Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Discovery and development of cephalosporins wikipedia , lookup
NK1 receptor antagonist wikipedia , lookup
Nicotinic agonist wikipedia , lookup
Drug design wikipedia , lookup
Pharmacokinetics wikipedia , lookup
Drug discovery wikipedia , lookup
Neuropsychopharmacology wikipedia , lookup
Discovery and development of tubulin inhibitors wikipedia , lookup
Discovery and development of non-nucleoside reverse-transcriptase inhibitors wikipedia , lookup
Discovery and development of angiotensin receptor blockers wikipedia , lookup
Discovery and development of proton pump inhibitors wikipedia , lookup
MTOR inhibitors wikipedia , lookup
Discovery and development of dipeptidyl peptidase-4 inhibitors wikipedia , lookup
Discovery and development of cyclooxygenase 2 inhibitors wikipedia , lookup
Discovery and development of HIV-protease inhibitors wikipedia , lookup
Discovery and development of integrase inhibitors wikipedia , lookup
Discovery and development of neuraminidase inhibitors wikipedia , lookup
Metalloprotease inhibitor wikipedia , lookup
Discovery and development of direct thrombin inhibitors wikipedia , lookup
Discovery and development of ACE inhibitors wikipedia , lookup
Discovery and development of direct Xa inhibitors wikipedia , lookup
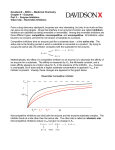
ATVB In Focus Emerging Anticoagulant Drugs Series Editor: Jeffrey I. Weitz Previous Brief Reviews in this Series: • Turpie AGG. Oral, direct factor Xa inhibitors in development for the prevention and treatment of thromboembolic diseases. Arterioscler Thromb Vasc Biol. 2007;27:1238 –1247. • Howard EL, Becker KCD, Rusconi CP, Becker RC. Factor IXa inhibitors as novel anticoagulants. Arterioscler Thromb Vasc Biol. 2007;27:722–727. • Weitz J. Emerging anticoagulant drugs. Arterioscler Thromb Vasc Biol. 2007;27:721. Downloaded from http://atvb.ahajournals.org/ by guest on May 2, 2017 Inhibitors of Factor VIIa/Tissue Factor Rebecca A. Shirk, George P. Vlasuk Abstract—The formation of the proteolytic complex composed of the serine protease Factor VIIa and the cell-associated glycoprotein tissue factor (FVIIa/TF) initiates a cascade of amplified zymogen activation reactions leading to thrombus formation. The critical role of the coagulation cascade in pathological thrombosis has been the basis for significant efforts to design selective inhibitors of the protease components as new anticoagulant alternatives for the treatment of thrombotic diseases. However, for the new generation of anticoagulant drugs in development that primarily target protease complexes distal from FVIIa/TF, the differential between efficacy and safety as defined by bleeding is unresolved. Targeting the FVIIa/TF complex has several theoretical advantages that exploit the amplified nature of the coagulation cascade. However, progress on the development of clinical-stage FVIIa/TF-based anticoagulants has not been as successful to date. This review summarizes recent efforts in the discovery of synthetic inhibitors of FVIIa/TF. (Arterioscler Thromb Vasc Biol. 2007;27:1895-1900.) Key Words: anticoagulants 䡲 factor VIIa (FVIIa) 䡲 tissue factor 䡲 serine protease inhibitors 䡲 coagulation H emostasis is achieved through a highly regulated and coordinated interplay of protein and cellular components designed to respond quickly to vascular insult. Critical in this rapid response is the initiation of blood coagulation by a proteolytic complex composed of the serine protease factor VIIa (FVIIa) and its cell-associated cofactor tissue factor (TF). Tissue factor is a transmembrane glycoprotein that is normally found in subendothelial structures throughout the vasculature. Exposure of TF to blood, through physical damage to the lining of the blood vessel or increased expression in endothelial and monocytic cells in response to certain inflammatory signals, permits an interaction with FVIIa, which is present at trace levels in the circulation. The formation of the FVIIa/TF complex is the critical step that initiates the sequentially amplified cascade of proteolytic activation steps that ultimately leads to the generation of the protease thrombin and thrombus formation.1 The critical nature of the FVIIa/TF complex in the process of blood coagulation makes it an attractive candidate for the development of antithrombotic agents that can inhibit complex formation or catalytic activity. An antithrombotic agent based on the inhibition of FVIIa/TF may have theoretical advantages over the inhibition of downstream coagulation proteases such as factor Xa (FXa) or thrombin based on the several thousand–fold amplification that occurs after initiation by FVIIa/TF. Original received May 18, 2007; final version accepted June 15, 2007. From the Department of Cardiovascular and Metabolic Diseases Research, Wyeth Research, Collegeville, Pa. Correspondence to Rebecca