Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Ellipsometry wikipedia , lookup
Atomic absorption spectroscopy wikipedia , lookup
Thomas Young (scientist) wikipedia , lookup
Retroreflector wikipedia , lookup
Gamma spectroscopy wikipedia , lookup
Mössbauer spectroscopy wikipedia , lookup
Optical coherence tomography wikipedia , lookup
Scanning tunneling spectroscopy wikipedia , lookup
Nonlinear optics wikipedia , lookup
Optical aberration wikipedia , lookup
Super-resolution microscopy wikipedia , lookup
Upconverting nanoparticles wikipedia , lookup
Ultrafast laser spectroscopy wikipedia , lookup
Surface plasmon resonance microscopy wikipedia , lookup
Harold Hopkins (physicist) wikipedia , lookup
Scanning joule expansion microscopy wikipedia , lookup
Magnetic circular dichroism wikipedia , lookup
Atomic force microscopy wikipedia , lookup
Auger electron spectroscopy wikipedia , lookup
Confocal microscopy wikipedia , lookup
Chemical imaging wikipedia , lookup
Photon scanning microscopy wikipedia , lookup
Vibrational analysis with scanning probe microscopy wikipedia , lookup
Phase-contrast X-ray imaging wikipedia , lookup
Gaseous detection device wikipedia , lookup
Ultraviolet–visible spectroscopy wikipedia , lookup
Powder diffraction wikipedia , lookup
Diffraction topography wikipedia , lookup
Reflection high-energy electron diffraction wikipedia , lookup
Transmission electron microscopy wikipedia , lookup
Scanning electron microscope wikipedia , lookup
Low-energy electron diffraction wikipedia , lookup
SEM
TEM
EBSD
“If you quit now you will soon be back to where you
started. And when you started you were wishing to be
where you are now”
Study Guide 2016-2017
Karen Louise De Sousa Pesse
Taught by: Prof. dr. ir.
Roumen Petrov
1
Table of Contents
1 GENERAL PART............................................................................................................................................................5
1.1 QUESTION 1................................................................................................................................................................5
1.1.1 GENERAL CONCEPT OF MICROSTRUCTURE
5
1.1.2 DEFINITION
5
1.1.3 GENERAL PRINCIPALS OF MICROSTRUCTURE CHARACTERIZATION
5
1.1.4 ELASTIC AND INELASTIC SCATTERED SIGNALS
6
1.1.5 STRUCTURE - PROPERTY RELATIONSHIPS
6
1.1.6 MICROSTRUCTURAL SCALE
6
1.2 QUESTION 2................................................................................................................................................................8
1.2.1 RESOLUTION OF THE IMAGING SYSTEMS
8
1.2.2 FACTORS WHICH INFLUENCE THE RESOLUTION OF THE IMAGING SYSTEM
8
1.2.3 RAYLEIGH CRITERION
9
1.3 QUESTION 3.............................................................................................................................................................. 10
1.3.1 INTERACTION OF THE RADIATION WITH THE MATTER
10
1.3.2 THE PENETRATION DEPTH
10
1.3.3 MATERIAL DAMAGE
13
1.4 QUESTION 4.............................................................................................................................................................. 14
1.4.1 SAMPLE PREPARATION - GENERAL REQUIREMENTS
14
1.4.2 SPECIFIC STEPS IN MACRO STRUCTURE SAMPLE PREPARATION
14
1.4.3 SPECIFIC STEPS IN MICRO STRUCTURAL SAMPLE PREPARATION
14
1.4.4 SURFACE EFFECTS AFTER GRINDING AND POLISHING
15
1.4.5 MECHANICAL POLISHING
16
1.4.6 ELECTROLYTIC POLISHING
17
1.4.7 CHEMICAL POLISHING
17
1.4.8 ETCHING, CLEANING AND KEEPING THE SAMPLES
18
2 LIGHT OPTICAL MICROSCOPY (LOM) .......................................................................................................................... 19
2.1 QUESTION 5.............................................................................................................................................................. 19
2.1.1 IMAGE FORMATION, PATH OF THE BEAM/LIGHT
19
2.1.2 LIMITATIONS
20
2.1.3 BRIGHT FIELD
21
2.1.4 DARK FIELD
21
2.1.5 DIFFERENTIAL INTERFERENCE CONTRAST (DIC)
21
2.1.6 POLARIZED LIGHT CONFIGURATION
22
QUESTION 6 ...................................................................................................................................................................... 23
2.1.7 LIGHT OPTICAL MICROSCOPY
23
2.1.8 RESOLUTION
23
2.1.9 NUMERICAL AND ANGULAR APERTURE
23
2.1.10 USEFUL MAGNIFICATION OF THE MICROSCOPE
25
2.1.11 LENS DEFECTS AND METHODS TO BE CORRECTED
26
3 QUANTITATIVE METALLOGRAPHY (QM) .................................................................................................................... 29
3.1 QUESTION 7.............................................................................................................................................................. 29
3.1.1 QUANTITATIVE METALLOGRAPHY (STEREOLOGY)
29
3.1.2 GRAIN SIZE DETERMINATION - VISUAL EVALUATION
29
3.1.3 GRAIN SIZE DETERMINATION – JEFRIES METHOD
30
3.1.4 GRAIN SIZE DETERMINATION (SALTICOV)
30
3.1.5 GRAIN SIZE DETERMINATION (LINEAR INTERCEPTION METHOD)
31
3.1.6 PHASE QUANTIFICATION
31
Karen Louise De Sousa Pesse
2
3.1.7
AUTOMATIC QUANTITATIVE ANALYSIS
31
4 X-RAY DIFFRACTION .................................................................................................................................................. 32
4.1 QUESTION 8.............................................................................................................................................................. 32
4.1.1 GENERAL THEORY (BRAGG’S LAW)
32
4.1.2 RECIPROCAL LATTICE
33
4.1.3 EWALD SPHERE
34
4.1.4 GENERATION OF X-RAYS
35
4.1.5 PENETRATION DEPTH
36
4.1.6 ABSORPTION
36
4.1.7 SAMPLE PREPARATION
37
4.2 QUESTION 9.............................................................................................................................................................. 38
4.2.1 APPLICATION OF X-RAY DIFFRACTION
38
4.2.2 METHODS FOR XRD MEASUREMENTS
39
4.2.3 DETERMINATION OF THE TYPE OF THE CRYSTAL LATTICE, PHASE ANALYSIS, DETERMINATION OF THE LATTICE PARAMETER
42
4.3 QUESTION 10 ............................................................................................................................................................ 44
4.3.1 APPLICATION OF X-RAY DIFFRACTION - QUANTITATIVE PHASE ANALYSIS QPA
44
4.3.2 INTERNAL STRESSES MEASUREMENT (RESIDUAL STRESS)
46
4.4 QUESTION 11 ............................................................................................................................................................ 49
4.4.1 TEXTURE
49
4.4.2 REPRESENTATION OF TEXTURE AND INDIVIDUAL CRYSTALLOGRAPHIC ORIENTATION
50
4.4.3 ORIENTATION DISTRIBUTION FUNCTION ODF
53
4.4.4 POLE FIGURE
54
4.4.5 INVERSE POLE FIGURE
54
4.5 QUESTION 12 ............................................................................................................................................................ 57
4.5.1 PRACTICAL ASPECTS OF TEXTURE MEASUREMENTS BY XRD - GEOMETRY OF THE MEASUREMENT SCHEME
57
4.5.2 SAMPLE PREPARATION
58
4.5.3 EXAMPLES
59
4.5.4 EXAMPLES OF ROLLING, TEXTURES, RECRYSTALLIZATION TEXTURES AND TRANSFORMATION TEXTURES IN FCC AND BCC CRYSTAL
STRUCTURES
60
5 SCANNING ELECTRON MICROSCOPY (SEM) ................................................................................................................ 62
5.1 QUESTION 13 ............................................................................................................................................................ 62
5.1.1 ARCHITECTURE OF SEM
62
5.1.2 TYPES OF FILAMENTS - ADVANTAGES AND DISADVANTAGES
63
5.1.3 INTERACTION OF THE PRIMARY BEAM WITH MATERIAL - EFFICIENCY OF SE AND BSE
64
5.2 QUESTION 14 ............................................................................................................................................................ 65
5.2.1 EDX AND WDX ANALYSIS IN SEM CHARACTERISTIC X-RAYS
65
5.2.2 DETECTORS PRINCIPLE
67
5.2.3 COMPARISON BETWEEN EDX AND WDX SPECTROSCOPY
69
6 ELECTRON MICROSCOPY TEM.................................................................................................................................... 70
6.1 QUESTION 15 ............................................................................................................................................................ 70
6.1.1 THE SAMPLE PREPARATION TECHNIQUES FOR TEM
70
6.1.2 GIVE SCHEMATIC DESCRIPTIONS OF DIFFERENT METHODS
71
6.2 QUESTION 16 ............................................................................................................................................................ 73
6.2.1 IMAGE FORMATION AND CONTRAST FORMATION IN A TEM
73
6.2.2 RESOLUTION IN TEM
73
6.2.3 BRIGHT AND DARK FIELD IMAGING
74
6.3 QUESTION 17 ............................................................................................................................................................ 75
6.3.1 OBJECTIVE APERTURE
75
6.3.2 WHAT IS SAD
75
Karen Louise De Sousa Pesse
3
6.3.3
HOW ARE SAD IMAGES ANALYSED FOR CUBIC MATERIALS?
76
7 ELECTRON BACKSCATTERED DIFFRACTION (EBSD) ...................................................................................................... 80
7.1 QUESTION 18 ............................................................................................................................................................ 80
7.1.1 DEFINITION
80
7.1.2 ARCHITECTURE
80
7.1.3 FORMATION OF KIKUCHI PATTERN.
81
7.1.4 BAND DETECTION
81
7.1.5 HOUGH TRANSFORM
82
7.2 QUESTION 19 ............................................................................................................................................................ 84
7.2.1 EVOLUTION OF ELECTRON BACK-SCATTER DIFFRACTION EBSD
84
7.2.2 ORIENTATION IMAGE ANALYSIS.
84
7.2.3 SPATIAL RESOLUTION AND ANGULAR RESOLUTION OF THE EBSD.
85
7.2.4 WHAT IS IQ, (BC) CI (MAD)
85
7.2.5 EXPERIMENT DESIGN PHILOSOPHY - WHAT KIND OF INFORMATION CAN BE OBTAINED FROM AN EBSD MEASUREMENT? (EXAMPLES) 88
7.2.6 SAMPLE PREPARATION FOR THE EBSD MEASUREMENT
88
7.2.7 COMPARE THE EBSD WITH THE XRD METHOD FOR TEXTURE CHARACTERIZATION.
89
8 3D MICROSTRUCTURE CHARACTERIZATION (3D-EBSD) ............................................................................................... 90
8.1 QUESTION 20 ............................................................................................................................................................ 90
8.1.1 OVERVIEW OF SPECIAL TECHNIQUES
90
8.1.2 3D-EBSD WITH FOCUSED ION BEAM
90
8.1.3 3D-XRAY DIFFRACTION
92
9 AFM/APM ................................................................................................................................................................ 93
9.1 QUESTION 21 ............................................................................................................................................................ 93
9.1.1 APM
93
9.1.2 FIELD ION MICROSCOPE
93
9.1.3 ATOM PROBE
93
9.1.4 SAMPLE PREPARATION
94
9.1.5 APPLICATIONS
94
9.1.6 AFM
95
9.1.7 CONTACT MODE AFM
95
9.1.8 NON-CONTACT MODE AFM
96
9.1.9 TAPPING MODE AFM
96
9.1.10 ADVANTAGES OF AFM
96
9.1.11 DISADVANTAGE OF AFM
96
PRACTICAL CLASSES ........................................................................................................................................................ 97
9.2 EXTRA ON EBSD ........................................................................................................................................................ 97
10 SOURCES................................................................................................................................................................. 98
Karen Louise De Sousa Pesse
4
Materiaalkundige Micro-Analyse en Structuurbepaling:
Vragen voor examen 2016/2017 studiejaar
(Reading guide)
By Karen Louise de Sousa Pesse
1 General part
1.1 Question 1
General concept of the microstructure. Definition, General principles of microstructure
characterization. Elastic and inelastic scattered signals. Structure –properties relationships.
Microstructural scales.
1.1.1 General Concept of Microstructure
1.1.2 Definition
Microstructure is the identical arrangement in 3D space of atoms and all types of non -equilibrium
defects (Book – Physical Methods);
The small scale structure of a material, defined as the structure of a prepared surface of material as
revealed by a microscope above 25x magnification (Wikipedia).
Totality of all thermodynamic non -equilibrium lattice defects in a space scale that ranges from Å to
meters.
The arrangement of phases and defects within a material
1.1.3 General Principals of Microstructure Characterization
The characterization of microstructures is mostly divided into 4 steps:
Probe Source (light, X-rays, Electrons) – Chose the correct method to analyse your material;
Different probe signals can be used for characterizing the microstructure in different scale and
different aspects.
Specimen Sample (polished and etched surface, thin films) – Chose the type of material and pre pare
it adequately to the method you chose;
Image Signal – Elastically or Inelastically scattered radiation, secondary signals)
Data Collection and processing, image interpretation
Karen Louise De Sousa Pesse
5
1.1.4 Elastic and Inelastic Scattered Signals
Light scattering is one of the ma jor physical processes that contribute to the visible appearance of most
objects. Ex.: Backscattered electrons.
1.1.4.1 Elastic Scattered Signal – Equal amount of energy
They have the same wavelength as the incident light, and are divided in:
Optical Imaging (real space): The distance of the image is directly
proportional to the distance of the object, with a constant equal
the magnification.
to
Diffraction Spectra (reciprocal space): The scattering angle
the diffracted radiation is inversely proportional to the
scale of the features of the object, so the distances in the
diffraction pattern are inversely proportional to the
separation of the features in the object.
for
1.1.4.2 Inelastically Scattered Signal – Lower final energy
Scattered photons have a change in ener gy of a molecule due to a transition to another energy level: Ex.:
X-Ray (XRD)
Energy Loss Spectra – electrons will undergo inelastic scattering, lose energy and have their paths
slightly and randomly deflected;
Secondary Signals – Secondary Electrons are loosely bound outer shell electrons from the atom,
which receive sufficient kinetic energy during inelastic scattering of beam electrons to be ejected
from the atom and set into motion.
1.1.5 Structure - Property Relationships
There are two types of structure properties:
Insensitive to Structure: Elastic Moduli, Thermal expansion coefficient, Specific Gravity
Sensitive to Structure: Yield Strength, Thermal conductivity and electrical resistivity and Fracture
toughness;
1.1.6 Microstructural Scale
The typical magnification lies on x10 4 for Microstructure; Common Techniques are Optical Microscopy,
Electronic Microscopy, Transmission Electron Microscopy, and Atomic Force Microscopy; Characteristic
Features (what you can see): Dislocations, Substructure, Grain and Phase boundaries, Precipitations
phenomena;
Karen Louise De Sousa Pesse
6
Scale
Macrostructure
Mesostructure
Typical
magnification
x1
x10
x10
x10
Common
techniques
Visual
inspection Xray
radiography,
Ultrasonic
inspection
Optical
microscopy
SEM
OM, EM,TEM,
AFM
X-ray diffraction
Scanning
Tunnelling
Microscopy,
HRTEM
Characteristic
features
Production
defects
Porosity cracks
and inclusions
Grain
and
particles size
Phase
morphology
and
anisotropy
Dislocation
substructure
Grain
and
phase
boundaries,
precipitations
phenomena
Crystal
and
interface
structure
Point
defects and point
defects clusters
2
Microstructure
4
Nanostructure
6
Resume:
Examples of non-equilibrium; lattice defects are:
Interstitial and Substitutional foreign atoms
Vacancies
Edge and Screw dislocations
Incoherent and coherent precipitations
Grain boundaries
Determination of the microstructure:
Which phase
Shape of the phase
Composition of the phase
Every characterizing system consists of four parts:
A source
A specimen
A signal resulting in the interaction between the source and the sample
A way to process the collected data
Karen Louise De Sousa Pesse
7
1.2 Question 2
Resolution of the imaging systems. Describe the factors which influence the resolution of the
imaging system. Rayleigh criterion.
