively little is knou 11 about the molecular corrclates of these events. Although procedures tor the cr the (Ca’+-Mg’+)-ATPascin three-dimensioiis are riou being developed 10- 1 1 ] and some structural information is becoming available from these crystals [ 121. most of our current understanding about r . + +