A. Shirk, PhD, Wyeth Research, P.O. Box 42528, Philadelphia, PA 19101-2528. E-mail [email protected] © 2007 American Heart Association, Inc. Arterioscler Thromb Vasc Biol. is available at http://atvb.ahajournals.org 1895 DOI: 10.1161/ATVBAHA.107.148304 1896 Arterioscler Thromb Vasc Biol. September 2007 Downloaded from http://atvb.ahajournals.org/ by guest on May 2, 2017 The success of several small protein- and antibody-based inhibitors of the FVIIa/TF pathway in preclinical models and clinical trials1,2 has led to substantial interest in the development of orally active small molecule inhibitors of the enzyme active site that could be used for the long-term prevention or treatment of thrombosis. Vitamin K antagonists such as warfarin, which have been used clinically for more than 50 years and target multiple vitamin K– dependent coagulation proteases, are the only orally active anticoagulants in clinical use today. Although vitamin K antagonists are inexpensive, their use is complicated by a slow onset of action, food and drug interactions that make it difficult to maintain stable exposure, and thus requiring regular monitoring. Therefore there is a critical unmet medical need for improved oral anticoagulants with a better therapeutic window. Direct comparisons of specific FVIIa/TF inhibitors with other anticoagulants in preclinical thrombosis models have shown a reduced bleeding tendency in several studies,3– 8 suggesting that targeting the upstream proteolytic complex FVIIa/TF may provide a safety advantage. Such a drug would need to be potent, orally bioavailable, and highly selective for FVIIa/TF versus other closely related serine proteases involved in coagulation and fibrinolysis, as well as demonstrate other optimal pharmacokinetic parameters. Despite considerable research effort over the past decade or so, to our knowledge, no small molecule FVIIa/TF inhibitors have advanced to the clinical trial testing phase to date. A number of research groups have reported the successful discovery of small molecule active-site FVIIa/TF inhibitors, the best of which show excellent potency (single digit or low double digit nmol/L Ki or IC50) in in vitro activity assays with purified enzyme complex and demonstrate greater than several hundred– to thousand-fold selectivity versus thrombin, FXa and trypsin. Several excellent reviews have been published that detail the diverse chemical scaffolds, in vitro potency, and protease selectivity (when available) of inhibitors reported in the patent and research literature.9 –11 The goal of the current review will be to summarize general strategies and describe recent advances in the field of small molecule FVIIa/TF inhibitors. Several recent reviews have covered the latest advances in the development of peptide and protein inhibitors of FVIIa/TF and will not be covered here.1,2,12 State-of-the Art The X-ray structure of the FVIIa/TF complex was first published in 1996 by Banner et al13 and facilitated the use of a structurebased design approach to guide optimization of FVIIa/TF active site inhibitors by using X-ray structural analysis and molecular modeling to improve binding affinity and protease selectivity. FVIIa is a trypsin-like serine protease. It shares with thrombin, factor Xa, factor IXa, etc the characteristic catalytic triad residues Ser195-His57-Asp102 (chymotrypsinogen numbering is used through-out) and a high degree of homology in amino acid sequence and spatial arrangement in the protease domain and active site. The characteristic feature of the trypsin-like serine proteases is a deep S1 primary specificity binding pocket with an acidic Asp189 at the bottom, which forms a salt bridge with the basic Arg or Lys side chain of the scissile bond in natural substrates. The size Figure 1. Comparison of FVIIa, thrombin, and FXa active sites occupied by small molecule ligands. A, FVIIa complexed with D-Phe-Phe-Arg-CMK (pdb file: 1DAN). B, Thrombin complexed with D-Phe-Pro-Arg-CMK (pdb file: 1PPB). C, FXa complexed with Razaxaban (pdb file: 1Z6E). To assist identification of subsites S1–S3, peptide ligands are depicted with a different color to denote each amino acid: gray, P1 Arg; copper, P2 Phe (FVIIa/ TF, panel A) or P2 Pro (thrombin, panel B); and gold, P3 D-Phe. Razaxaban (FXa, panel C) is color-coded by atom type. Smooth approximations of the solvent accessible surfaces, based on van der Waals radii, were generated using “Soft Surface”, Discovery Studio 1.6 (Accelerys). The surfaces are color coded as follows: red, side chains of catalytic His57 and Ser195 left to right; blue, side chain of Lys192 (FVIIa/TF, panel A), Glu192 (thrombin, panel B), or Gln192 (FXa, panel C); green, side chain of Thr99 (FVIIa/TF, panel A), Leu99 (thrombin, panel B), or residues Tyr99, Trp215, Phe174 top to bottom (FXa, panel C); yellow, the side chain of Asp60 (FVIIa/TF, panel A), insertion loop 60A– 