1.2.1 Resolution of the imaging systems
To describe the microstructure in different scales, we need an appropriate resolution – not always
the highest resolution is the best solution;
Def: Resolution is the minimum distance between two points from which they still can be recognized
as 2 points;
One method is to illuminate the object over its entire surface by using a
suitable source of radiation (photons, electrons or ions) and use a lens
arrangement to form an image, by focusing the radiation that is either
reflected or emitted from the object. A point on the object is f ocused to
equivalent point on an image plane.
an
1.2.2 Factors which influence the resolution of the imaging system
One of the most powerful tools to improve the resolution is the probe’s wave length (λ):
The resolution normal to the direction of the incident beam, often called spatial resolution, is influenced
by the diameter of the incident beam , the wavelength of the incident radiation and the mean free path of
the incident beam in the material.
This can be seen in the equation that defines resolution:
o
Minimum separation distance between two points d
o
Focal Length, the distance between observer and the
points L (Lens system)
o
Angle of resolution () which is obtained from d and L;
o
Diameter of circular opening or diameter of lens aperture D;
o
- Wavelength;
q min = 1.22
l
D
If one uses a second method, in which you direct a very narrow beam of radiation onto the object and
detect the absorbed or reflected radiation, the reflected radiation allows an image of the surface to be
build up. The spatial resolution will then be deter mined by the:
Diameter of the incident beam
Wavelength of the incident radiation
Scattering of the incident radiation within the object surface.
Karen Louise De Sousa Pesse
8
1.2.3 Rayleigh Criterion
Airy disc: when one passes a laser beam through a pinhole aperture; it creates a specifi c
diffraction pattern of light the Airy pattern, which has a bright region in the centre
the Airy disk.
According to Rayleigh Criterion, two point sources cannot be resolved if their
separation is less than the radius of an Airy Disk. The bigger the aperture (D), the
smaller the angle you can resolve (formula);
The Rayleigh criterion for barely resolving two objects that are point sources of light, such as stars seen
through a telescope, is that the centre of the Airy disk for the first object occurs at the first minimum of
the Airy disk of the second.
q min = 1.22
l
D
However, while the angle at which the first minimum occurs (which is sometimes described as the radius of
the Airy disk) depends only on wavelength and aperture size D, the appearance of the diffraction
pattern will vary with the intensity (brightness) of the light source. Because any detector (eye, film,
digital) used to observe the diffraction pattern can have an intensity threshold for detection, the full
diffraction pattern may not be apparent.
1.2.3.1 Rayleigh Scattering
When light is incident on a material, certain resonance frequencies are absorbed in raising the atom to an
excited state.
When the atom decays, that same frequency may be re -emitted in a random direction and not necessarily
the direction of the incident beam.
Karen Louise De Sousa Pesse
9
1.3 Question 3
Interaction of the radiation with the matter. Penetration depth and material damage caused by
photons, electrons and ions.
1.3.1 Interaction of the radiation with the matter
To characterize a microstructure, it is necessary to perturb the material by interacting in some way
with it – to see a surface you have to bombard it with photons.
Objective: Obtain maximum information from material while causing the least amount of damage ;
When the radiation from the probe source strikes on the sample, interaction with the matter occurs.
This interaction is measured and reveals the characteristics of the microstructure. Of course the
intention is to obtain maximum information with the least amount of damage to the sample . A
general rule is to initiate the examination of the sample with the lowest possible intensity. There
are different kinds of radiation sources. The lowest intensity is obtained by using low energy
photons. To obtain more information the source energy may be increased by using X-rays. Electrons
have an even higher energy while ions have the highest energy. The more microstructural
information one wants to obtain, the higher energy source needs to be used . Indeed, the higher the
energy, the lower the wavelength and thus the h igher the resolution. However, this also damages
the sample the most, because of the different penetration depths from the above radiation sources.
1.3.2 The penetration depth
Also known as the mean free path of the incident beam, determines the depth and volume of material that
will be sampled. You probe w ith one type of radiation – for example a beam of X-ray photons - and detect a
second type – like emitted electrons.
The depth that a photon can penetrate in the bulk of a sample depends on the wavelength of the incident
radiation and the material of the sample (more specifically the absorption coefficient of the material µ).
This penetration depth determines the depth and volume of material that will be sampled. Of course a
wavelength is needed that is of comp arable size to the features being studied. Several kinds of radiation
are applied: photons, electrons, neutrons, ions and atoms.
Karen Louise De Sousa Pesse
10
1.3.2.1 Photons
Are discrete quanta of electromagnetic radiation and identified by their wavelength , energy E and
frequency .
𝐸 = ℎ = ℎ𝑐/
The penetration of photons shows considerable and dramatic variations between different types of
material and photon energy or wavelength.
The most important wavelengths for material characterization are
The infrared radiation (long wavelengths) investigate how specific wavelengths are
absorbed;
The visible light shorter wavelengths used to obtain a visual image of the surface,
penetration depth of 50 to 300 nm equal to a several hundred atom layers);
Ultraviolet radiation (shorter wavelength than previous, used to obtain information about
the electron distribution in the surface atoms );
X-rays are even shorter wavelengths and maybe most used. The penetration depth of X -rays
varies both with wavelength and material, and it is typically a few micrometers. The
absorption coefficient µ increases with atomic number and determines the penetration
depth. X-rays are produced by bombarding a metal target with high energy electrons to
produce a band of white radiation.
Superimposed on the white rad iation are a series of discrete maxima whose wavelength and
intensity is determined by the electron binding energies of the atom making up the metal
target being bombard. These characteristic X-ray photons result from electrons falling into
holes created in core electron levels by the incident electron beam with the emission of a
photon whose energy is given by the energy difference between the electron shells.
The shortest wavelengths are obtained with gamma rays (10 - 2 nm), and also the higher
energies. These rays can penetrate considerable distances through a material but the
penetration depth varies inversely with the atomic number.
Karen Louise De Sousa Pesse
11
1.3.2.2 Electrons
The penetration depth varies with energy of
the electron and atomic number of the material
the mean free path of electrons in elements of low
atomic number is large and vice -versa; this is
important because often the material is composed
of different elements with different atomic
numbers;
Electrons are widely used for analysi s because of
their wide energy spectrum (10 eV - 1 MeV), the
flexibility of the electron optics , the strong
interaction between the electron and the solids and
the diffraction of the electrons (figure 2.6 from
physical methods). The penetration depth of electrons varies dramatically wit h both the energy E of
the electron beam and the atomic number Z of the material that is being examined. The energy of
the electrons is produced by the used voltage to accelerate the electrons in the microscope. The
higher the voltage applied, the higher the electrons energy. Thus, the lower their wavelength, and
therefore the higher the resolution, the higher the penetration depth.
Energy = Voltage; = Resolution; Penetration Depth
The penetration depth increases with decreasing atomic number . This has important consequences
for any microstructural characterization since materials will invariably be composed of element s
with different atomic numbers:
Images can present differences in terms of penetration of the electrons into the bulk and the
backscattering of electrons by atoms of different atomic number. The obtained surface images will
differ as the beam energy (and thus the penetration depth) is changed and different anal ysis will be
obtained.
Many techniques detect electrons with low energies in the region of 0 to 2 keV where the effect of
the material on the penetration depth is reduced. Up to 10 eV the very low energy causes the
electrons to move very slowly and instead of colliding with the atoms of the material, they pass
between them. Therefore the penetration depth is very high (and increasing with decreasing
electron energy). These changes in penetration depth can be used in many ways to obtain additional
microstructural information concerning a surface, but also indicates the great ca re that must be
exercised when using electrons to probe a material.
1.3.2.3 Neutrons
Have a mass that is 1000 times more than the mass of electrons . They also have a wave character
and diffract, but they are not electrically charged which means that their penetration depths will be
much larger than those of electrons or X -rays (several millimeters) because they do not interact
with the electron cloud surrounding the nucleus , only with the nucleus. Therefore neutrons are used
to study the microstructure within th e bulk of the material. A disadvantage is the fact that a nuclear
reactor is needed to produce neutrons.
Karen Louise De Sousa Pesse
12
1.3.2.4 Ions or atoms
If ions penetrate a material, so much damage is caused that is more accurate to address the
stopping distance than the penetration di stance;
Ions have an even larger mass. Depending on the energy the ions will either produce elastic (low
energy) or inelastic (high energy) interactions. The elastic ones are limited to imaging the materials
surface – in Ion Scattering Spectroscopy, the io ns reflect from the surface and do not perturb it as
much as a high energy photon or electron . The high energy ions give rise to complex reactions and
penetrate deep into the material . These ions can push out electrons, atoms, ions and ion clusters.
Of course that the higher the energy, the deeper the penetration . With this type of radiation a lot
of damage occurs. When an ion enters the material it will follow a path which is not necessarily
normal to the surface and travel a distance b efore coming to rest at a point. The distance
penetrated is determined by the kinetic energy of the incident ion , the atomic number of the ion
and the atomic number of the material. Although extremely damaging, this method provides
microstructural information that outweighs t he disadvantage.
1.3.3 Material Damage
When radiation interacts with the material, damage always occurs.
1.3.3.1 Photon
A photon source is regarded as the least damaging of the analytical probes. The damage caused is the
result of heating and the degree and extent of this damage is determined by the penetration into the
material, the energy of the radiation and the photon flux.
1.3.3.2 X-Rays
As an example X-rays can cause the surface of certain oxides to be reduced and laser beams can burn holes
through metal by heating to temperatures that result in the instantaneous melting and evaporation in the
immediate vicinity of the beam. In some techniques this phenomenon is used as an advantage. However, as
a general rule, if the results for a microstructural investigation can be obtained using a photon source,
then this should be used.
1.3.3.3 Electrons
Electrons are more damaging than photons because of their greater momentum. Again the resultant
damage is related to the amount of energy or heat transferred to the material and to the thermal
conductivity of the material. Even after a few minutes of observation, changes can be seen. In conventional
instruments, the incident beam energy does not exceed 100 to 200 keV, however some instruments with
more energetic electrons can cause atoms to be displaced from normal lattice positions by the transfer of
momentum.
1.3.3.4 Ions/atoms
When ions or atoms penetrate a material they either interact in essentially a totally non-damaging manner
where they interact elastically with the surface or they cause severe damage where they interact inelastic
with the surface. The damage is done by displacing atoms from their normal lattice positions , and a
minimum energy is required by the ion to exceed the binding energy of the atom. The greatest damage is
caused by this type of radiation. With ions it is even possible to cut a zone by “damaging”.
Karen Louise De Sousa Pesse
13
1.4 Question 4
Sample preparation. General requirements. Specific steps in micro- and macro-sample
preparation. Surface effects after grinding and polishing. Mechanical, chemical, electrolytic
polishing. Etching, cleaning and keeping the samples.
Metallographic Analysis: Analysis of the structure of metals (phases, morphology and distribution) to link
to the properties and manufacturing process, by means of preparing the sur face of the material; can be a
macro/micro structural analysis;
1.4.1 Sample Preparation - General Requirements
Specimen must be representative;
All structural elements must be retained
No scratches/deformations on surface
No foreign matter must be introduced in the specimen surface
Specimen must be plane and highly reflective
Optimal price $$ per sample should be obtained
All preparations must be 100% reproducible
Goal: Prepare for observation the zone of the material that we are interested in! Do not change the
microstructure in this zone during preparation procedure;
1.4.2 Specific Steps in MACRO structure sample preparation
i.
CUT the sample – size is dependent on the needs of the researcher; there is no size limitations
ii.
GRINDING the samples with mechanical grinding papers up to #1000 grit SiC
iii.
ETCHING – the etchants reveal the chemical heterogeneities in macrostructure
1.4.3 Specific Steps in Micro structural sample preparation
i.
Documentation of the material;
ii.
SELECTION of the place that contains zone of interest
iii.
CUT the sample – size depends on the needs of the researcher; surface usually do es not exceed 1,52cm 2 ; depending upon the material, sectioning operation can be obtained by abrasive cutting disc
(metals and metal matrix composites) or diamond cutting disc (ceramics, electronics, biomaterials,
minerals); proper cutting required the correct selection of disc type (hardness) as well as proper
cutting speed, load and coolant.
iv.
PACKAGING or MOUNTING the samples depending on the shape and zone that must be observed –
mould in polymer (cold or hot embedding) or metal sample holders (clamping); small samples are
generally mounted in plastic compound (thermosets, epoxies or thermoplastics) for convenience in
Karen Louise De Sousa Pesse
14
handling and to protect the edges from the specimen being prepared; En grave identification with
electro pen;
v.
GRINDING – Mechanical grinding papers or honeycomb type diamond discs up to #4000 grit; purpose
of grinding is to generate a FLAT SURFACE necessary for the next steps; It goes from COARSE
grinding to FINE grinding, by implying a series of abrasives; Samples must be rotated 90˚ between
stages
vi.
POLISHING (mechanical, electrolytic, chemical, electromechanical, electrochemical); the purpose is
to obtain a final surface free from marks; Diamond abrasives are the most utilize d in polishing (3µm
and 1µm), aluminium oxide powders are also applied for general purposes.
vii.
ETCHING (chemical – most common - or electrolytic); is used to highlight and identify
microstructural features or phases present on the sample. Etchants are usuall y dilute acid or dilute
alkalis in water, alcohol or some other solvent. The acid or base is placed on specimen surface for
seconds or minutes depending on the rate attack on the phases and their orientation.
1.4.4 Surface Effects after Grinding and Polishing
i.
During grinding and polishing, you remove the deformed surface from your material which does not
represent the microstructure; the idea is to see the non -deformed layer of the sample;
ii.
GRINDING removes saw marks and levels and cleans the specimen surface . Polishing removes the
artefacts of grinding but very little stock. Grinding uses fixed abrasives —the abrasive particles are
bonded to the paper or platen—for fast stock removal. Polishing uses free abrasives on a cloth; that
is, the abrasive particles are suspended in a lubricant and can roll or slide across the cloth and
specimen.
iii.
As you decrease the average particle diameter (increasing SiC grit designation), you reduce the
depth of disturbed material (roughness and deformation) as you increase from 100 to 800 and
then 1000 your grinding paper, this means the particles inside it are smaller, thus it will make a
more refine polish on the surface, removing previous deformations and also removing deformations
caused by grinding itself.
iv.
At first you remove the deformed smear zone, then the fragmented zone and contours of equal
deformation until you reach the non -deformed region (figure);
Karen Louise De Sousa Pesse
15
1.4.5 Mechanical Polishing
Parameters: Speed, pressure, Time, Lubrication and Cooling (SPTLC)
Labour is a major variable in the process
The process is to do grinding first, polishing second, and buffing third. In general, grinding permits far
more aggressive abrading action than polishing. Likewise, polishing is a far more aggressive abrading action
than buffing.
In grinding, polishing and buffing, labour is a major variable in the process. The requirement is for highly
skilled labour with years of experience and a thorough knowledge of the art of their craft.
The basic mill plate and sheet metal finishes for stainless steel include five grades that have finishes that
are produced mechanically by using abrasive compositions and buffing wheels. There is also available on
the market what is generically known as 'non -directional No. 8."
Special mechanical polishing procedures ar e required for preparing metal surfaces, such as stainless steel,
for electropolishing. Mechanical polishing and buffing cannot be viewed as an adequate substitute for
electropolishing in most applications due to the embedded abrasives and compounds, exposed grain
structure of the metal, and the lack of the non -particulating, non-contaminating, and non-outgassing
characteristics of an electropolished surface.
A mechanically polished metal surface yields an abundance of scratches, strains, metal debris and
embedded abrasives. Mechanical polishing fails to remove inclusions, but also tends to push them further
into the surface and even increase them by further pickup of abrasive materials which can lead to future
points of corrosion. In contrast, the electropolishing process results in a surface which is completely
featureless. It reveals the true crystal structure of the metal without the distortion produced by the cold working process that always accompanies mechanical finishing methods.
Karen Louise De Sousa Pesse
16
1.4.6 Electrolytic Polishing
Excellent method for deformation-free polishing, but restricted
mainly to single phase materials;
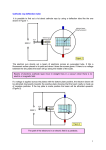
In electropolishing, the metal is removed ion by ion from the
surface of the metal object being polished;
In basic terms, the object to be electropolished is immersed in an
electrolyte (typically phosphoric and sulphuric acid) and subjected
to a direct electrical current. The object is maintained anodic,
with the cathodic connection being made to a nearby metal
conductor (see diagram). In electropolishing, the metal is
removed ion by ion from the surface of the metal object being
polished.