60D (Tyr-Pro-Pro-Trp in thrombin, panel B), or Leu60 (FXa, panel C). The S1⬘/2⬘ subsite is located “northeast” of the catalytic Ser195. and shape of the FVIIa S1 pocket is very similar to the S1 pocket in thrombin, factor Xa, and trypsin, yet small S1 differences are observed. However, the main differences are in the S2 proximal and S3 distal hydrophobic pockets and the S1⬘ site, which show size and hydrophobicity differences between enzymes (Figure 1). Shirk and Vlasuk Downloaded from http://atvb.ahajournals.org/ by guest on May 2, 2017 Figure 2. Structures of selected FVIIa/TF inhibitors. L-arginine and small molecule active site inhibitors of FVIIa/TF: PHA-927 from Suleymanov et al7; “9” from Young et al18; “22” from Zbinden et al19; “10” from Rai et al33; “17d” from Miura et al.34 To achieve potency and selectivity, researchers have exploited key residues and differences between proteases in the S1–S4 and S1⬘ binding pockets of the FVIIa active site, typically interacting with at least 3 of the subsites. The vast majority of the FVIIa inhibitors reported so far have a benzamidine or other amidine P1 moiety that mimics the Arg side chain guanidinium group in natural substrates and can form a strong salt bridge with Asp189 in the bottom of the S1 pocket (Figure 2). A serine at position 190 in the S1 pocket is unique to FVIIa (versus Ala190 in thrombin and FXa), and some FVIIa inhibitors have been shown to form key hydrogen bonds with Ser190.14,15 Interaction with the catalytic Ser195 via addition of a -amino group on the benzamidine contributes significantly to potency in a number of inhibitors.8,16 In contrast, a series of compounds was shown to form a unique network of hydrogen bonds to Ser195 via a hydroxyl group on a non-P1 central ring.17,18 Another P1 substitution reported is the addition of an o-OH on -aminobenzamidine to increase selectivity. The crystal structure shows the binding of o-OH is associated with a conformational change at Gln217–Gly219 near the bottom of the S1 pocket that is possibly stabilized by a hydrogen bond with the side chain of Thr221, a residue not found in other proteases examined.8 At the top of the S1 pocket FVIIa has a distinct binding site, called the S1 side pocket or S1 subsite, which contains a unique Lys at position 192 versus Glu192 in thrombin and Gln192 in FXa (Figure 1, blue surfaces). Acid moieties have been exploited by several groups to interact with Lys192 near the oxyanion hole, enhancing both potency and selectivity.16,18 –22 Some FVIIa inhibitors have been shown to induce a flipped conformation of the amide bond between Lys192 and Gly193 which results in an enzymatically incompetent oxyanion hole.8,19,20 The 180° rotation of the Lys192-Gly193 bond is stabilized by a hydrogen bond with Gln143, but other tyrpsin-like proteases do not appear to have a suitable partner in that location for hydrogen bond formation, which may explain the observed selectivity for FVIIa. Key differences in Factor VIIa/Tissue Factor Inhibitors 1897 the S2 pocket have been used to improve selectivity as well. FVIIa has a large S2 pocket with a unique Asp60 (Figure 1A, yellow surface) that has been targeted by basic P2 moieties in FVIIa inhibitors,14,22 whereas other coagulation proteases lack an acidic residue in that position. In contrast, the thrombin and FXa S2 pockets are markedly smaller, with the thrombin S2 pocket partially blocked by a rigid insertion loopTyr60A⫺ Trp60D (Figure 1B, yellow surface) and FXa S2 occluded by the side chain of Tyr99 (Figure 2C, top green surface). Therefore, large P2 moieties improve selectivity against these proteases. And lastly, several groups have targeted the S1⬘ pocket to improve both potency and selectivity, using hydrophobic moieties with specific substitutions for key hydrogen bond interactions.17,23,24 Some of these small molecule FVIIa/TF inhibitors have been tested in vivo and shown to be efficacious in preclinical models. The first reports emerged in 2001 and showed efficacy in a guinea pig arterial thrombosis model and that a 2 week parenteral dosing regimen was well tolerated in the same species.25,26 Separation of antithrombotic efficacy and bleeding tendency with a selective small molecule FVIIa/TF inhibitor was first reported in 2003. PHA-927F (Figure 2) was reported to inhibit FVIIa/TF with an IC50 of 25 nmol/L and more than 3500-fold selectivity against thrombin and FXa.7 Intravenous administration of PHA-927F in a nonhuman primate model of arterial injury model demonstrated dose-dependent inhibition of thrombus formation with markedly less bleeding when compared with a FXa inhibitor or thrombin inhibitor at the minimally efficacious dose or up to 4.4-fold multiples thereof. Other reports have followed of small molecule FVIIa/TF inhibitors demonstrating efficacy after intravenous dosing in monkeys8,18,27 and lower species as well.28,29 Finally, compound CRA-027483 has been shown to have ⬇100% bioavailability after subcutaneous delivery, inhibiting thrombus formation in a baboon arterial injury model30 and suppressing the lipopolysaccharide (LPS)induced inflammatory response in a mouse model of endotoxemia.31 The Challenge of Making a Drug Although a number of potent and selective FVIIa/TF inhibitors have been reported, some demonstrating in vivo efficacy after parenteral administration, a significant challenge remains in the discovery of an orally bioavailable drug. A successful oral drug will require a careful balance of optimal inhibitor characteristics and drug-like or pharmacokinetic properties, demands that can have contradictory effects on the molecule. The FVIIa inhibitors described so far have a highly basic amidine or benzamidine P1 moiety that can form a salt bridge with Asp189 in the bottom of the S1 pocket. These basic functional groups, although contributing significantly to binding affinity, are protonated at physiological pH and thus are associated with poor oral bioavailability because of limited absorption in the gastrointestinal tract. Two general approaches have been taken to attempt to bypass this problem in FVIIa/TF inhibitors. The first is to replace the basic amidine with a less basic or neutral P1 moiety. This approach represents a significant challenge, because the mode of binding in the S1 pocket dramatically impacts overall 1898 Arterioscler Thromb Vasc Biol. September 2007 Downloaded from http://atvb.ahajournals.org/ by guest on May 2, 2017 binding. The second approach is to use a prodrug approach to reversibly mask the basic moiety. Two groups have recently reported efforts to find a suitable replacement for the P1 amidine moiety. Researchers at Celera Genomics began with potent FVIIa/TF inhibitors that contained biaryl amidine P1 moieties, such as 5-amidinoindole and 5-amidinobenzimidazole, and replaced these scaffolds with less basic heterocyclic scaffolds that could potentially interact with key residues in the S1 pocket.24,32,33 These inhibitors lose substantial potency initially but typically gained significant selectivity against thrombin, FXa, and trypsin, even before further modification. By further optimizing the inhibitor to interact with the S1⬘ pocket or the loop separating the S2 and S1⬘ pockets, a highly potent (20 nmol/L Ki) and selective inhibitor with a 5-aminopyrrolo-pyridine P1 scaffold was generated (compound “10”, Figure 2).33 Similarly, a P1 5-azaindole inhibitor with a Ki of 800 nmol/L was further optimized to exploit the S1⬘ pocket, and the best of these inhibitors showed Ki values of 30 to 60 nmol/L for FVIIa/TF and ⬎1000-fold selectivity against FXa, thrombin, and trypsin.24 Crystallography data indicate that both of these P1 scaffolds bind in the S1 pocket via multiple H-bonds and by causing a conformational rearrangement relative to structures obtained with amidine-containing inhibitors. The 5-aminopyrrolo-pyridine and 5-azaindole P1 scaffolds described in these reports are markedly less basic, with a calculated pKa of ⬇8.8 and ⬇7.8, respectively, versus pKa ⬇11 to 12 for benzamidine.32,33 However, it is not clear whether these modifications are sufficient to significantly improve oral bioavailability, as no pharmacokinetic data are reported. Researchers at Astellas Pharma Inc took a different approach, replacing the amidine on the P1 benzene ring of a 90 nmol/L FVIIa/TF inhibitor with “neutral” 3-aminocarbonyl or 4-aminomethyl groups.34 Precipitous loss of activity resulted, but after further optimization of the P2 group, the best compound inhibited TF/FVIIa with moderate potency (IC50⫽690 nmol/L) and ⬎300-fold selectivity (“17d”, Figure 2). In a more recent report, a P1 4-aminomethylphenyl FVIIa/TF inhibitor with moderate potency (IC50⫽590 nmol/L) was reported to be orally available.35 After oral administration to mice of a high dose 100 mg/kg, the inhibitor reached and sustained plasma levels of ⬇2 mol/L for 0.5 to 2 hours, as estimated by ex vivo plasma activity against human FVIIa/ TF. The oral availability was estimated at ⬇10%. Reviewing the results from these 2 groups, it appears that discovery of a potent FVIIa/TF inhibitor with a neutral P1 moiety remains a challenge. A prodrug approach has shown the most preclinical progress to date in the search for an orally active FVIIa/TF inhibitor. Prodrugs are agents that are delivered in inactive or markedly less active form and then converted to the active form by metabolic processes in vivo; therefore good oral bioavailability of active drug is dependent on both absorption of the prodrug from the intestine into the circulation and subsequent cleavage of the masking group to generate the active parent compound. Highly basic amidine functional groups can be masked with N-hydroxyl or N-hydroxyalkyl groups to form amidoximes or alternatively with N-carboxyalkyl