During electropolishing, the polarized surface film is subjected to
the combined effects of gassing (oxygen), which occurs with
electrochemical metal removal, saturation of the surface with
dissolved metal and the agitation and temperature of the
electrolyte. Electropolishing sel ectively removes microscopic high points or "peaks" faster than the rate of
attack on the corresponding micro -depressions or "valleys."
Source: Delstar.com
1.4.7 Chemical Polishing
https://en.wikipedia.org/wiki/Chemical -mechanical_planarization
Chemical mechanical polishing/planarization is a process of smoothing surfaces
with the combination of chemical and mechanical forces. It can be thought of as a
hybrid of chemical etching and free abrasive polishing.
The process uses an abrasive and corrosive chemical slurry (commonly a colloid) in
conjunction with a polishing pad and retaining ring, typically of a greater diameter
than the wafer. The pad and wafer are pressed together by a dynamic polishing
head and held in place by a plastic retaining ring. The dynamic polishing head is
rotated with different axes of rotation (i.e., not concentric). This removes material
and tends to even out any irregular topography, making the wafer flat or planar.
Typical CMP tools, such as the ones seen on the right, consist of a rotating and
extremely flat platen which is covered by a pad. The wafer that is being polished is
mounted upside-down in a carrier/spindle on a backing film. The retaining ring
(Figure 1) keeps the wafer in the correct horizontal position. During the process of
loading and unloading the wafer onto the tool, the wafer is held by vacuum by the
carrier to prevent unwanted particles from building up on the wafer surface.
Karen Louise De Sousa Pesse
17
1.4.8 Etching, Cleaning and Keeping the Samples
Non-metallic inclusions are better observable in non -etched samples;
i.
Etching is an important step for adequate further visualization of the sample. This technique uses
chemical action to produce lines on metal samples, in order to view the metal specimen under an
optical microscope. On figure 1(a), Titanium sample was not etched and observed in Keyence Light
Optical Microscope, using bright field illumination, and figure (b) shows the etched sample with
obvious microstructure.
ii.
Etching is used to highlight and identify microstructural features or phases present . Etchants
are usually dilute acid or dilute alkalis in water, alcohol or some other solvent.
iii.
Etching occurs when the acid or base is placed on the specimen surface because (for seconds
or several minutes) of the difference in rate of attack of the various phases present and their
orientation. The etching process is usually accomplished by merely applying the appropriate
solution to the specimen surface.
iv.
The most common technique for etching is the chemical etching. Other techniques such as
electrolytic, thermal and plasma etching have also found specialized applic ations.
Figure 1: Difference in
a) non-etched and b)
etched sample of
Titanium.
Cleaning the samples the samples are first cleaned with ethanol. After polishing they are also cleaned
with acetone. After this cleaning the sample must be dried. Without this cleaning there is a possibility of
corrosion of the sample.
http://www.asminternational.org/documents/10192/3460742/06785G_Sample.pdf/ad6f8964 -40da-4ff3-a2f5c4647e2a94d8
Karen Louise De Sousa Pesse
18
2 Light Optical Microscopy (LOM)
2.1 Question 5
Discuss the image formation, path of the beam/light and
limitations (resolution, in-depth sharpness) of light
optical microscopy. How does the image form during
the observation in bright field, dark field and
differential interference contrast?
2.1.1 Image Formation, Path of the Beam/Light
2.1.1.1 First Explanation
The microscope increases the angle of observation;
The Reflected Light Optical or Metallurgical Microscope is ideal for samples in which light is unable to pass
through. Thus, light is directed onto the surface and eventually returns to the microscope’s objective lens
by specular or diffused reflection.
On the first step, a Tungsten-Halogen Lamp emits light through the Collector Lens, Condenser
Aperture Diaphragm and Field Diaphragm. The condenser concentrates light onto the specimen while its
diaphragm regulates resolution, contrast and depth of field;
The beam splitter (or half mirror) is an optical device that splits a beam of light in two, reflecting and
transmitting light, and can be used to recombine separate light beams into a single path. Deviated light
goes out of phase, causing destructive int erference with the direct light that has passed through non deviated. (Some of the light passes undisturbed in its path – non-deviated light - and some light is
diffracted when it encounters parts of the specimen);
The objective lens gathers light from the object being observed and focuses the light rays to produce a real
and inverted image; this lens is the one at the bottom near the sample;
Then, light reflected from the surface of the specimen re -enters the objective and is directed to the eye pieces – ocular lens placed near the focal point of the objective – magnifying the intermediate image.
The eye lens of the eyepiece magnifies this image which is then projected onto the retina;
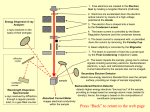
Reflected Light Microscopy Illuminator. Light
passes through collector lens, being controlled
by aperture and field diaphragms. Afterwards,
a half mirror reflects the light through the
objective to illuminate the specimen. Source:
Olympus
Karen Louise De Sousa Pesse
19
2.1.1.2 Another Explanation
i.
An object O of height h is being imaged on the
retina of the eye O’’.
ii.
The objective lens (L o b ) projects a real and inverted
image of O magnified to the size O’ and height h’ into the
intermediate image plane of the microscope.
iii.
This occurs at the eyepiece diaphragm, at the fixed
distance fb + z’ behind the objective.
iv.
In this diagram, fb represents the back focal length
of the objective and z’ is the optical tube length of the
microscope.
v.
The aerial intermediate image at O’ is further
magnified by the microscope eyepiece (L e y ) and produces
an erect image of the object at O’’ on the retina, which
appears inverted to the viewer.
vi.
The magnification factor of the object is calculated
by considering the distance a between the object O and
the objective (L ob ) and the front focal length of the
objective lens (f).
vii.
The object is placed a short distance ( z) outside of the objective’s front focal length ( f), such that z
+ f = a.
viii.
The intermediate image of the object, O’, is located at distance b, which equals the back focal
length of the objective (fb) plus (z’), the optical tube length of the microscope.
ix.
Magnification of the object at the intermediate image plane equals h’. The image height at this
position is derived by multiplying the microscope tube length ( b) by the object height (h), and
dividing this by the distance of the object from the objective: h’ = (h x b)/a.
x.
Source: http://www.olympusmicro.com/primer/microscopy.pdf
2.1.2 Limitations
Resolution At very high magnifications with transmitted light, point objects are seen as fuzzy
discs surrounded by diffraction rings (Airy Disks). The resolving power of a microscope is taken as
the ability to distinguish between two closely spaced AIRY DISKS.
Diffraction Limit Finite limit beyond which it is impossible to resolve separate points . The
diffraction patterns are affected by both the wave length of the light, the refractive material used
to manufacture the lens and the numerical aperture of the objective lens .
Karen Louise De Sousa Pesse
20
2.1.3 Bright Field
*It is important to know how to draw all of them
Among the illumination modes, Bright Field (BF) is the simplest of them. The light beam strikes the
sample perpendicularly, relating bright areas to horizontal zones in which the beam returns unaffected,
and darker areas to tilted zones where the recurrent beam is scattered (Figure 4, a). Although the Bright
Field image suffers from lack of contrast details, it supplies a general outline of the overall features on the
specimen. Grain boundaries have darker colours since less light goes through the objective lens.
2.1.4 Dark Field
A very important technique in reflected light microscopy is the dark field, which allows, through and
oblique illumination, to obtain a bright contrast in regions with a small inclination regarding the surface.
As you increase the tilt of vertical illuminator, waves are directed away from the objective . The waves go
through the mirror assembly and oval mirror (Figure 4, b), passing through an outer sleeve next to the
objective lens towards a concave mirror and then finally hit the sample surface at highly incident angle .
Bright features are formed by areas with relief contour that direct light back through the objective lens,
however most of the light is not reflected back, hence the dark background.
On this technique, it is advised that the field and aperture diaphragms located in the vertical
illuminator remain opened to their widest points, avoiding that the light beam illumina ting the mirror
assembly is blocked.
2.1.5 Differential interference contrast (DIC)
Material Scientists typically employ the reflection mode, also known as episcopic light differential
interference contrast (DIC), in opaque specimens that are highly reflective and do not absorb or transmit
significant amount of incident light. This te chnique yields more complete analysis of the surface structure.
Topographical differences like slopes, depressions and other discontinuities on the surface of the
sample create optical path differences in the reflected beam, which will further be transfor med to
amplitude or intensity variation by the illumination mode. The image can often be interpreted as a three
dimensional representative, although depth may be misleading. The rainbow patterns along the features is
caused as various colours destructively interfere at different locations on the surface, since the formation
of final image is the result of interference between two distinct wave fronts that reach the image plane out
of phase.
A birefringent prism (also known as Nomarski prism) is placed in th e space above the objective and
a polarizer is installed in the vertical illuminator. The prism will then divide the polarized wave lengths into
two orthogonal polarized beams that will hit the specimen, creating a lateral displacement in regions of
surface relief. Flat surfaces do not display any features. Once the beam returns through the objective and
prism, it goes through a second polarizer ( analyser). The interference produces an intermediate image that
is captured by the eyepiece and then image is ma gnified.
Karen Louise De Sousa Pesse
21
2.1.6 Polarized Light Configuration
Polarizers can be inserted into the vertical illuminator before the mirror unit, as well as before light
enters the objective (Figure 4, c), enhancing contrast and improving the quality of the image obtained.
Optically anisotropic samples alter the state of polarization during the reflection process. The reflected
wave goes through the objective and is projected onto a second polarizer (the analyser) which filters
depolarized wave fronts, letting them pass. The tech nique is important to distinguish isotropic and
anisotropic materials.
Learn how to draw:
Karen Louise De Sousa Pesse
22
Question 6
Light optical microscopy. Resolution. Numerical and angular aperture. Useful magnification of
the microscope. Lens defects and methods to be corrected.
2.1.7 Light Optical Microscopy
Simpler method for the analysis of solid materials
Two modes are typically employed, based on the measurement of transmitted or reflected light,
from transparent to opaque sample respectively.
For metallurgy, samples are mostly opaque, hence the usage of the Reflec ted Light Optical
Microscope (also called Metallurgical Microscope)
The Reflected Light Optical or Metallurgical Microscope is ideal for samples in which light is unable
to pass through (opaque materials). Thus, light is directed onto the surface and eventually returns
to the microscope’s objective lens by specular or diffused reflection, using a system of mirrors,
prisms and semi-mirrored glasses which allows the light beam to pass in one direction and refl ects
in the other.
Due to the inherent difference in intensity or wavelength of the light absorption characteristics of
the different phases, contrasts are observed. These contrasts can be enhanced by etching.
2.1.8 Resolution
The ability to discern fine details within a magnified image is referred to as the resolution of a
microscope.
Since light is used as the illumination source in optical microscopy, the resolution is expressed in
the same unit as the wavelength of the light (nm ). The theoretical resolution, d, of any optical
system may be calculated using Abbe’s equation.
n sinµ is the numerical aperture with sinµ being the angular aperture. N is the refracting index of the
medium in which the lens operates (mostly 1).
2.1.9 Numerical and Angular Aperture
Numerical aperture is a number that characterizes a range of angles, and within these angles a system can
receive or emit light. NA = n sinµ
Karen Louise De Sousa Pesse
23
It is clear that the resolution depends mainly on the numerical aperture .
The bigger its value, the higher the resol ution (the smaller d).
This numerical aperture can be changed by changing the medium, for example using oil (n=1.54)
instead or air (n=1).
The numerical aperture can also be enlarged by a bigger collecting angle; this angle is bigger when
the focal distance is lower or the width of the objective lens is higher. Of cours e these changes are
restricted by geometrical factors. Theoretically the maximum angular aperture is equal to one, but
in practice it is restricted to a value of 0.95. This of course also l imits the resolution of the optical
microscope.
The best resolution is obtained with the highest numerical aperture and the lowest wavelength. The
shortest wavelength for visible light is blue: 450 nm. The best lense s have a collecting angle of 70 °
which means that the best angular aperture is equal to 0.94. The highest resolution lenses work in
an oil medium with a refractive index of 1.56. Together this gives a maximum resolution of about
200 nm.
d m i n = 1.22 * 450 nm/(2*1.56*0.94) = 202 nm
When two points on the visualized sample are closer than 202 nm, they will not be distinguished by optical
microscopy.
http://www.olympusmicro.com/primer/anatomy/numaperture.html
Karen Louise De Sousa Pesse
24
2.1.10 Useful Magnification of the Microscope
Whereas the resolution is influenced by the objective,
magnification is influenced by the ocular .
the
It is important to see the difference between
and magnification:
resolution
Resolution involves the visibility of details o n
doesn’t mean
Magnification only enlarges the view, but this
that more details are obtained. The ocular only
magnifies the
intermediate image, without giving the additional details in it. For the latter, the objective needs to
be adapted, so that the resolution is better and details are revealed.
The objective characteristics thus determine the main characteristics of the microscope, namely the
useful magnification limits and the global (general) magnification because you can magnify the
image all you want but when the resolution is not good enough at those magnifications, you will not
see a thing. Therefore, it is important that the real magnification lies in the range of the minimal
and maximal magnification determined by the objective.
the sample.
W re a l = W o bj e c t i ve * W o c u la r
W o bj e c t ive , m i n = 500 * numerical aperture
W o bj e c t ive , ma x = 1000 * numerical aperture
W o bj e c t ive , m i n < W re a l < W o bj e c t iv e , ma x
If W re a l is smaller than the minimal objective magnification, details visible for the objective are lost
because the magnification is not high enough. When W re a l is bigger than the maximal objective
magnification, a blurred image will be obtained, because the magnification is out of the range for a good
resolution. Recapitulatory the objective determines the minimal and maximal magnification at which a
good image (good resolution) is obtained. When the real magnification does not lie within this range, the
image will be blurred. (For an example see lecture 2, slide 39)
Example: If a mignification of 500x is needed, this means Wmin < 500 < Wmax
This can be written as 500*A < 500 < 1 000*A we needed A between 0,5 - 1
If there is one objective with this condition: A is 0,65 with a magnification of 50 , the magnification of the
ocular is between 10 (to have 500x magnification) and 13 (because the magnification cannot be bigger than
650) which left only the ocular with magnification of 10 .
Karen Louise De Sousa Pesse
25
2.1.11 Lens Defects and Methods to be corrected
Lenses used in optical systems do not give perfect images because of defects and aberrations. Luckily
correction methods are available (see also figure 5.2 in physical methods and figures in lecture on lens
defects).
Spherical aberration: The rays which are deviated from the optical centre
have a focal point which is different from the one of the central rays . As a
consequence, the image is not sharp. This defect can be corrected by
applying lens corrections and/or placing a diaphragm in front of the le ns,
but this reduces the numerical aperture, which in turn reduces the
resolution. Another solution is the use of aspheric lenses.
Spherical aberration occurs because spherical surfaces are not the ideal shape with which to make a lens,
but they are by far the simplest shape to which glass can be ground and polished and so are often used.
Spherical aberration causes beams parallel to but away from the lens axis to be focused in a slightly
different place than beams close to the axis. This manifests itself as a blurring of the image. Lenses in
which closer-to-ideal, non-spherical surfaces are used are called aspheric lenses. These were formerly
complex to make and often extremely expensive, although advances in technology have greatly reduced
the cost of manufacture for these lenses. Spherical aberration can be minimised by careful choice of the
curvature of the surfaces for a particular application: for instance, a plano -convex lens which is used to
focus a collimated beam produces a sharper focal spot when used with the convex side towards the beam.
Coma formation: The surroundings of a point are distorted like a comet. When a lens is corrected for
spherical aberration, coma formation can still occur. This is a type of aberration that affects rays which lie
off the axis of the lens. Coma arises from differences in refraction indices of the rays passing through the
inner and outer zones of the lens. Under these conditions the point images as a comet shape. This effect
can be reduced by the use of a suitable l ens aperture.
Another type of aberration is coma, which derives its name from the comet -like appearance of the
aberrated image. Coma occurs when an object off the optical axis of the lens is imaged, where rays pass
through the lens at an angle to the axis θ. Rays which pass through the centre of the lens of focal length f
are focused at a point with distance f tan θ from the axis. Rays passing through the outer margins of the
lens are focused at different points, either further from the axis (positive
coma) or closer to the axis (negative coma). In general, a bundle of
parallel rays passing through the lens at a fixed distance from the centre
of the lens are focused to a ring-shaped image in the focal plane, known
as a comatic circle. The sum of all these c ircles results in a V-shaped or
comet-like flare. As with spherical aberration, coma can be minimised
(and in some cases eliminated) by choosing the curvature of the two lens
surfaces to match the application. Lenses in which both spherical
aberration and coma are minimized are called bestform lenses.