groups to generate carbamate prodrugs.36 Amidoxime and carbamate-modified amidines are not protonated under physiological conditions and therefore show improved oral absorption. This approach was first shown to have promise for antithrombotic drugs in the clinical development of oral GPIIb/IIIa inhibitors and later with the direct thrombin inhibitor ximelagatran (discussed below). Researchers at Hoffman–La Roche have shown the most progress with amidoxime prodrugs of FVIIa/TF inhibitors. In the first report, Zbinden et al demonstrated that masking the P1 benzamidine moiety as an amidoxime prodrug increased the oral bioavailability of the active drug from 2% to 30% in the rat, and a double prodrug containing both the amidoxime modification and an ester modification of an acidic group (“22”, Figure 2) resulted in a surprising 100% oral bioavailability.19 However, the active parent inhibitor had only moderate in vitro potency (Ki⫽190 nmol/L) and poor activity in plasma as measured in the PT clotting assay; no in vivo activity was reported. In a subsequent report, Zbinden et al demonstrated antithrombotic activity after oral administration of a double amidoxime/ester prodrug of another FVIIa/TF inhibitor with improved potency (81 nmol/L) and markedly better activity in plasma.20 This prodrug showed moderate oral bioavailability (20%) in the rat and dose-dependent antithrombotic efficacy in a guinea pig recurrent arterial thrombosis model at high oral doses (20 to 500 mg/kg, necessitated by species differences in plasma activity). Antithrombotic activity was accompanied by a dose-dependent increase in the PT clotting assay but no effect on the APTT or bleeding time except at the highest dose tested. These findings are quite significant because this is the first report of an orally active, potent, and selective FVIIa/TF inhibitor, achieved through the masking of a benzamidine P1 moiety with an amidoxime prodrug. However, not all FVIIa/TF inhibitor prodrug efforts have met with success. Researchers at Celera Genomics attempted to improve oral bioavailability of compounds containing a biaryl P1 moiety, 5-amidinoindole.37 A series of hydroxyl-, alkoxy-, and carbamate-amidine prodrugs were synthesized that showed moderate to good oral absorption of the prodrug in rat (typically 40% to 100%) but poor conversion to parent amidine, resulting in overall oral availability of ⬍1% to 2% for the amidine drug. The authors noted that previous successes with amidine prodrugs were obtained with mono aryl structures (eg, benzamidine) or alkyl-amidines and hypothesized that the biaryl-amidine prodrugs were not recognized by the enzymatic machinery required for prodrug conversion. It remains to be seen whether other more successful FVIIa/TF prodrugs will emerge and whether they perform well in the clinic. In contrast to the current status of small moleclule FVIIa/TF inhibitors, several oral active-site thrombin and FXa inhibitors have advanced to clinical trials.38,39 Both amidine prodrugs and nonamidine P1 approaches have been used. Ximelagatran is an amidoxime/ester double prodrug that is converted in vivo to the active drug melagatran, a potent thrombin inhibitor that contains a P1 benzamidine moiety.40 The prodrug was rapidly absorbed and converted to melagatran with an oral bioavailability of ⬇20% in human subjects. Ximelagatran was initially marketed in Europe in Shirk and Vlasuk Downloaded from http://atvb.ahajournals.org/ by guest on May 2, 2017 2004 for short-term prevention of thrombosis after orthopedic surgery, but the drug was withdrawn in 2006 because of concerns over liver toxicity. Other prodrugs of amidinecontaining thrombin inhibitors include dabigatran etexilate, a carbamate prodrug which is in phase III trials, and AZD 0837, a follow-on to ximelagatran which contains an alkylamidoxime P1 moiety and is currently in phase II clinical trials. Oral FXa inhibitors are being evaluated in advanced clinical studies as well, and a number of them do not contain amidine P1 moieties.38,39 For example, rivaroxaban (BAY 59 to 7939), apixaban (BMS 562247), and LY-517717 have successfully completed phase II trials with subcutaneous low molecular weight heparin as the comparator in patients undergoing total knee or hip replacement surgery, and they were found to be safe and efficacious.41 Rivaroxaban and apixaban have advanced to Phase III trials in a comparable patient population and are also being tested for additional indications in phase II /III. The binding mode to FXa has not been disclosed for most oral FXa inhibitors currently in development. However, crystal structures for rivaroxaban (with a neutral P1 chlorthiophene moiety),42 razaxaban (DPC-906, halted in Phase II),43 and other nonamidine FXa inhibitors44 – 46 demonstrate a common binding theme. The inhibitors typically bind to FXa in a bent or L-shaped extended conformation, making essential interactions deep in the S1 pocket with a nonbasic moiety and in what the authors term the S4 pocket, a narrow hydrophobic channel defined by the side chains of Tyr99, Phe174, and Trp215 (Figure 1C, green surface). For these inhibitors, strong binding occurs in the absence of a highly basic P1 moiety that can interfere with oral bioavailability. Conclusions Despite significant efforts and some success in the discovery of potent synthetic inhibitors of FVIIa/TF, no compounds have advanced into clinical trials. The issues as discussed above are difficult and will likely take considerable innovation to overcome the barriers to the successful development of a suitable orally bioavailable drug targeting FVIIa/TF. Despite these challenges, the FVIIa/TF complex remains a validated and important target for the development of the next generation of antithrombotic agents. Acknowledgments The authors thank Dr Bruce A. Malcolm for providing the figures, useful discussions of the literature, and critical evaluation of the manuscript. Disclosures None. References 1. Steffel J, Lüscher TF, Tanner FC. Tissue factor in cardiovascular disease. Molecular mechanisms and clinical implications. Circulation. 2006; 113:722–731. 2. De Caterina R, Husted S, Wallentin L, Agnelli G, Bachman F, Baigent C, Jespersen J, Kristensen SD, Montalescot G, Siebahn A, Verheugt FWA, Weitz J. Anticoagulants in heart disease: current status and perspectives. Eur Heart J. 2007;28:880 –913. 3. Harker LA, Hanson SR, Wilcox JN, Kelly AB. Antithrombotic and antilesion benefits without hemorrhagic risk by inhibiting tissue factor pathway. Haemostasis. 1996;26(Suppl 1):76 – 82. Factor VIIa/Tissue Factor Inhibitors 1899 4. Himber J, Kirchhofer D, Riederer M, Tschopp TB, Steiner B, Roux SP. Dissociation of antithrombotic effect and bleeding time prolongation in rabbits by inhibiting tissue factor function. Thromb Haemost. 1997;78: 1142–1149. 5. Golino P, Ragni M, Cirillo P, D’Andrea D, Scognamiglio A, Ravera A, Buono C, Ezban M, Corcione N, Vigorito F, Condorelli M, Chiariello M. Antithrombotic effects of recombinant human, active site-blocked factor VIIa in a rabbit model of recurrent arterial thrombosis. Circ Res. 1998; 82:39 – 46. 6. Himber J, Whohlgensinger C, Rouz S, Damico LA, Fallon JT, Kirchhofer D, Nemerson Y, Rieder MA. Inhibition of tissue factor limits the growth of venous thrombosis in the rabbit. J Thromb Haemost. 2003;1:889 – 895. 7. Suleymanov OD, Szalony JA, Salyers AK, LaChance RM, Parlow JJ, South MS, Wood RS, Nicholson NS. Pharmacological interruption of acute thrombosis formation with minimal hemorrhagic complications by a small molecule tissue factor/factor VIIa inhibitor: Comparision to factor Xa and thrombin inhibition in a nonhuman primate thrombosis model. J Pharmacol Exp Ther. 2003;306:1115–1121. 8. Olivero AG, Eigenbrot G, Goldsmith R, Robarge K, Artis DR, Flygare J, Rawson T, Sutherlin DP, Kadkhodayan S, Beresini M, Elliott LO, DeGuzman GG, Banner DW, Ultsch M, Marzec U, Hanson SR, Refino C, Bunting S, Kirchhofer D. A selective, slow binding inhibitor of factor VIIa binds to a nonstandard active site conformation and attenuates thrombus formation in vivo. J Biol Chem. 2005;280:9160 –9169. 9. Lazarus RA, Olivero AG, Eigenbrot C, Kirchhofer D. Inhibitors of tissue factor-factor VIIa for anticoagulant therapy. Curr Med Chem. 2004;11: 2275–2290. 10. Frédérick R, Pochet L, Charlier C, Masereel B. Modulators of the coagulation cascade: focus and recent advances in inhibitors of tissue factor, factor VIIa and their complex. Curr Med Chem. 2005;12:397– 417. 11. Kranjc A, Kikelj D, Peterlin-Masic L. Recent advances in the discovery of tissue factor/factor VIIa inhibitors and dual inhibitors of factor VIIa/ factor Xa. Curr Pharm Design. 2005;11:4207– 4227. 12. Lee AYY, Vlasuk GP. Recombinant nematode anticoagulant protein c2 and other inhibitors targeting blood coagulation factor VIIa/tissue factor. J Internal Med. 2003;254:313–321. 13. Banner DW, D’Arcy A, Chène C, Winkler FK, Guha A, Konigsberg WH, Nemerson Y, Kirchhofer D. The crystal structure of the complex of blood coagulation factor VIIa with soluble tissue factor. Nature. 1996;380: 41– 46. 14. Schweitzer BA, Neumann WL, Rahman HK, Kusturin CL, Sample KR, Poda GI, Kurumbail RG, Stevens AM, Stegeman RA, Stallings WC, South MS. Structure-based design and synthesis of pyrazinones containing novel P1 ‘side pocket’ moieties as inhibitors of TF/VIIa. Bioorg Med Chem Lett. 2005;15:3006 –3011. 15. Kohrt JT, Filipski KJ, Cody WL, Cai C, Dudley DA, Van Huis CA, Willardsen JA, Rapundalo ST, Saiya-Cork K, Leadley RJ, Narasimhan L, Zhang E, Whitlow M, Adler M, McLean K, Chou Y-L, McKnight C, Arnaiz DO, Shaw KJ, Light DR, Edmunds JJ. The discovery of fluoropyridine-based inhibitors of the factor VIIa/TF complex. Bioorg Med Chem Lett. 2005;15:4752– 4756. 