Karen Louise De Sousa Pesse
26
Chromatic aberration: A light source consists of different wavelengths. Rays with different wavelength
have a different refraction index, which results in a different focal point. This is called chromatic
aberration. It can be solved by using double or multiple lenses.
Chromatic aberration is caused by the dispersion of the lens material, the variation of its refractive index n
with the wavelength of light. Since from the formulae above f is dependent on n , it follows that different
wavelengths of light will be focused to different positions. Chromatic aberration of a lens is seen as fringes
of color around the image. It can be minimised by using an achromatic doublet (or achromat) in which two
materials with differing dispersion are bonded together to form a single lens.
This reduces the amount of chromatic aberration over a certain range of
wavelengths, though it does not produce perfect correction. The use of
achromats was an important step in the develop ment of the optical
microscope. An apochromat is a lens or lens system which has even better
correction of chromatic aberration, combined with improved correction of
spherical aberration. Apochromats are much more expensive than
achromats.
Astigmatism: If a lens does not have perfect axial symmetry , the image plane for objects lying in one
direction differs from the image plane for objects lying in another direction (the image is formed
asymmetrically). Consequently, vertical components of the image focus in a different plane compared with
the horizontal components and no sharp image plane exists, only a plane of least confusion between two
sharply focused images. In optical systems this is inherent and relates to the manufacturing quality of the
glass lens.
Complex and expensive objectives are available which improve the correction for formation of undistorted
images and colours.
Karen Louise De Sousa Pesse
27
There are two types of corrections: achromatic and apochromatic. The resolution in optical microscopes is
thus determined/restricted by the lenses of the optical system.
Achromatic doublet (or achromat) in which two materials with differing dispersion are bonded together to
form a single lens.
apochromat is a lens or lens system which has even better correction of chromatic a berration, combined
with improved correction of spherical aberration
Sources: Lecture 2, slides 34 to 39,
Flewitt:”Physical methods for materials characterization -Second Edition”, Chapter 5. Parts 5.2 and 5.3 .
Karen Louise De Sousa Pesse
28
3 Quantitative Metallography (QM)
3.1 Question 7
Quantitative metallography (Stereology). Grain size determination (visual evaluation, Jeffries,
Salticov and linear interception method) Phase quantification. Automatic quantitative analysis.
3.1.1 Quantitative metallography (stereology)
Stereology is a group of statistical methods (several measurements are necessary to obtain a reliable
result) to obtain the size of the structural constituents and elements of a material. Also the quantity of the
phases can be calculated. The main problem with m etals is them being opaque (non-transparent). Thanks
to appropriate mathematical assumptions, extension to 3D characterization is possible. These methods are
based on the Kavalieri principle: If the cross sections are equal or proportional then also the objects are
equal or proportional (see figure below). For all the methods, it is important to know the magnification.
3.1.2 Grain size determination - visual evaluation
The American Society for Testing of Materials (ASTM) has introduced a grain
size number N which is defined as
n = 2 N- 1
where n is the number of grains in 10 - 4 square inch, or in 0.0645 mm 2 , or in 1
in 2 under 100x magnification. (Verify that these three definitions are
equivalent). N provides an excellent characterization of the grain size of the
material. For most microstructures N has a value between 0 and 8, but n can
be negative or greater than 8.
If we recalculate for 1mm², then
𝒏𝟎 = 𝟐𝑵+𝟑
Where n 0 is the number of grains in 1mm 2 .
n 0 is actually two times n where n is the number of grains at a magnification of 100:1.
The average surface of the grains S = 1/ n 0 .
The average grain diameter d = 1/√𝑛0
The ASTM number is then given by G = -6.64*log10 (d) -2.95 where d is the grain diameter given in mm.
Karen Louise De Sousa Pesse
29
3.1.3 Grain size determination – Jeffries method
This method is also based on counting the number of grains
in a predetermined view field (mostly a circle with a certain
diameter). It is important to work with an appropriate
magnification. When the number of grains inside the view
field is too low, the calculation will not be representative.
60 to 70 grains is good. When the magnification is set, the
number of grains completely inside the view field is
counted. This value is called p. Also the number of grains
partially in the view field is counted. This value is called q.
Next n x is determined; this is the number of grains at the
current magnification Mx.
n x = p+k*q with k a correction coefficient (the lower p, the
smaller k)
Now n 0 (the number of grains in 1 mm²) can be calculated:
n 0 = (2*n x * M² x )/M² with M the standard magnification equal to 100:1
n 0 is also equal to two times n: the number of grains at the standard magni fication M (view field is 0.5
mm²). Then N can be calculated from the above relationship between N and n 0 . Again the average surface
of the grain S and the average grain diameter d can be calculated as before.
3.1.4 Grain size determination (Salticov)
This method is based on counting the number of triple junction points inside a predetermined view field
(mostly a circle with a certain diameter). A triple junction point is a point where 3 grains meet. K is the
number of these triple junction points inside the view field. K is then equal to 2 times n for a standard
magnification M of 100:1. At the current magnification M x , K x is equal to 2 times n x . n 0 is then calculated as
follows.
K=2n for M=100:1 and
(K x =2n x ) for M x
=M
n 0 = Kx * M²x/M²
Again from this, grain size number N, the average surface of the grains
S and the average grain diameter d can be calculated.
Karen Louise De Sousa Pesse
30
3.1.5 Grain size determination (linear interception method)
This method is based on counting the grains that intersect with a
predetermined line. P L is the number of intercepts of grain
boundaries per unit length of the test line (the exact length of the
test line must be known) and the average or mean intercept length
(or average grain diameter) is then:
L 3 = d/P L
For example, if you cross a line of 15cm, use the scale bar to
adjust to the size of measurement. If 20 intercepts were counted,
the average size diameter will be N A = size of line/number of
intercepts.
The ASTM grain size number n can be obtained from the Hillard relation.
n = -3.36-2.88*ln(L 3 ) with L 3 given in mm
The amount of grain boundary surface per unit volume is called Sv and is equal to two times P L .
Attention: Best method if the question is in diameters (and not in surface, which would be Jeffries)
3.1.6 Phase quantification
The Rosival method is a linear method to quantify th e average phase
fraction. For this some lines are drawn and the total number of scale
divisions is called L. Here it is thus not important to know the length
of the test lines but rather the fractions. Li is the number of divisions
that lay in the black co nstituent (second phase). Lav is then equal to
the sum of all Li divided by i with i the number of test lines. The
volume fraction is then given by V = Lav/L. Sometimes for high
anisotropic materials, a circle is used instead of a line.
For example: cross 4 lines on your picture of 15 cm each. Make
traces every 0.5cm. How many times a trace passed the white phase? Get this number and find the
percentage of white phase in your material.
3.1.7 Automatic quantitative analysis
This method is done automatically by image processing. The phases are quantified by the delineation of
the pixels that belong to different phases based on the light intensity or colour differences. Starting from
an optical microscopy image or a SEM image, a masked image is produced and then a binary image (2
colours). From this binary image, different phases are quantified. The advantage is the easy and fast data
collection and the repeatability of the results. This method is however very sensitive to the sample
preparation (which is operator dependent). This is an important disadvantage. The best option for
quantitative characterization is the orientation mapping with EBSD but this requires highly specialized
equipment and sample preparation.
Karen Louise De Sousa Pesse
31
4 X-ray diffraction
4.1 Question 8
Give the general theory about X-ray diffraction (Bragg law, reciprocal lattice, Ewald sphere).
Generation of X-rays. Discuss also the penetration depth, absorption and sample preparation for
XRD examination.
4.1.1 General theory (Bragg’s Law)
Related to scattering of waves that h it a crystal Bragg diffraction occur when radiation (with
comparable to atomic spacing d h k l ) is scattered by atoms of a system and undergoes constructive
interference.
𝑛𝜆 = 2𝑑𝑠𝑖𝑛𝛳 →when x-rays are scattered from a crystal lattice, peaks of scattered intensity are
observed which correspond to the angle of incidence = angle of scattering;
X-Ray Diffraction (XRD) is based on the diffraction of incident X -rays on a sample. The beam of X -rays has a
thickness of a few mm which means that XRD is a macroscopic technique. No local information is obtained
and the technique is less sensitive to imperfections . X-rays are photons with a wavelength of the order of a
fraction of a nanometer.
A sample, for example a metal, consists of different atomic layers corresponding to its crystal structure
(see figure below). When X-rays are incident on the metal, they are reflected by the different atomic layers
of the sample (the sample somewhat acts like a mirror).
Thomson effect: scattering of X-Rays by electrons: diffraction
Actually the Thomson effect occurs: The X-rays are elastically scattered by the electrons of the atoms. The
atoms are polarized by the X -rays, acting like separate emitters. Only the waves with a common tangent to
the wave front (coherent waves) can leave the material, the other waves interfere destructively. The path
difference between the reflected X -rays of different atomic layers corresponds to the interplanar distance
d h k l , between the atomic layers which is characteristic to the crystal stru cture. The reflected waves are
detected only when constructive interference occurs (when the reflected wave lengths are coherent, in
phase). This means that Braggs law needs to be fulfilled.
Karen Louise De Sousa Pesse
32
n ∗ λ = 2 ∗ dhkl ∗ sin θ
n is the order of diffraction, θ is the diffraction angle and actually determines the direction of the crystal
planes with respect to the X -rays at which interference occurs. This angle is changed by rotating the
sample. Constructive interference only occurs when the difference in path length equals an even number
of times the wavelength of the source . If not the waves cancel out and no signal is detected. By measuring
theta and knowing λ, the interplanar spacing can be determined and the crystal spacing identified. Also the
lattice parameter or the miller indices can be obtained.
𝑑 =
𝑎
√(ℎ2 + 𝑘 2 + 𝑙 2 )
𝑓𝑜𝑟 𝑎 𝑐𝑢𝑏𝑖𝑐 𝑐𝑟𝑦𝑠𝑡𝑎𝑙
These values are compared to measurements of samples and with random orientations (powde r). This
means that XRD is a relative measurement.
4.1.2 Reciprocal Lattice
The reciprocal lattice is constructed to aid the
interpretation of diffraction from crystal lattices.
In real space crystal planes are defined by their intercepts
on coordinate axis, usually with axis units being defined as
the Miller Indices hkl.
1/dhkl
Planes with intercepts hkl have families of planes nh, nk, nl
that are parallel to hkl and contribute to a diffracted beam.
These planes are separated by a distance
𝑑ℎ𝑘𝑙
𝑛
.
Check https://www.youtube.com/watch?v=fZ0m8wustVk
In the reciprocal space reciprocal lattice is constructed for a defined crystal lattice by drawing a line
from the origin, normal to the lattice plane hkl . This will be of length g = 1/d h kl = d* h kl and is equal to the
reciprocal of the interplanar spacing d h k l . The reciprocal lattice points correspond both to planes with
miller indices hkl and those with indices nh, nk, nl which al so contribute to diffraction.
Thus, the reciprocal lattice defines a range of potential lattice sites that may lead to diffraction. A
particular lattice type may be characterized by absent diffraction positions and the corresponding points in
the reciprocal lattice will be missing. For example the reciprocal lattice of BCC is FCC and vice versa. Also
an hkl vector in the reciprocal space is perpendicular to an hkl vector in the real space. The diffraction of
an X-ray beam can be predicted from the reciproca l lattice using the Ewald construction or Ewald sphere.
Each spot is a set of planes in our unit cell;
The intensity of each spot is related to the amount of scattering matter on that set of planes
The distance g between spots is 1/d h k l ;
Karen Louise De Sousa Pesse
33
4.1.3 Ewald Sphere
The Ewald Sphere is used to predict the reciprocal lattice of an X -ray diffraction. The Ewald circle
represents in reciprocal space all the possible points where planes (reflections) could satisfy the
Bragg equation.
The incident X-ray beam is considered to pass through the origin (0 0 0) in both real and reciprocal
space. A sphere is then drawn with a radius of 1/λ (diffraction sphere) inside a limiting sphere of
radius 2/.
The centre of the diffraction sphere is on the incident beam direction and position as well, as such
that the surface of the sphere passes through the origin .
This sphere is known as the Ewald sphere or the diffraction sphere. Diffraction of the X -ray beam will
only occur if the Ewald sphere passes throu gh a reciprocal lattice point r* h k l when another
reciprocal point (that is not the origin) touches the sphe re, like r*, the Bragg condition is satisfied;
k 1 represents the diffracted wave vector, k 0 the incident wave vector and g the reciprocal lattice
vector corresponding to the diffracting planes.
The condition here for diffraction to occur is thus that the change in wave vector k 1 must be equal to
a vector of the reciprocal lattice (which is perpendicular to the hkl planes in real space and has a
length of the order of the reciprocal of the interplanar spacing).
g=1/dhkl
When the sample is rotated (and hence the r eciprocal lattice points and the Ewald sphere), different
crystal planes can be analysed. Since the wavelength stays the same, so does the diameter of the Ewald
sphere and no reciprocal lattice points outside a sphere of radius 2/λ can pass through the Ewald sphere
and therefore cannot diffract the X -ray beam so that this is called the limiti ng sphere.
Please check https://www.doitpoms.ac.uk/tlplib/reciprocal_lattice/ewald.php
Karen Louise De Sousa Pesse
34
4.1.4 Generation of X-rays
X-rays can most easily be produced by bombarding a
material surface with relatively high energy electrons ,
which were accelerated by a certain voltage.
A preheated filament (mostly Tungsten) is used as a
cathode and emits electrons which are accelerated to the
anode by a positive voltage of 20 -50 kV. The electrons
generate X-rays after collision with the anode . The higher
the atomic number of the material of the anode, the
lower the wavelength of the produced X -rays. The X-rays
escape through a Beryllium window.
In practice it is normal to use metals to produce X -rays
for use in XRD because an intense beam of X -rays is
desired and the good thermal conductivity of the metal allows the heat produced
during bombardment with an intense high energy electron beam to be readily
removed thus avoiding damage to the source.
When a high energy electron collides on the material, X-rays are produced in two
ways. Because the incident electrons are decelerated by the material they emit a
continuous spectrum of bremsstrahlung (inelastic scattering). The intensity of
these X-rays is a function of the electrons energies and the atomic number of the
target. The incident electrons also cause the surface atoms to be ionized by the removal of an inner shell
electron. As the atom rearranges with electrons from outer orbitals falling into the hole created, energy is
released in the form of a photon. The probability that these characteristic X -rays will be emitted increases
with atomic number of the target. Normally these characteristic peaks are used to
produce diffraction patterns in XRD.
Another explanation:
Sometimes the electron comes very close to a nucleus in the target and is
deviated by the electromagnetic interaction. In this process, which is called
bremsstrahlung (braking radiation), the electron loses much energy and a photon
(X-ray) is emitted.
The high energy electron can also cause an electron close to the nucleus in a
metal atom to be knocked out from its place. This vacancy is filled by an
electron further from the nucleus. The difference in binding energy is emitted
as a photon. This photon is detected as an x -ray line in the energy spectrum.
Karen Louise De Sousa Pesse
35
4.1.5
Penetration depth
The electrons generate X-rays after collision with the anode
Since the wavelength of the produced X -rays depends on the material of the anode on which
the electrons are bombarded, a higher atomic number Z of this material causes the X -rays to
have a lower wavelength.
Z =
This means that the X-rays have a higher energy which influences not only the resolution but
also the penetration depth.
The penetration depth is higher when the energy of the X -rays is higher. The penetration
depth also depends on the absorption coefficient of the material. (I recommend to read
further on the third question 1.3.2.2).
Energy = Voltage; = Resolution; Penetration Depth
4.1.6 Absorption
During the measurement a diffractogram is obtained.
This is a graph of the intensity in function of two times theta {I(2)}. The
peaks on this graph correspond to constructive interference and the angle
at which they occur is a characteristic for the material examined. This
means that for the angle, the conditions of measurement do not have an
influence on the dominant peaks, because they are only dependent on
(and characteristic for) the crystal lattice type of the material . The
intensity does depend on the conditions of measurement though; more
precisely it depends on the source, scan speed and increment:
The scan speed is the speed at which the sample is scanned. When this speed is higher, the
intensity of the peaks will be lower.
The increment is the step at which two theta is changed. The lower its value, the higher the
resolution. The intensity of transmitted waves is never the same as the intensity of the
incident X-ray beam because a part of the intensity is always absorbed by the m aterial. This
absorption is described by the absorption coefficient of the material.
A high absorption coefficient is associated with a high atomic number.