16. Zbinden KG, Banner DW, Ackermann J, D’Arcy A, Kirchhofer D, Ji Y-H, Tschopp TB, Wallbaum S, Weber L. Design of selective phenylglycine amide tissue factor/factor VIIa inhibitors. Bioorg Med Chem Lett. 2005;15:817– 822. 17. Shrader WD, Kolesnikov A, Burgess-Henry J, Rai R, Hendrix J, Hu H, Torkelson S, Ton T, Young WB, Katz BA, Yu C, Tang J, Cabuslay R, Sanford E, Janc JW, Sprengeler PA. Factor VIIa inhibitors: gaining selectivity within the trypsin family. Bioorg Med Chem Lettr. 2006;16: 1596 –1600. 18. Young WB, Mordenti J, Torkelson S, Shrader WD, Kolesnikov A, Rai R, Liu L, Hu H, Leahy EM, Green MJ, Sprengeler PA, Katz BA, Yu C, Janc JW, Elrod KC, Marzec UM, Hanson SR. FVIIa inhibitors: chemical optimization, preclinical pharmacokinetics, pharmacodynamics, and efficacy in an arterial baboon thrombosis model. Bioorg Med Chem Lett. 2006;16:2037–2041. 19. Zbinden KG, Obst-Sander U, Hilpert K, Kühne H, Banner DW, Böhm H-J, Stahl M, Ackermann J, Alig L, Weber L, Wessel HP, Riederer MA, Tschopp TB, Lavé T. Selective and orally bioavailable phenylglycine tissue factor/factor VIIa inhibitors. Bioorg Med Chem Lett. 2005;15: 5344 –5352. 20. Zbinden KG, Banner DW, Hilpert K, Himber J, Lavé T, Riederer MA, Stahl M, Tschopp TB, Obst-Sander U. Dose-dependent antithrombotic activity of an orally active tissue factor/factor VIIa inhibitor without 1900 21. 22. 23. 24. 25. Downloaded from http://atvb.ahajournals.org/ by guest on May 2, 2017 26. 27. 28. 29. 30. 31. 32. 33. Arterioscler Thromb Vasc Biol. September 2007 concomitant enhancement of bleeding propensity. Bioorg Med Chem. 2006;14:5357–5369. Kolesnikov A, Rai R, Young WB, Mordenti J, Liu L, Torkelson S, Shrader WD, Leahy EM, Hu H, Gjerstad E, Janc J, Katz BA, Sprengeler PA. Factor FVIIa inhibitors: Improved pharmacokinetic parameters. Bioorg Med Chem Lett. 2006;16:2243–2246. Kadona S, Sakamoto A, Kikuchi Y, Oh-eda M, Yabuta N, Koga T, Hattori K, Shiraishi T, Haramura M, Kodama H, Ono Y, Esaki T, Sato H, Watanabe Y, Itoh S, Ohta M, Kozono T. Structure of human factor VIIa/tissue factor in complex with a peptide-mimetic inhibitor: high selectivity against thrombin by introducing two charged groups in P2 and P4. Acta Cryst. 2005;F61:169 –173. Kohrt JT, Filipski KJ, Cody WL, Cai C. Dudley DA, Van Huis CA, Willardsen JA, Narasimhan LS, Zhang E, Rapundalo ST, Saiya-Cork K, Leadley RJ, Edmunds JL. The discovery of fluoropyridine-based inhibitors of the factor VIIa/TF complex – Part 2. Bioorg Med Chem Lett. 2006;16:1060 –1064. Riggs JR, Hu H, Kolesnikov A, Leahy EM, Wesson KE, Shrader WD, Vijaykumar D, Wahl TA, Tong Z, Sprengeler PA, Green MJ, Yu C, Katz BA, Sanford E, Nguyen M, Cabuslay R, Young WB. Novel 5-azaindole factor VIIa inhibitors. Bioorg Med Chem Lett. 2006;16:3197–3200. Himber J, Burcklen L, Tschopp T, Fingerle J, Bunting S, Steiner B, Riederer MA. Efficacy and safety of selective inhibitors of thrombin, FXa, and FVIIa in a guinea pig arterial thrombosis model. Thromb Haemost. 2001;Supplement:abstract #P1387. Fingerle J, Himber J, Tschopp T, Bunting S, Steiner B, Riederer MA. Minimal bleeding risk after chronic application of factor VIIa inhibitor in a guinea pig model. Thromb Haemost. 2001;Supplement:abstract #OC1766. Szalony JA, Suleymanov OD, Salyers AK, Panzer-Knodle SG, Blom JD, LaChance RM, Case BL, Parlow JJ, South MS, Wood RS, Nicholson NS. Administration of a small molecule tissue factor/factor VIIa inhibitor in a non-human primate thrombosis model of venous thrombosis: effects on thrombus formation and bleeding time. Thromb Res. 2003;112:167–174. Arnold CS, Parker C, Upshaw R, Prydz H, Chand P, Kotian P, Bantia S, Babu YS. The antithrombotic and anti-inflammatory effects of BCX-3607, a small molecule tissue factor/factor VIIa inhibitor. Thromb Res. 2006;117:343–349. Wong P, Crain E, Watson C, Staus A, Luettgen J, Rendina A, Abell L, Knabb R, Baomin X, Zhou J, DeLucca I, Priestley E. Evaluation of a novel direct factor VIIa inhibitor in rabbit models of arterial thrombosis and hemostatis. J Thromb Haemost. 2005;3(supplement 1):abstract #P0162. Marzec UM, Sukbuntherng J, Dalrymple SA, Young WB, Vijaykumar D, Young PR, Hanson SR. Subcutaneous dosing of CRA-027483, a small molecule FVIIa inhibitor, prevents arterial thrombosis in a baboon model. Blood. 2005;106:abstract#1867. Dalrymple SA, San Pablo FD, Sukbentherng J, Janc JW, Young WB, Vijaykumar D. A selective small molecule factor VIIa inhibitor, CRA027483, suppressed acute inflammation in mice. Blood. 2005;106: abstract#3089. Hu H, Kolesnikov A, Riggs JR, Wesson KE, Stephens R, Leahy EM, Shrader WD, Sprengeler PA, Green MJ, Sanford E, Nguyen M, Gjerstad E, Cabuslay R, Young WB. Potent 4-amino-5-azaindole factor VIIa inhibitors. Bioorg Med Chem Lett. 2006;16:4567– 4570. Rai R, Kolesnikov A, Sprengeler PA, Torkelson S, Ton T, Katz BA, Yu C, Hendrix J, Shrader WD, Stephens R, Cabuslay R, Sanford E, Young 34. 35. 36. 37. 38. 39. 40. 41. 42. 43. 44. 45. 