I = I0 ∗ exp(−µ ∗ t)
This absorbed intensity is used for example the Thompson effect. Indeed the elec trons inside the material
elastically scatter the X-rays by emitting X-rays themselves in the electromagnetic field created by the
incident X-rays. These X-rays have the same frequency and therefore the same energy as the incident X rays. These created X-rays are then used for diffraction measurements.
Karen Louise De Sousa Pesse
36
4.1.7 Sample preparation
Easy For XRD the sample preparation is less intensive than for optical or other kinds of microscopy,
since not an image is required for the sample analysis but only diffraction is meas ured. This means that
there is no need for etching the sample. However the sample should be flat since diffraction occurs on
parallel planes at different diffraction angles so polishing is still needed.
Solid Samples from unprepared pieces of metal to cu t and polished metal samples. The
ideal is a perfect flat surface. Irregular sample surfaces change the distance from the sample
to the x-ray source and introduce error. All X -rays are calibrated based on a fixed sample -tosource distance, and changing thi s distance can vary your intensity.
Loose powder must be put into a plastic sample holder with a plastic support film; the
more finely ground and homogeneous, the better is the analysis.
For X-ray diffraction we must have a single crystal. The crystals should be transparent and appear to
contain no flaws when viewed under the microscope. Crystals that are cloudy, have cracks, appear to have
other crystals buried inside or intergrown crystals protruding from the side should be rejected.
Extra
Condition for planes (you better know this table):
H+K+L=2n BCC
H,K,L =>all or 2n or 2n+1 FCC
Karen Louise De Sousa Pesse
37
4.2 Question 9
Application of X-ray diffraction. Methods for XRD measurements (Laue, Debay-Sherrer).
Architecture of the X-ray diffractometer and focusing schemes. Determination of the type of the
crystal lattice, phase analysis, determination of the lattice parameter.
4.2.1 Application of X-Ray Diffraction
Qualitative, Quantitative and Texture
XRD is a very useful tool to identify crystalline phases and the orientation of a material. It is also a non destructive method which is a large benefit.
XRD can also be used to determ ine some structural properties of a material. The lattice parameter, residual
stresses, strain, grain size, phase composition and thermal expansion can be determined by using XRD. You
can also measure the thickness of thin films and multilayers with XRD. T he texture can also be determined
by using XRD.
Identify phases and their
composition
Orientation
Atomic Arrangement
(Structure of Crystals) Lattice parameter
Residual Stress
Grain Size
Thermal Expansion
Texture
Measure thickness
XRD can be used in two different ways:
It can be used to determine the wavelength of the incident X -rays. This is done by using a
known sample where you know the interplanar spacing d h k l . When you measure the
diffraction angle θ you can calculate the wavelength λ from the Brag g’s law.
A second and more frequently used way to use the XRD is to calculate the interplanar spacing
d h k l of a sample. Therefore, you use an X-ray source with known wavelength λ. The diffraction
angle θ is measured by XRD and then you can calculate the in terplanar spacing from the
Bragg’s law. When you know the interplanar spacing of the peaks you can calculate to which
phase each peak belongs. In that way you can determine which phases are present in the
material.
Karen Louise De Sousa Pesse
38
4.2.2 Methods for XRD measurements
There are two main methods for XRD measurements (Laue and Debye -Scherrer).
4.2.2.1 Laue Method
Determine orientation of large single crystal
White radiation is reflected from or transmitted through a fixed crystal;
Variation of wavelength
Each plane (that is suitable) diffracts, turning into a different dot (diffraction pattern)
The Laue method is for the determination of the orientation of large single crystals and the type of lattice.
Therefore white radiation is used. This implies that d h kl is fixed and λ changes. The white, polychromatic
radiation provides the range of wavelengths necessary to ensure that the Bragg’s Law is satisfied for all
planes. Each set of planes picks out and diffracts the particular wavelength from the white radiation. Each
curve therefore corresponds to different wavelength. The spots lying on any one curve are reflections from
planes belonging to one zone. Reflections from planes of the same zone all lie on the surface of an
imaginary cone whose axis is the zone axis.
The Laue method can be done in two different ways: back-reflection Laue or Transmission Laue (see
picture).
Zone axis
In the back-reflection method the film is placed between the source and the sample . The beams
which are diffracted in a backward direction are recorded. The film intersects the imaginary cone,
with the diffraction spots generally lying on a hyperbola
In Transmission Laue the film is placed behind the sample to record beams which are transmitted
through the crystal. The film intersects the cone with the diff raction spots generally lying on an
ellipse.
Crystal orientation is determined from the position of the spots. Each spot can be indexed using special
charts each spot is a particular plane. You can also use these methods to check if the crystal was bent or
tilted, because the spots will be distorted.
Karen Louise De Sousa Pesse
39
4.2.2.2 Debye-Scherrer Method
Incident beam of monochromatic X -ray interacts with
specimen.
This specimen must contain sufficient particles with the
correct orientation to allow diffraction from diffracting
planes when rotated in the x-ray beam;
The Debye-Scherrer method is also called the powder method.
The Debye-Scherrer camera is a flat cylinder. On the walls of the
cylinder a photographic film is placed. Through illumination of
the diffracted X-rays after developing segments of the
diffraction cones shows.
Determination of lattice parameter and type of lattice
Here monochromatic X-ray source interacts with fine grained polycrystalline (notice the rings instead of
dots) powders with random texture.
In a powder sample typically all possible orientations are
present, so that always some crystals will have lattice planes
that are oriented at the corresponding Bragg angle with respect
to the incident beam.
The angle between the diffracted radiation and the transmitted
radiation is always 2θ (2x the incident angle). The diffracted
beam intensity provides a measure of the distribution and
position of atoms within the crystal.
Comparison of Debye-Scherrer ring and
diffractogram
Karen Louise De Sousa Pesse
40
4.2.2.3 Bragg’s Method
Not asked on the question*
Determination of different phases in the material
Diffractogram {I(2)}
The mostly used method is the Bragg-Brantano set-up (see
picture). Here the source is fixed, the sample rotated with a
speed θ/∆t and the detector moves with a speed of 2θ/∆t. The
step size of turning determines the resol ution of the
measurement. The source is an X-ray tube with a filter (mostly
Kβ-filter) that makes the beam monochromatic. It also has
Soller-slits; these are some kind of apertures that focus the
beam by cutting it.
With this method you can determine the l attice parameters and
the type of lattice of the sample. The detector can be an
ionization detector or and Scintillation detector (see EDX). We
also have to watch out for fluorescence ; this problem can be
solved by placing a filter in front of the detector .
Karen Louise De Sousa Pesse
41
4.2.3 Determination of the type of the crystal lattice, phase analysis, determination of the lattice parameter
*This is often asked on the exam
One way to determine the type of crystal lattice is by analysis of a diffractogram. You can obtain
quantitative phase fraction and lattice parameter information. In the next figure you can see a
diffractogram of single crystal (one family of peak) and polycrystal (several peaks):
Now that we know the different methods, we try to determine the type of crystal lat tice. Therefore, we
need the Bragg’s Law and the definition of d h kl :
If some information is given (n=1, 2 (rad), =0.7E-10m); you are able to find the Diameter and relate to
the diffraction data tables:
28.6°
2
= 14.3° 𝑥 180° = 𝜋 𝑟𝑎𝑑 14.3° = 0.249 𝑟𝑎𝑑 𝑠𝑒𝑛 0.249 𝑟𝑎𝑑 =
0.2464 𝑟𝑎𝑑
𝑑 = 0.7 ∗ 10
−10
⁄2 ∗ 0.2464 = 1,42 Å
A table relating d and hkl can be given, or you will have to calculate it yourself.
This table will have information about which phase you are dealing with and
your Miller Indices.
Karen Louise De Sousa Pesse
42
And if you have to calculate it yours elf? We can play with these two formulas
When we substitute one formula in another and calculate sin²θ we get:
In this method the wavelength is kept constant and for the same type of lattice the interatomic distance a
is off course constant as well. Consequently, for each two peaks of the same type of lattice is valid:
Here k is the reference peak. These ratios tell us to which phase the different peaks belong because each
phase has different ratios that can be extracted from tables. Now that you know this, you can make the
indexation of the diffraction pattern. Therefore you use the second part of the equation above and the fact
that for BCC crystals the sum of h,k,l has to be even. For FCC structures h,k,l all have to be odd or even .
To get the right angle 2θ you have to pay attention to some details. The background has to be taken into
account. You have to be careful with overlap of different peaks. Next to these two also a defocusing error
and an error caused by absorption can be taken into account. Probably the best method to calculate the
right θ is by determining at which angle the gravity centre of the peak is situated.
The lattice parameter can also be determined. Therefore, we use of:
*Past exam question
Interpolation
We can calculate a for all different θ h kl .
To calculate the extrapolated a we plot
all the calculated a’s versus the
function f. The zero axis gives us the
extrapolated a.
The lattice parameter can cha nge with
introducing of stresses or by changing
the temperature.
Karen Louise De Sousa Pesse
43
4.3 Question 10
Application of X-ray diffraction. Quantitative phase analysis, internal stresses measurement.
4.3.1 Application of X-Ray Diffraction - Quantitative Phase Analysis QPA
X-Ray powder diffraction is one of the most powerful methods to perform QPA.
Each crystalline phase of the material gives a characteristic diffraction pattern independent
from other phases
The intensity of a peak is proportional to its fraction
XRD can measure quantitative phase analyses. Therefore, you have to know the density ρ, absorption
coefficient μ and the intensity of the phase I when it’s 100%. Then, by measuring the real intensity and the
use of some formulas you can calculate the volume fraction of the phase in the material.
It has to be noted that you first have to clean up the spectra before starting the calculations ( subtract
background, eliminate α 2 -peaks …)
The phase fraction of austenite in iro n can be calculated in this way:
K is a factor (instrumental factor constant) depending on the nature of the phase, selected
diffraction line and geometry of the diffractometer.
Source:
Rigaku
Karen Louise De Sousa Pesse
44
To calculate the volume fraction, one has to know the miller indices of your sample. Afterwards apply in
the formula:
For example, for retained austenite:
Karen Louise De Sousa Pesse
45
4.3.2 Internal Stresses Measurement (Residual Stress)
For this measurement, one will get a graph similar to this one:
In case of an unstressed sample, the graph would be a line with no change in y axis (n o variation in d); this
graph is a change in d caused by compressive test, which can be seen because the interplanar spacing d of
the plane (311) is varying according to the angle (sin 2 ψ) reduction in size.
To determine the residual stress on the practical class, the following formula was used:
E and are given. m is obtained from the curve plotted. If m is negative compressive.
Internal stresses are static multiaxial stresses and related strains, in an
isolated material that isn’t subjected to an external force. These stresses
cause a change in the i nterplanar spacing d h k l . In an XRD-measurement
this can be measured by the change in diffraction angle 2θ in comparison
to the stress-free state.
It has to be noted that this method is only valid for crystalline materials.
For a crystal that is free from stresses the Bragg’s law is valid. But
influenced by an interal stress σ , the interplanar spacing d h k l chances from
d 0 to d in a uniform way. This means that the peak in the XRD -spectra
shifts. (Learn how to draw this figure )
The influence of a non-uniform residual stress on microscale gives rise to
a peak broadening whereas equal areas are in compression as there are in
a tensile condition. Therefore there is no shift of the peak. This peak
broadening can be calculated by differentiation of the Bragg’s Law.
Uniform residual strain – peak shift
Non-uniform residual strain – peak broadening
Karen Louise De Sousa Pesse
46
4.3.2.1 Sin2ψ method
Goal – find Residual Stress σ Φ
Link the force applied on the lattice (and the change in d) to the stress that is left even when
you are not applying a force anymore;
It is clear that the change in d h k l and so also the strains can be measured b y XRD. To make the link between
these strains and the residual stresses we make use of the elasticity theory (Hooke’s Law: [σ ij ]=[E ij ][ε ij ]). In
the general case of internal, homogenous, residual stress a spherical volume unit is changed to an
ellipsoid. The axes of this ellipsoid are the main axi s of the σ’s and ε’s. Planes that are perpendicular to
these axes aren’t subjected to shear stresses (shear normal to the surface = 0) .
So when a material is anisotropic the mechanical properties are not depende nt on the direction, so at the
surface of the material the following equations are valid (Hooke’s Law):
To determine the residual stress, we have to determine the residual stress σ Φ in an arbitrary direction in
reference to the main axes. Theories have proven that therefore only two measurements have to be done;
we have to calculate ε 3 and ε ψ :
Karen Louise De Sousa Pesse
47
A first measurement of d h k l is taken when the (hkl)-planes are parallel to the surface.
Hereby we can calculate ε 3 .
In a second measurement, d h kl is measured when the normal of same (hkl) -planes and
the surface have angle of ψ between them (practically ψ is often 45°). In this way ε ψ can
be determined.
Strain along a Specific Direction
When you know these two strains, you can easily calculate the residual stress. This method is called the
sin² ψ-method.
Assuming that the stress normal to the surface is zero, the biaxial stresses σ x = σ y and taking into
consideration the strain along a specific direction,
Sources:
Lecture 9, ”X-ray diffraction” slides 47 to 51,
Flewitt:”Physical methods for materials characterization -Second Edition”, Chapter 4. Parts 4.3.8, (file
IP556_CH04.pdf)
B.C.De Cooman “Materiaalkundige observatietechniken” File: “Chapter 18.pdf” Chapters 3.6.1.”Meting van
interne spanningen”
Practical classes report
Karen Louise De Sousa Pesse
48
4.4 Question 11
*Hot Question
Texture measurements: What is texture and how do we represent individual crystallographic
orientations and textures. What is a pole-figure, inverse pole figure and an ODF?
4.4.1 Texture
Polycrystalline materials
Preferential Orientation (planes are not random)
Texture is a term that is related to polycrystalline materials. Polycrystalline materials are an aggregate of
single crystallites with all their individual orientation with respect to the sample reference system.
Textured materials are materials in which the individual crystallites have a preferential orientation with
respect to the sample reference system.
The opposite of a textured material is a textureless or random textured material.
By crystallographic orientation I actually mean the comparison of two different coordinate systems: the
crystal reference system K C and the sample reference system K S . When the sample is a rolled material the
sample reference system is often the rolling direction (RD), the transversal direction (TD) and the normal
direction (ND).
Other specimens, such as a tensile test piece, a rod or a wire have only uniaxial symmetry and hence it is
only necessary to specify one axis in the specimen coordinate system, the other two axes can be chosen
arbitrarily.
The crystal coordinate system (K c ) is specified by directions in the crystal. The choice of directions is in
principle arbitrary, although it is convenient to adapt it to the crystal symmetry. For example, for
orthogonal symmetry, [100],[010] and [001] al ready form an orthogonal frame and are often adopted as
the crystal coordinate system.
Karen Louise De Sousa Pesse
49
4.4.2 Representation of Texture and Individual Crystallographic Orientation
The representation of individual crystallographic orientations and textures can be done in diffe rent ways.
4.4.2.1 Rotational Matrix
The first option is by the rotation orientation matrix [g] (see figure). Each column of the matrix [g]
displays the direction cosines of a sample reference axis with respect to the crystal reference system K c .
Both rows and columns of the matrix are unit vectors, that is, the matrix is orthonormal and the inverse of
the matrix is equal to its transpose. Since a crystal orientation needs only three independent variables to
specify it, it is clear that the matrix, having nine n umbers, contains non-independent elements. In fact the
cross product of any two rows (or columns) gives the third and for any row or column the sum of the
squares of the three elements is equal to one.
4.4.2.2 Miller Indices
(hkl) crystallographic plane, paral lel to rolling plane
uvw crystallographic direction parallel to Rolling Direction
A second way of notating the orientation is by using Miller Indices (hkl)[uvw]. In this notation (hkl) is the
crystallographic plane which is parallel to the rolling plane. Direction [uvw] is the direction that is parallel
to the rolling direction. By using formulas it is possible to go from the rotation (orientation) matrix to the
Miller Indices and vice versa. A very famous orientation is the Goss orientation (110)[100]. It is known for
its very good electrical properties.
Karen Louise De Sousa Pesse
50
4.4.2.3 Euler Angles
A third way to represent the orientation is by using Euler Angles. The Euler angles refer to three rotations
which, when performed in the correct sequence, transform the sample coordinate system SCS (Xs Ys Zs)
onto the crystal coordinate system CCS (Xc Yc Zc). There are several different conventions for expressing
Euler angles but the most commonly used are those 3 steps formulated by Bunge:
i.
Rotate the SCS around the Z S axis with an angle 1 . The X S axis will then be in the X C Y C -plane of the
CCS.
ii.
Rotate the SCS around the X S axis with an angle Φ until Z S coincides with Z C .
iii.
Rotate the SCS around Z S again, but with an angle 2 until X S coincides with X C and Ys coincides with
YC.
These rotations can be written analytically in the form of a matrix.
By multiplying these three matrices you obtain t he rotation
orientation matrix.
These three angles can be plotted in the Euler Space.
Symmetry reduces Euler Space
An Euler space is a 3D representation of orientations expressed in
terms of its Euler angles. This red dot in the image is an
orientation expressed in terms of Euler angle , and its
coordinates are ( 1 ,Φ, 2 ).
Karen Louise De Sousa Pesse
51
Each point in the Euler space determines only one crystallographic orientation , and each 3 Euler angles
determine a position in the 3d space . The opposite is not true: the same crystallographic orientation can
be represented with different Euler an gles. The Euler space does not have linear borders. It‘s not a linear
space.
Properties of the Euler Space:
In the most general case the Euler angles are defined in the range 0°≤ 1/ 2 ≤2π and 0°≤Φ≤π which defines
the maximum size (volume = 8π²) of the Eul er space, the so-called asymmetric unit (the maximum size of
Euler space). However symmetries lead to a reduction in the size of the Euler space.
Crystal symmetry can reduce the Euler space. In general each n -fold symmetry axis reduces the Euler space
by a factor of n. For example, in a Cubic crystal there are 24 equivalent ways of attaching a right -handed
orthogonal reference system to a cube. This means that there are 24 equivalent points to represent one
single cubic orientation in Euler space. So the fundamental zone V’=V/24.
Therefore:
0° ≤ 1 ≤ 2π
0° ≤ Φ ≤ π/2
0° ≤ 2 ≤ π/2
Here the three-fold axes aren’t included because they would lead to a too complex subspace. So for cubic
crystals each orientation appears three times in the reduced Euler space.
Next to crystal symmetry also sample symmetry can reduce the Euler space. In samples deformed by rolling
or more generally under a plane -strain deformation state, it is usually assumed that there is a two -fold
symmetry axis parallel to each of the three sample axes (orthorhom bic sample symmetry). Therefore the
Euler space is again reduced by a factor of 4.
Therefore:
0° ≤ 1 ≤ π/2
0° ≤ Φ ≤ π/2
0° ≤ 2 ≤ π/2
Step by step:
Different sample symmetries affect the range of the angle 1 reduce Euler Space
When there is no symmetry, 0° ≤ 1 ≤ 360°.
If one symmetry element is present, 0° ≤ 1 ≤ 180°.
If a two-folded symmetry axis is present, then we have 0° ≤ 1 ≤ 90°.
Crystal symmetry further reduces the size of the Euler Space by affecting the range of Φ and 2 .
N-fold symmetry axis reduces Euler space by a factor of n
Karen Louise De Sousa Pesse
52
4.4.3 Orientation Distribution Function ODF
A representation of the Euler space can be done with ODF (Orientation Distribution Function). ODF give a
complete image of all the texture components that are present.
ODF f(g) is a probability density function describing the probability of finding a grain with an orientation g
within a given distance in orientation space (∆g) of a specified orientation g 0 or alternatively the volume
fraction of material oriented within ∆g of g 0 .
The units of an ODF are time the random intensity.
A texture can be described by the Orientation
Distribution Function (ODF);
Orientation g of each crystal in the sample is quantified
The orientation of this crystal is determined by the
sample reference system
ODF = volume fraction of crystals with orientation g+dg
Random texture Constant f(g) = 1/8π 2 .
Texture function has a determined maximum
When you measure the texture grain by grain, and plot each orientation in the Euler space
Obtain ODF.
Karen Louise De Sousa Pesse
53
4.4.4 Pole Figure
Orientation can also be plotted as two dimensional projections in pole figures. Such figures can be useful
for simplifying the analysis of the orientation distribution. A pole figure shows the position of a pole (a
normal to a lattice plane) relative to the sample reference frame (RD, TD, ND). So it’s a stereographic
projection that gives the distribution of one crystallographic direction in the material.
For rolling materials ND is typically chosen to be the North Pole. The RD-TD plane is the projection plane.
To project a pole onto the pole figure you have to link the pole with the South Pole. The intersection of
this line with the projection plane gives you the projection of the pole onto the pole figure.
So far, only individual poles have been considered. However one pole does not give the entire orientation
information, since the crystal can still rotate about this particular pole. The orientation is fully
characterized by three poles. Sometimes two poles are enough depending on the symmetry of the crystal
and/or the poles.
So the pole figure is a semi-quantitative method. It shows the d istribution of <hkl> crystallographic poles
with respect to the sample reference system. In a pole figure the sample reference system and the crystal
pole <hkl> must be represented. A pole figure displays the sample symmetry (orthorhombic horizontal and
vertical symmetric/ monoclinic only vertically). To conclude you have to know that one pole figure cannot
represent the complete texture. You need in general three pole figures.
4.4.5 Inverse Pole Figure
Instead of a pole figure you can also use the Inverse Pole Figure to
display the orientation. In the inverse pole figure the orientation of the
sample coordinate system is projected into the crystal coordinate
system. So the reference system of the inverse pole figure is the crystal
coordinate system and the ‘orientation’ is defined by the axes of the
sample coordinate system (RD, TD, ND). According to the crystal
symmetry it is not necessary to show the entire pole figure, but just
one unit triangle. In an inverse pole figure the crystal reference system
and the sample direction must be represented. Just like a normal pole
figure an inverse pole figure displays the crystal symmetry and doesn’t
represent the complete texture.
Pole Figure Sample Reference System
Inverse Pole Figure Crystal Reference System
Karen Louise De Sousa Pesse
54
This is a Pole Figure. It’s colours indicate a direction that can be seen in the triangle: Blue is 111, Red is
001 and green is 101. RD stands for Rolling Direction, ND is Normal Direction and TD is Transverse
direction.
Also very important:
4.4.6 Kernel Average Misorientation (KAM)
For KAM calculation, the misorientation between the
centre point of the kernel and all surrounding points
in the kernel are calculated and averaged, which gives
the local misorientation value of the centre pixel.
Substructures, having misorientation lower
than 5°, are identified by kernel average
misorientation (KAM).
This map has a value for each pixel equal to the
average disorientation that pixel has with its
neighbours.
While the Grain Average IQ-method is an average method that compares image qualities of
different grains, the KAM-method is more local and compares orientation gradients between
different points. This implies that the area selected based on the Grain Avera ge IQ-method
will always correspond to the junction of several grains, whereas the area selected based on
the KAM-method not.
Karen Louise De Sousa Pesse
55
Observe the relationship between colours.
Knowing this graph is very important.
Karen Louise De Sousa Pesse
56
4.5 Question 12
Practical aspects of texture measurements by XRD (geometry of the measurement scheme).
Sample preparation. Examples of rolling, textures, recrystallization textures and transformation
textures in FCC and BCC crystal structures.
4.5.1 Practical aspects of texture measurements by XRD - Geometry of the Measurement Scheme
Bragg’s Law is the law that governs XRD = 𝑠𝑖𝑛 = 𝑛/2𝑑
To discuss the practical aspects of a texture measurement we start with discussing
the geometry of the arrangement. A texture measurement is most of the time done
with a special diffractometer, equipped with a texture Goniometer (Euler-Cradle).
Texture Goniometer X-Ray source and X-Ray counter positioned
according to the Bragg angle 2
Eulerian Cradle Important piece of the Goniometer, that combined
create multi-circle (four-angle) diffractometers.
With this equipment, one measure the texture using x-ray diffraction through a method
called “Schulz Reflection Method”
Check:https://www.doitpoms.ac.uk/tlplib/crystallographic_texture/t
exture_measaurement.php
The method that is most commonly used is called the Schulz
reflection method. In this method we have a sample holder that can
make 4 different movements:
1) A translation (omega) to average the orientations by
measuring more grain orientat ions usually constant at 0°;
2) A rotation (phi) about the normal of the sample surface
3) A rotation (chi) χ about the orthogonal axis
4) The classical θ-2θ rotation
Karen Louise De Sousa Pesse
57
During a measurement there is a continuous rotation of the -angle and a step wise change of the χ -angle
(for example 5°). Meanwhile the intensity of an (hkl) -plane reflection is measured for a fix ed Bragg angle.
The χ-angle normally only goes from 0 to 70° so we get an incomplete pole figure. Therefore you need to
take three or four incomplete pole figures to get the total information of the total texture, orientation.
Typically is kept at 0°, because if you change it, there will be no more symmetry between the X -Ray
source and X-Ray counter (which are perfectly positioned to satisfy the Bragg-Brentano arrangement, at
the Bragg angle ). When you do change , is to measure the quality of the crys tal, and to derive exact
peak shapes.
Azimuth
4.5.2 Sample Preparation
It is clear that the surface of the sample that you want to investigate has to be as flat as possible. So you
always have to be careful during the sample preparation that you don’t make any deep sc ratches on the
surface during grinding. You also have to use a special kind of sample holder. Otherwise you would
measure the mounting material and not the sample itself. Next to this there is no special sample
preparation necessary for an XRD-measurement.
A sample with a flat surface is mounted on the sample holder with its normal direction parallel to the axis
of the rotation.
Detector
Sample
Source
Karen Louise De Sousa Pesse
58
4.5.3 Examples
This sample is rotated in its plane around the axis. This angle corresponds to the azimuth of a pole in
a pole figure.
After a full rotation, the sample is tilted on the axis. and (pole figure radial angle) are defined in the
opposite direction = 90° - .
The limiting value of in the Bragg-Brentano focusing condition is when both incident and ref lected beam
are parallel to the sample (90°), but usually 60 -85° are used. The limiting factor is mostly because the
intensity of a peak decreases with increasing .
By rotating the sample, the projection of the beam onto the samples surface becomes incre asingly
elongated. Furthermore, different incident angles θ add an additional distortion of the projected beam.
These things should also be kept in mind when doing a texture measurement. On the figures you can see
the different angles and .
In general, absorption of the X-rays increases with the atomic number of the analysed sample material and
with the wavelength of the X -rays. Shorter wavelengths are less absorbed than large ones. However, there
are some combinations of sample and target materials whic h yield anomalously high absorption.
Absorption is undesirable in texture measurements so these combinations should be avoided.
For texture measurements the incoming X -rays have to be monochromatic. An X-ray source most of the
time sends out continuous wavelengths with a sharp Kα -peak. Next to this peak it may also have a sharp
Kβ-peak. This peak has to be taken out when we want monochromatic X -rays. Therefore the source has to
be equipped with a Kβ-filter to reduce the intensity of this Kβ -peak.
Continuous contributions to the X-ray spectrum remain at wavelengths
larger than Kα and at very short wavelengths. Although these intensities
are relatively small compared to the Kα -intensity, they may become
significant in the case of very sharp textures.
In this case we make use of truly monochromatic radiation. This can be
obtained by using single crystal monochromators. The monochromator is
best inserted in the primary beam between the X -ray tube and the
sample.
Karen Louise De Sousa Pesse
59
4.5.4 Examples of rolling, textures, recrystallization textures and transformation textures in FCC and BCC crystal
structures
Orientation Distribution Function ODF calculated from XRD measurement.
Transformation from FCC to BCC structure
ODF is a statistical way to plot an image Measurement of preferred orientations grain-by-grain gives
discrete points in the Euler Space that can be plotted.
Some transformations and recrystallization processes can be simulat ed, and then compared to the ODF
experiment to ensure its quality (b y using XRD you can check if these simulations are good or not).
For example the transformation from FCC to a BCC structure can be seen as a Kurdjumow-Sachs (K-S
mechanism) (<112>90°) or Bain (<001>45°) transformation. By simulation you can see what the Orientation
Distribution Function ODF should be. You can compare this image then with the actual image that you
become.
Rolling The KS mechanism consists of distortion of matrix by a first shear and a second shear . From the
lattice distortion matrix, one can obtain invariant normal pla nes.
Bain mechanism uniaxial tension in z direction and uniform expansion in x and y axis BCC material
subjected to high strain, the lattice may be elastically distorted towards an FCC lattice, by activation of slip
systems.
Transforming Austenite into Ferrite
For the bcc-fcc phase, these are the only plans that are more or less identical in each crystal.
So you can see that the K-S transformation texture is a good approximation for describing the FCC to BCC
structure. You can also get difference s in deformed and recrystallized st ructures as seen in the picture.
Karen Louise De Sousa Pesse
60
The deformation texture of low carbon steel is based on the Taylor Theory. This theory is based on two
assumptions:
1) Macroscopic strain = microscopic strain
2) Dissipated plastic power is minimized
So a displacement is accommodated by a combination of 5 slip systems (out of 24).
This theory can also be checked by using simulations and XRD and as seen in the picture below you can se e
that this theory is quite accurate .
Also the effect of recrystallization on the texture as the change in R -values (for
deep drawing – those cups that we did in OCAS) can be determined by using XRD.
Significant effect on the deformation as a consequence of the - phase transformation
deep drawing properties of TRIP steels;
KS relationship is used to convert crystallization textures to textures
Karen Louise De Sousa Pesse
61
5 Scanning Electron microscopy (SEM)
5.1 Question 13
Scanning Electron Microscopy (SEM). Architecture of SEM. Types of filaments-advantages and
disadvantages. Interaction of the primary beam with Material-Efficiency of SE and BSE.
5.1.1 Architecture of SEM
Scanning electron microscopes are composed of several parts. Based on the vacuum, 2
parts can be distinguished: an ultra-high vacuum chamber on top (starting pump,
diffusion pump and possibly 2 ion pumps (IP)) and a normal vacuum chamber below
(starting pump and diffusion pump).
The ultra-high vacuum is to protect the electron source and to have as little interaction
with air as possible.
In this vacuum room, an electron gun and the Wehnelt cap are
located. The Wehnelt cap is used to centre and locate the emitted electrons as good as
possible (focusing and control of the electron beam), to group the electrons and to form a parallel beam
(minimize cross-over).
The normal vacuum room contains: lenses, an x -ray detector, a BSE and a SE -detector, a stage, coils,
apertures, valves...
Liquid Nitrogen cooling
Electron Gun
Vacuum Column
Condenser Lenses
IP – Lamps
SE – Secondary Electron Detector
Karen Louise De Sousa Pesse
62
5.1.2 Types of filaments - Advantages and Disadvantages
Different types of electron sources are currently available: W (Wolfram
or Tungsten), LaB 6 (Lanthanum Hexaboride) and FEG (Field Emission
Gun). Both W and LaB 6 are thermo-ionic emitters; FEG emits electrons
by field emission (room temperature).
W is a small wire and from the top of the curve, the electrons are emitted. This is a cheap source, but is
not very efficient. The brightness is the lowest, and it has the highest cross-over diameter (decrease
resolution) and the highest energy width ∆E. An advantage is the relatively low vacuum (10 −3 Pa).
The
optimal temperature is 2800 K. Higher temperatures don’t give higher electron densities so there is no use
in raising the temperature since raising the temperature lo wers the lifetime (≈ 100 hr) of the wire . Without
the benefit of using a corrector, the energy width degrades resolution because of chromatic aberration .
Cheap (100-600$)
Not very efficient
Low Vacuum
Needs very high temperature
Low Lifetime (evaporates)
LaB 6 is a small cone-shaped single crystal (110)-plane of LaB 6 . The electrons are captured by a Wehnelt cup. Optimal working temperature is 1400 -2000K. The properties of this kind of source are better than the
properties of the tungsten wire, but worse than the FEG. It is used for about 500 hr (long lifetime). To
create the vacuum of 10 −4 an extra IP is added.
Better lifetime than Tungsten W (lower
evaporation)
Chemically reactive when hot Poison
cathode
Expensive (1300 – 3000$)
Needs a high vacuum
Really half-way W and FEG
The FEG (Field Emission Gun) is super expensive
(second hand is 7k$). No high temperatures are
needed (RT) since anodes are strong enough to suck
the electrons out of the tip. A first anode is used to
suck the electrons out; a second anode is used as a
Wehnelt-cap. The properties of the FEG are very
good. High current stability (< 5%), excellent brightness, lower cross-over diameter, lower energy width,
large lifetime (>1 year). The biggest difficulty is the ultra-high vacuum (< 10 −8 Pa). To establish this kind of
vacuum, at least 4 pumps are used.