46. WB. Discovery of novel heterocyclic factor VIIa inhibitors. Bioorg Med Chem Lett. 2006;16:2270 –2273. Miura M, Seki N, Koike T, Ishihara T, Niimi T, Hirayama F, Shigenaga T, Sakai-Moritani Y, Kawasaki T, Sakamoto S, Okada M, Ohta M, Tsukamato S. Potent and selective TF/FVIIa inhibitors containing a neutral P1 ligand. Bioorg Med Chem. 2006;14:7688 –7705. Miura M, Seki N, Koike T, Ishihara T, Niimi T, Hirayama F, Shigenaga T, Sakai-Moritani Y, Tagawa A, Kawasaki T, Sakamoto S, Okada M, Ohta M, Tsukamato S. Design, synthesis and biological activity of selective and orally available TF/FVIIa complex inhibitors containing non-amidine P1 ligands. Bioorg Med Chem. 2007;15:160 –173. Peterlin-Masic L, Cesar J, Zega A. Metabolism-directed optimization of antithrombotics: the prodrug principle. Curr Pharm Des. 2006;12:73–91. Riggs JR, Kolesnikov A, Hendrix J, Young WB, Shrader WD, Vijaykumar D, Stephens R, Liu L, Pan L, Mordenti J, Green MJ, Sukbenterng J. FVIIa inhibitors: A prodrug strategy to improve oral bioavailability. Bioorg Med Chem Lettr. 2006;16:2224 –2228. Preželj A, Štefanič P, Peternel L, Urleb U. Recent advances in serine protease inhibitors as anticoagulant agents. Curr Pharm Design. 2007; 13:287–312. Turpie AGG. Oral direct factor Xa inhibitors in development for the prevention and treatment of thromboembolic diseases. Arterioscler Thromb Vasc Biol. 2007;27:1238 –1247. Gustafsson D, Elg M. The pharmacodynamics and pharmacokinetics of the oral direct thrombin inhibitor ximelagatran and its active metabolite melagatran: a mini-review. Thromb Res. 2003;109:S9 –S15. Hampton T. New oral anticoagulants show promise. J Am Med Assoc. 2006;295:743–744. Roehrig S, Straub A, Pohlmann J, Lampe T, Pernerstorfer J, Schlemmer K-H, Reinemer P, Perzborn E. Discovery of the novel antithrombotic agent 5-chloro-N-({(5S)-2-oxo-3-[4-(3-oxomorpholin-4-yl)phenyl]-1,3oxazolidin-5-yl}methyl)thiophene-2-carboxamide (BAY-59-7939): an oral, direct FXa inhibitor. J Med Chem. 2005;48:5900 –5908. Quan ML, Lam PYS, Han Q, Pinto DJP, He MY, Li R, Ellis CD, Clark CG, Teleha CA, Sun J-H, Alexander RS, Bai S, Luettgen JM, Knabb RM, Wong PC, Wexler RR. Discovery of 1-(3⬘-Aminobenzisoxazol-5⬘-yl)-3trifluoromethyl-N-[2-fluoro-4-[(2⬘-dimethylaminomethyl)imidazol-1yl]phenyl]-1H-pyrazole-5-carboxyamide hydrochloride (Razaxaban), a highly potent, selective, and orally bioavailable factor Xa inhibitor. J Med Chem. 2005;48:1729 –1744. Adler M, Kochanny MJ, Ye B, Rumennik G, Light DR, Biancalana S, Whitlow M. Crystal structures of two potent nonamidine inhibitors bound to factor Xa. Biochemistry. 2002;41:15512–15523. Choi-Sledeski YM, Kearney R, Poli G, Pauls H, Gardner C, Gong Y, Becker M, Davis R, Spada A, Liang G, Chu V, Brown D, Collussi D, Leadley R, Rebello S, Moxey P, Morgan S, Bentley R, Kasiewski C, Maignan S, Guilloteau J-P, Mikol V. Discovery of an orally efficacious inhibitor of coagulation factor Xa which incorporates a neutral P1 ligand. J Med Chem. 2003;46:681– 684. Pruitt JR, Pinto DJP, Galemmo RA, Alexander RS, Rossi KA, Wells BL, Drummond S, Bostrom LL, Burdick D, Bruckner R, Chen H, Smallwood A, Wong PC, Wright MR, Bai S, Luettgen JM, Knabb RM, Lam PYS, Wexler RR. Discovery of 1-(2-Aminomethylphenyl)-3-trifluoromethylN-[3-fluoro-2⬘-(aminosulfonyl)[1,1⬘-biphenyl)]-4-yl]-1H-pyrazole-5carboxyamide (DPC602), a potent, selective, and orally bioavailable factor Xa inhibitor. J Med Chem. 2003;46 –5298 –5315. Downloaded from http://atvb.ahajournals.org/ by guest on May 2, 2017 Inhibitors of Factor VIIa/Tissue Factor Rebecca A. Shirk and George P. Vlasuk Arterioscler Thromb Vasc Biol. 2007;27:1895-1900; originally published online June 28, 2007; doi: 10.1161/ATVBAHA.107.148304 Arteriosclerosis, Thrombosis, and Vascular Biology is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231 Copyright © 2007 American Heart Association, Inc. All rights reserved. Print ISSN: 1079-5642. Online ISSN: 1524-4636 The online version of this article, along with updated information and services, is located on the World Wide Web at: http://atvb.ahajournals.org/content/27/9/1895 Permissions: Requests for permissions to reproduce figures, tables, or portions of articles originally published in Arteriosclerosis, Thrombosis, and Vascular Biology can be obtained via RightsLink, a service of the Copyright Clearance Center, not the Editorial Office. Once the online version of the published article for which permission is being requested is located, click Request Permissions in the middle column of the Web page under Services. Further information about this process is available in the Permissions and Rights Question and Answer document. Reprints: Information about reprints can be found online at: http://www.lww.com/reprints Subscriptions: Information about subscribing to Arteriosclerosis, Thrombosis, and Vascular Biology is online at: http://atvb.ahajournals.org//subscriptions/