Not as stable as the others
Better brightness
Needs a super high vacuum
Long lifetime
Not good for large specimen (empty magnification)
Small crossover d better resolution
Very fine current density
Works in room temperature
Karen Louise De Sousa Pesse
63
Because the emitted electrons in the various types of guns are heated, their energy distribution is not a
sharp peak. Instead, they have a Boltzmann distribution that can vary widely depending on the type of
filament. For a good microscope, you want ∆E/E to be as small as possible, that is, the energy distribution
to be a small fraction of the average energy of the electrons.
5.1.3 Interaction of the primary beam with Material - Efficiency of SE and BSE
Interaction with material
The electron beam suffers direction change, energy loss and absorption
The material is excited, ionised, create lattice defects, there is heat production and
secondary radiation (electrons and x -rays);
There are two types of interaction: elastic an d inelastic (scattering).
o
Elastic: electron-nucleus interaction, low energy release, diffraction
o
Inelastic: Interaction electron-electron, large energy variation;
SE Secondary Electrons
Emitted by the surface of the material after impact
Inelastic interaction Collision between two electrons and variation in energy E;
Low energy (less than 50 eV), low penetration depth
BSE Backscattered electrons;
High energy electrons originating in the electron beam.
They are reflected (or back scattered) by elastic scattering interactions with specimen atoms.
Small energy loose has high Energy deeper
penetration depth
Interaction between electron and nucleus
#SE
The efficiency of SE and BSE: 𝛿 = #PE 𝜂 =
#BSE
#PE
#PE = Ratio of Primary Electron emitted
η is Z dependent the heavier the element
the brighter the signal in the microscope ;
𝛿 shows no dependence with Z low
penetration depth
SE efficiency is higher when the sample is
tilted in the direction of the detector
BSE is not really influenced by the angle
Karen Louise De Sousa Pesse
64
5.2 Question 14
EDX and WDX analysis in SEM Characteristic X-rays. Detectors-principle. Comparison between
EDX and WDX spectroscopy.
5.2.1 EDX and WDX analysis in SEM characteristic X-Rays
EDX or EDS Energy Dispersive X-Ray Analysis
Goal: Identify the elemental composition of a specimen. The EDX analysis system works as an integrated
feature of a Scanning Electron Microscope and cannot operate its own without it.
During EDX Analysis, the specimen is bombarded with an electron beam inside the scanning electron
microscope.
The bombarding electrons collide with the specimen atoms’ own electrons, knocking some of them
off in the process;
A position vacated by an ejected inner shell electron is eventually occupied by a higher-energy
electron from an outer shell. The outer electron will give up some energy and emit an x -ray.
This energy depends on which shell it is transferring from, and which
shell is going to.
The atom of every element re leases X-rays with unique amounts of
energy this is the identity of the atom from which the x -ray was
emitted.
The output of an EDX analysis is an EDX spectrum Peaks corresponding
to energy levels, each peak being unique to an atom (corresponding to a
single element); the higher the peak in a spectrum, the more
concentrated the element is in the specimen;
The EDX spectrum not only identifies the element corresponding to
each peak. The type of x-ray can also be identified. An x -ray from the L
to the K shell is a K peak, and from MP to K shell is a K peak, and so
on.
WDX Analysis or WDS Wavelength Dispersive X-Ray Analysis
The detector counts the X-rays in terms of characteristic wavelengths , and not energy.
It is very similar to EDX, but more expensive, time consuming (slower) and causes higher sample
damage/chamber contamination because of the high beam currents required;
So why would someone use it? The analysis is better. Better energy resolution prevents peak
overlap errors (picture) that are frequent in EDX and has lower background noise (more accurate
quantitative analysis).
Karen Louise De Sousa Pesse
65
5.2.1.1 Quantification via X-ray - ZAF correction
(It is not highly necessary to be mentioned)
Different atoms give different responses to x-rays:
• Z Atomic Number Effect:
Backscattered coefficient increases with atomic number Z premature loss of beam electrons
before ionization resulting in x-ray production
Stopping power the rate of energy loss due to inelastic interaction increases with decreasing
atomic number (leading to the same result; these two factors tend to cancel one another)
Z Energy Loss (inelastic) lower Z = lower backscattered coefficient and high energy loss;
The higher the Z, the better the Intensity.
Two factors must be considered regardi ng atomic number: the backscatter coefficient and “stopping
power”. The backscatter coefficient increases with atomic number —leading to the premature loss of beam
electrons prior to ionization resulting in X -ray production. The rate of energy loss due to i nelastic
interaction increases with decreasing atomic number —leading to the same result. These two factors tend
to cancel one another.
• A X-Ray absorption Effect
Intensity lowers due to absorption of x-rays in the sample. Absorption is usually the biggest factor that
must be considered in the measurement of composition by x -ray microanalysis. As an X-ray travels through
the sample, it may be absorbed, giving up its energy entirely to an electron and ejecting the electron from
its orbital. The probabilit y that an X-ray will be absorbed depends on its energy and the energy with which
the electron is bound to its nucleus. The probability of absorption increases as the X -ray energy
approaches this binding energy from above and reaches a maximum when the X -ray energy is just greater
than the binding energy. At this point there is a discontinuity (absorption edge) in the probability curve.
Lower energy X-rays no longer have sufficient energy to overcome the binding energy and the probability
of absorption drops to a lower value. The probability of absorption then increases again as the X -ray
energy approaches the binding energy of a more loosely bound electron. An absorption curve [1] for a
given element includes an absorption edge for each electron shell.
Absorption edges can be directly observed when the X -ray spectrum energy range spans the critical
excitation energy for the absorber element in the sample [1]. At the absorption edge, the continuum
background abruptly decreases for X -ray energies slightly above the edge because the mass absorption
coefficient increases abruptly at the absorption edge.
As X-rays are generated deeper in the specimen, progressively greater fractions are lost to absorption. The
ratio of the measured X-ray intensity to the generate d X-ray intensity at some position in the sample is
dependent on the: mass absorption coefficient; specimen density; and path length. The probability of X -ray
absorption as a function of path length through the sample is given by Beer’s Law:
I/Io = exp (-μ M ρd) where:
I/Io = fraction of X-rays transmitted;
d = thickness;
ρ = material density;
μ m = mass absorption coefficient ( available in published tables).
Karen Louise De Sousa Pesse
66
F: X-Ray Fluorescence Effect:
When an X-ray is absorbed by a sample atom, the absorbing atom is left in an excited state. It subsequently
relaxes, emitting its own characteristic X-rays (secondary fluorescence). Since an X -ray can be absorbed
only in an interaction with an electron having a binding energy less than the energy of the absorbed X -ray,
the energy of the secondary fluorescence is necessa rily less than the energy of the primary X -ray. For
example, in a Cu-Fe sample, Cu Kα radiation (8.04 keV) is of sufficient energy to excite Fe K radiation (K a b =
7.11 keV). As a consequence, the measured iron intensity would be enhanced due to fluorescenc e by
copper, while the copper intensity would be suppressed due to strong absorption by iron. In practice,
secondary fluorescence is only significant if the characteristic energy is within approximately 3 keV of the
critical ionization energy. The fluoresc ence effect can be calculated with sufficient accuracy and it is
usually the least important of the three factors.
5.2.2 Detectors Principle
WDS Principle
A beam of electrons is aimed at the sample. X -rays escape. On a certain
distance on the imaginary Rowland circle or focusing circle, a well-known
analytical crystal is placed. The crystal has specific lattice spacing d, wellknown. When x-rays encounter the analytical crystal at a specific angle θ,
only those x-rays that satisfy Bragg’s law are reflected an d a single
wavelength is passed onto the detector . When the measurement is done,
the intensities (*) are compared with those of standards containing known values of the elements of
interest. Therefore, the fractions can be calculated.
The sample is often fixed, so the crystal and the detector have to move around the Rowland circle.
(*) Actually, not the intensities, but the x -rays are counted. This is done with a proportional counter. In a tube of
90% Ar and 10% CH 4 , x-rays are attracted to the cathode a nd can create a voltage. By measuring the voltage, the
fraction of a certain element can be measured.
All the X-Rays originating from the point
source on the sample are diffracted over a
great percentage of the crystal surface and
are brought to focus at the same point on the
detector, thus maximising the collection
efficiency of the spectrometer.
Several different diffracting crystals with
different crystal lattice spacing are normally
used for WDS, in order to cover all of the
wavelengths (energies) of interest, as well as
to optimize performance in the different
wavelength ranges.
Further reading: https://www.oxford instruments.com/OxfordInstruments/media/nanoanalysis/broc
hures%20and%20thumbs/OI_AppNote_WDS_Explained.pdf
To maintain the correct geometrical relationship between specimen, crystal and detector for the full range
of diffracted angles, it is necessary to maintain all three on the Rowland circle. This is accomplished by a
mechanical goniometer which moves the crystal and detector so that correct diffraction conditions are
maintained.
Karen Louise De Sousa Pesse
67
Detectors used in WDS are usually of the gas
proportional counter type.
X-ray photons are diffracted into the detector
through a collimator (receiving slit) , entering the
counter through a thin window.
The photons are then absorbed by atoms of the
counter gas.
A photoelectron is ejected from each atom
absorbing an x-ray. The photoelectrons are
accelerated to the central wire causing further
ionization events in the gas, so that an
“avalanche” of electrons drawn to the wire
produces an electrical pulse the inert gas is
ionized by the radiation.
The detector potential is se t so that the amplitude
of this pulse is proportional to the energy of the X -ray photon that started the process. Electronic pulse
height analysis is subsequently performed on the pulses to filter out noise.
EDS
EDS detector is made of an electron trap, a
window, the crystal and a cryostat.
• Electron trap: To make sure the electrons
don’t damage the window, electrons are
’trapped’ by a magnetic field and reflected by
a very thin (20 nm) Al-film;
• Window: The window is made of Be or
polymer. A window is necessary to overcome
the pressure gradient between the vacuum
EDS and atmospheric pressure. If the window
would not exist, air molecules would
condensate on the detector and disturb the
measurements. Be is quite strong, but it
absorbs low energy x-rays. With a Be window,
elements below Na can’t be detected. A
polymer film is a lot thinner and therefor
better. It does not absorb x-rays, but a
supporting grid has to be attached. Mostly
polymer windows are used nowadays.
• The cryostat maintains a low temperature
and is not in contact with the detector, but the side of the detector is in contact with the cryostat. The
detector is cooled indirectly.
The detector is composed of Silicon (silicium). When an x-ray enters, a hole is created. This hole can be
detected. If the energy of the x-ray is too low, it is absorbed by Be, if it is too high, it passes through the
silicium. From Na till U are measura ble.
Karen Louise De Sousa Pesse
68
5.2.3 Comparison between EDX and WDX spectroscopy
EDS is more commonly employed data collection and analysis with EDS is a relatively quick and simple
process because the complete spectrum of energies is acquired simultaneously. Using WDX, the spectrum
is acquired sequentially (as the full wavelength is scanned).
Although it takes longer to acquire the full spe ctrum, WD technique has better resolution compared to EDS
the typical resolution of ED is 70 to 130 eV, and WD resolution is 2 to 20 eV (source Oxford
Instruments).
The ability to combine better resolution and higher count rates allow WDS to detect elements at an order
of magnitude lower concentration than EDS . EDS, is not able to detect pieces less than 10 wt%, elements
have to be heavier than beryllium. Even when measuring a precipitate, a part of the bulk will be measured
since the spatial resolution is approximately the penetration depth. Accu racy is between 99 and 95%.
WDS is better in resolution, but cost more (often 4 detectors with different crystals needed) and is more
time consuming. Since focusing is needed, mistakes are more easily made. Limit of detection is around 1
wt%.
While WDS technique has always been appreciated for its higher resolution and trace element capability, it
has been traditionally viewed as more complex to set up, and WDS data is more tedious to obtain and
interpret than EDS (says Oxford Instruments).
http://eesemi.com/edxwdx.htm
Karen Louise De Sousa Pesse
69
6 Electron Microscopy TEM
6.1 Question 15
Discuss the sample preparation techniques for TEM. Give schematic descriptions of different
methods.
6.1.1 The sample preparation techniques for TEM
In transmission electron microscopy (TEM), a high -energy electron beam (~ 200 keV) interacts with an
electron transparent (~ 100-150 nm thick) specimen in order to study the microstructure and
composition. Preparation of such a thickness is both an art and a science. It needs the devising of suitable
methods as well as realising/demonstrating them in a defined process with reproducibility. Also, utmost
care is necessary in preparing and handling the specimens, as they are extremely thin and hence prone to
bending and breaking.
Important things to notice when preparing the sample:
Electron transparent for a 100 keV, a thickness of less than 100 nm is required (depending on the
material)
Representative the sample has to contain what you want to
analyse
Stable under electron beam could be a problem in organic matter,
but no problem in metals.
Polishing is mostly done by chemical polishing. But safety is needed.
Polishing is often done with HCN, HF, HNO 3 and HClO 4 . All of those
liquids are dangerous, even explosive. The sample is placed on a supporting grid (lines, hexagonal, raster,
split,...) The supporting grid is placed in a sample holder (single entry
holder) that is actually a stick with a O -ring as seal and a platform and
a jewel bearing. The jewel bearing is to link the holder to the column;
the stick is to insert the specimen and to be able to rotate it in the
column. The O-seal is to make sure the vacuum is maintained. The
platform is very different everywhere. Rotation, heating, cooli ng,
double tilt, multiple specimens, bulk... If a special thing is needed, it
will be made. A top entry holder is also possible, but very rare. The
advantage is that tilting can be done in every direction, but the tilting
angle is limited. Karen Louise De Sousa Pesse
70
6.1.2 Give schematic descriptions of different methods
The method to prepare the specimens for TEM
depends on what information is required. In order
to observe TEM images with high resolution, it is
necessary to prepare thin films without
introducing contamination or defects. For this
purpose, it is important to select an appropriate
specimen preparation method for each material,
and to find an optimum condition for each
method.
The most common way to prepare a
sample possible for the TEM are:
replicas and foils (Twin-Jet
electropolishing). Replicas can be positive (the true surface ) or negative (the inverse surface). Mostly they are made by C,
Cr or Pt. Negatives are made by evaporating C, Cr, or Pt on the sample and dissolving the metal sample
is lost. A positive is made by adding a layer of plastic on top of the sample, removing the sample,
evaporating C, Cr or Pt on the plastic and dissolving the plastic. These techniques are working fine,
because the evaporation is stopped when the layer looks flat. This means that there are places only a few
atoms thick, while others are the thickness of the bumps, which is also small enough since the bumps are
not very well visible under the SEM.
A special kind of replica is the extractive replica
to see the inclusions. By deep selective etching,
the inclusions are almost loose. By evaporating
a thin film of C, Cr of Pt on the etched
specimen, the inclusions can be removed. This is in order to view the inclusions without background
and knowing that no inclusion is hidden behind another. The particles can also be tested for orientation
and composition, the shape can be seen and th e texture can be measured without interference from
the bulk material. In the replicas, diffraction is useless. The information about the orientation is lost. Only the shape and
some thicknesses can still be seen.
Other methods involve crushing the sample with an agate mortar and pestle
(the flakes obtained are suspended in an organic solvent like acetone and
dispersed with sonic bath), electropolishing, chemical polishing,
ultramicrotomy for sectioning, ion milling (Argon ions are used for
sputtering).
Focused Ion Beam A thin slice of the sample is cut by an ion beam o n a
scanning ion microscope. The main advantage of this method is that it allows
selective thinning at a desired location by cutting trenches in the sample.
6.1.2.1 Single Jet Method
Polishing of a material of one side at a time. The sample is an anode , and can
be polished by electrochemical action, by keeping it in an electrolyte and
applying potential. The electrolyte, its temperature and bias voltage are
important parameters in controlling the rate of dissolution of the sample.
Karen Louise De Sousa Pesse
71
6.1.2.2 Double Jet Method
The double-jet technique enables the polishing of metal disks simultaneously from both sides and
automatically stops the polishing operation when perforation occurs.
A twin jet electro-polisher can be used for diameter. This equipment has a sample holder which can
accommodate sample discs with 3 mm diameter. The sample disc acts as the anode. It is positioned
between two nozzles, which are also acting as cathodes. Both the sample disc and the nozzles are
submerged in a suitable electrolyte. A small submersible pump, wh ich is a part of the equipment, pumps
jets of the electrolyte through these nozzles on both sides
of the sample. The thinning rate in the central portions of
the disc is higher than at the edges that are relatively
unaffected. Therefore, the perforation oc curs preferentially
near the central region with a rim at the edges of the disc.
The perforation can be identified using a light source and a
light sensor that are placed on opposite sides of the sample.
The edges near the perforation will have a wedge -shape
with those next to the perforation having the desired
thickness for electron transparency.
Obtain a sample between 100 and 200 μm thick. Use
a holder to polish the sample
Thinning Cut 3 mm diameter discs of the foil
Electropolishing Pre-thin the central region (from 1 or 2 sides). The double-jet method is often
used;
Often, the double-jet is enough. Ion thinning can also be done. This method is very clean, precise and a
larger zone of flat material is obtained. Disadvantage is the production time: 6 -10h. A sample can also
be made with FIB (Focused ion beam, with Ga + - ions). Ions cut pieces of the sample till the right shape,
thickness, and place are reached. With the electron beam you can investigate the sample. By using the
FIB a very thin plate can be made. When this is welded to an ominprop (= needle) a small plate can be
produced. Again, the cutting with ions takes a lot of time (at least 5h and probably more).
Notification: When using TEM for dislocation density, make sure not to bend the specimen. This will
change the dislocation density. Source: Necip Ünlü - Preparation of high quality Al T EM specimens via a double -jet electropolishing
technique.
D. V. Sridhara Rao 1 , K. Muraleedharan 1 and C. J. Humphreys . TEM specimen preparation techniques .
Resume:
Prepare slices (cut with a blade)
Prepare TEM discs (disc punch or ultrasonic disc cutter, la pping/polishing)
Twin-jet electropolishing prepare electrolite, adjust temperature, specimen holder
Karen Louise De Sousa Pesse
72
6.2 Question 16
Discuss the image formation and contrast formation in a TEM. What determines the resolution in
a TEM and why? Explain the bright and dark field image formation in TEM and the way you can
obtain an image.
6.2.1 Image Formation and Contrast Formation in a TEM
Interpreting transmission images is tricky image has no depth sensitivity. You will not know what
is surface and what is in the bulk.
The instrument often has 5 lens illumination system and a trend increasing the number of lenses to
optimise the overall performance;
After leaving the source, electrons are formed into a cross -over and this demagnified source image
is projected on to the spec imen by two condenser lens.
The first condenser lens forms a demagnified image of about 1 m diameter that is projected on to
the specimen by a second condenser lens with magnification of about two. The final illumination
spot on the specimen is typically a s small as 2m, which is sufficient to fill the viewing screen at the
highest magnifications.
6.2.2 Resolution in TEM
Almost all TEM’s are build the same way. The only difference is the energy of the electrons, ranging from
100 keV till 1,25 MeV. Mostly energie s of 200-300 keV are used. 1,25 Mev is to powerful and damages the
sample. The electron sources are the same as in the SEM (W, LaB 6 or FEG). Again the wehnelt cup is used
to collect the electrons. The wehnelt cup is again a negatively loaded cup. The curre nt to load the cup
cannot be too high or the electrons aren’t focused but repelled. Therefore, a bias resistor is implanted to
change the current. A positivlely charged anode is behind the wehnelt cup to attract and accelerate the
electrons.
Then, electromagnetic coils are placed to aim the beam, 2 condenser lenses create the focus, 2
fixed apertures and 1 modifiable, an alignment coil at the beginning makes sure the electrons stay in the
tube after the anode and a stigmator to correct stigma tism are in front of the specimen holder. After the
specimen holder, 2 apertures are pla ced (1 is only used in diffrac tion mode), many lenses and a stigmator.
At the end is a fluorescent screen or CCD camera. All is done in high vaccuum.
The resolution of a TEM image is determined according to the Reighleig criterion.
Since λ is proportional to 1/E 1/2 , higher energetic electrons (lower λ) give better resolution. Although, the
resolution is also restricted due to aberr ation: spherical and chromatic (̸ = between red, green, blue). why
aberration?
When looking at the image of a TEM, contrast is made by absorption or diffraction. The thicker the sample
the more absorption (Lambert-Beer). Contrast can also be because of diffraction (electrons are too wide
scattered), or diffracted differently (phases).
Magnification is done by using lenses. An object of length l is magnified by a lens till L =q/p with q the
distance from the lens till the image plane. The focal distance (f) is different from q. f is lower than q.
In imaging mode, you want rays that started at the same place to come exactly together on the phosphor
screen. In diffracting mode, you want paral lel beams from different spots (= diffraction) to come together
Karen Louise De Sousa Pesse
73
at the same place on the phosphor screen. Both methods are quite the same, but the intermediate lens is
changed in strength to go from diffraction mode to imaging mode.
6.2.3 Bright and dark Field Imaging
Bright field and dark field images are the bas ic mode for viewing crystalline specimens in the transmission
electron microscope. They provide the essential microstructural information fro m a specimen prior to
having to resort to more specialist imaging or opera ting techniques. Although these images can be
obtained over the complete range of accelerating voltages, we address here the range that covers both the
conventional and medium
If an objective aperture intercepts all the diffracted
beams and allows only the direct beam to pass,
deficiency contrast occurs and a bright field image is
formed.
In addition, the objective aperture can be used to
select a single diffracted beam to produce a dark
field image. If this is produced by tilting the incident
electron beam, the astigmatism in the image is
reduced.
Karen Louise De Sousa Pesse
74
6.3 Question 17
What are the functions of the objective aperture in TEM and where it is positioned? What does
change in the image when you insert and objective aperture. What is SAD and how are the SAD
images analysed for cubic materials?
6.3.1 Objective aperture
Above the focal point or below it:
To reduce the effects of spherical aberration , apertures are
introduced into the beam path. Apertures are circular holes in
metal disks on the micron scale. The net effect of the aperture is
to reduce the diameter of the disk of minimum confusion .
However, that positive effect comes at the price of reduced beam
current. Also, a very small aperture will display diffraction
effects. The diameter of the aperture used will also affect the
convergence angle of the beam and this in turn will affect its
coherence as well as image properties such as depth of focus.
An objective aperture is situated within the beam path just
below the objective lens. The objective aperture is important
for several reasons. The aperture will:
Allow for signal selection (What you want to see)
Provide for contrast within the image
Decrease objective lens aberrations, spherical
and chromatic, which will degrade image
resolution.
Affect depth of field in the image – a smaller
aperture giving better depth of field.
In diffraction mode, we remove the objective aperture and
insert another aperture further down the column —a selected
area diffraction (SAD) aperture, to select a portion of the
sample from which the diffraction pattern arises.
6.3.2 What is SAD
SAD is referred to as "selected" because the user can easily
choose from which part of the specimen to obtain the
diffraction pattern. Located below the sample holder on the
TEM column is a selected area aperture, which can be inserted
into the beam path. This is a thin strip of metal that will block the beam. It contains several different sized
holes, and can be moved by the user. The effect is to block the entire electron beam except for the small
fraction passing through one of the holes; by moving the aperture hole to the section of the sample the
user wishes to examine, this particular area is selected by the aperture, and only this section will
contribute to the SADP on the screen. This is important, for exampl e, in polycrystalline specimens. If more
than one crystal contributes to the SADP, it can be difficult or impossible to analyse. As such, it is useful to
select a single crystal for analysis at a time. It may also be useful to select two crystals at a time , in order
to examine the crystallographic orientation between them.
Karen Louise De Sousa Pesse
75
6.3.3 How are SAD images analysed for cubic materials?
i.
In order to analyse a SAEDP, firstly a parallelogram with the smallest g 1 , g 2 and g 3 is chosen on the
spot electron diffraction patt ern.
a. Measurement of the size of g 1 , g 2 and g 3 and the scale bar is done with a ruler.
b. The values are then compared with the actual size on the scale bar.
c. The length |g i | in nm - 1 is determined using the scale bar ratio on the diffraction pattern;
Table 1: gi was calculated from the relation of scale bar on a ration 6cm 15 nm .
-1
ii.
Index i
⃗⃗⃗⃗⃗⃗⃗⃗
𝒈𝒉𝒌𝒍 𝒏𝒎−𝟏
⃗⃗⃗⃗⃗⃗⃗⃗
𝒈𝒉𝒌𝒍 𝐜𝐦
1
4.25
1.7
3
5.00
2.0
2
4.25
1.7
Afterwards, the inter planar distance d i is determined by the equation
𝑑𝑖 =
Karen Louise De Sousa Pesse
1
𝑔
⃗⃗⃗⃗⃗⃗⃗⃗
ℎ𝑘𝑙
76
These values are then compared with the given table in the exercise:
Index
i
⃗⃗⃗⃗⃗⃗⃗⃗
𝒈𝒉𝒌𝒍 𝒏𝒎−𝟏
d hkl nm
Family
plane
Plane
(hkl)
{hkl}
1
4.26
0.234
{111}
(111)
2
4.26
0.234
{111}
(1̅1̅1)
3
4.92
0.203
{002}
(002)
Table 2:
Determination of
inter planar
distances, d values,
family planes and
planes.
In order to find the plane (hkl), the following formula shall be applied:
ℎ1 + ℎ2 = ℎ3
𝑘1 + 𝑘2 = 𝑘3
𝑙1 + 𝑙2 = 𝑙3
Thus in {hkl}{111}, {111} and {002} we have:
1−1 = 0
1−1 = 0
1+1 = 2
And the plane will then be the column of numbers with the appropriate signal chosen .
i.
In order to measure the angles, th ere are two possibilities:
a. The family planes are inserted in the formula
b. Draw an angle (using the protractor template)
Figure 2: Angle masurement with protractor template.
Karen Louise De Sousa Pesse
77
The values obtained were the following:
1 = cos−1
2 = cos −1
(ℎ1 ℎ3 + 𝑘1 𝑘3 + 𝑙1 𝑙3 )
√(ℎ12
+
𝑘12
+
𝑙12 )(ℎ32
+
𝑘32
+
𝑙32 )
(ℎ2 ℎ3 + 𝑘2 𝑘3 + 𝑙2 𝑙3 )
√(ℎ22
+
𝑘22
+
𝑙22 )(ℎ32
+
𝑘32
+
𝑙32 )
= cos−1
= cos−1
(0 + 0 + 2)
√12
(0 + 0 + 2)
√12
= 54.7°
= 54.7°
Table 3: Interplanar angles for cubic system
𝝋𝟏
𝝋𝟐
Measurement
52°
56°
Calculation
54.7°
54.7°
The values correspond to each other and can be considered satisfactory.
On the second task, one must determine the crystal zone axis for the indexed diffraction spots (i.e. crystal
planes in a real space). This can be obtained with the following formula:
< 𝑢𝑣𝑤 > = {111}𝑥{002} =
𝑢
[1
0
𝑣 𝑤 𝑢𝑣
1 1 ] 1 1 = 2𝑢 − 2𝑣 + 0w = < 22̅0 >
0 2 00
General Family of Directions
If we calculate the direction parallel to the electron beam
one set of planes:
specifically for
< 𝑢𝑣𝑤 > = (1̅1̅1)𝑥(002) = 2̅20
𝑢
[1̅
0
𝑣 𝑤 𝑢𝑣
1̅ 1 ] 1̅ 1̅ = −2u + 2v + 0𝑤 = 2̅20 = 1̅10
0 2 00
This is a particular solution for particular indices of planes, thus
should be square.
Figure 3: The electron beam
is parallel to the zone axis
direction. Source: Google
brackets
The zone axis is a direction in the crystal which is parallel to the electron beam. Thus this axis is 1̅10.
Indices must be reduced to least numbers.
Karen Louise De Sousa Pesse
78
On another example:
(133)
(113)
2
1
(020)
On this one you might find families {002} and {113}, with {133} in the middle, however you cannot seem to
apply the rules because 3 and 2 can never equal 3, right? But here we are talking about families, so {002} =
(002), (020) and (200). If you use (020), should be able to find the r ight solution.
1+0 = 1
1+2 = 3
3+0 = 3
Thus the family of planes is (113), (020) and (133).
1 = cos−1
2 = cos−1
(ℎ1 ℎ3 + 𝑘1 𝑘3 + 𝑙1 𝑙3 )
√(ℎ12 + 𝑘12 + 𝑙12 )(ℎ32 + 𝑘32 + 𝑙32 )
(ℎ2 ℎ3 + 𝑘2 𝑘3 + 𝑙2 𝑙3 )
√(ℎ22
+
𝑘22
𝑢
[1
0
Karen Louise De Sousa Pesse
+
𝑙22 )(ℎ32
+
𝑘32
+
𝑙32 )
= cos−1
= cos−1
(0 + 6 + 0)
√18 ∗ 4
(1 + 3 + 9)
√19 ∗ 11
= 45°
= 25°
𝑣 𝑤 𝑖𝑗
3 3 ] 13 = −6𝑢 + 0𝑣 + 2𝑤 = 3̅01
2 0 02
79
7 Electron Backscattered Diffraction (EBSD)
7.1 Question 18
Explain the basic operational principles of the EBSD method. Formation of Kikuchi bands. Band
Detection. Hough Transform. Pattern Indexation
7.1.1 Definition
EBSD – Electron Backscattered Diffraction: is a SEM based technique that provides crystallographic
orientation data necessary to understa nd the micro-structure property relationships for alloy
design, materials characterization and failure analysis;
Steps necessary to produce an EBSD pattern in a SEM
Tilt the sample so that its surface makes an angle about 70° with the horizontal
Turn off the scan coils to obtain a stationary electron beam
Place a recording medium in front of the tilted specimen to capture the diffraction pattern:
phosphor screen
7.1.2 Architecture
EBSD system is attached to scanning electron microscope in one of the free ports us ually
perpendicular to the tilt axis of the microscope stage.
Operational System: Signal detector camera connected to a post -processing unit and a computer;
Specialized hardware and software is used for post -processing the signal from the camera, analysis
(indexation) of the acquired diffraction patterns and beam or stage control.
EBSD Cameras: detectors for forward scattered electrons – additional images with orientation or
elemental contrast
OIM computer (orientation imaging microscopy) asks Microscope C ontrol computer to place a fixed
electron beam on a spot on the
sample;
A cone of diffracted electrons is
intercepted by a specifically
placed phosphor screen;
Incident electrons excite the
phosphor, producing photons
A charge Coupled Device CCD
Camera detects and amplifies
the photons and sends the signal
to the OIM computer for
indexing;
Karen Louise De Sousa Pesse
80
7.1.3 Formation of Kikuchi pattern.
What influences a Kikuchi Pattern? Which of those can be avoided by the
researcher?
Is a SEM based technique → proper sample preparation is very important;
The patterns are observed when a fixed and focused electron beam is
positioned on a tilted specimen:
TILT – Reduce the path length of backscattered electrons – 70° is ideal
Backscattered electrons escape from 30 -40 up to 100nm underneath the
surface, hence there is a diffracting volume;
EBSD patterns (consisting of Kikuchi bands) are formed when a stationary
electron beam interacts with a crystalline lattice in a highly tilted sample in the
SEM.
The geometry of the band hold informatio n about crystal lattice in the diffracting volume;
The width and intensity of a band is related to the spacing of atoms in the corresponding crystal
plane;
The symmetry of the crystal lattice is reflected in the pattern
The orientation of the crystal latti ce with respect to a laboratory reference frame can be
determined from a pattern assuming the material is of a known crystal structure;
Remember → ↑atomic number Z↑ Backscattering
7.1.4 Band Detection
There are two distinct artefacts: BANDS (planes) and POLES (vectors);
Bands are intersections of diffraction cones with the phosphorous screen; they correspond to a
family of crystallographic planes
Bands widths are proportional to the inverse inter planar
spacing
Intersection of multiple bands (planes) correspond to a pole
of those planes (vector)
Karen Louise De Sousa Pesse
81
7.1.5 Hough Transform
Bands are transformed to peaks
p =x cos T +y sin T
x and y are the coordinates of a pi xel in the EBSD patter image (column and row)
p and T are the coordinates of lines that pass through the pixel
A PIXEL in the image space becomes a sinusoidal curve in the transform space
When applied to every pixel, the transform becomes a large set of si nusoidal curves
For each pixel in a line, possible values of p are calculated using the formula, with T ranging from 0
to 180 degrees
The curves will intersect at point that is the angle of the line and its position relative to the origin;
All the pixels in a band form a peak at the intersection of their individual sinusoidal curves;
A LINE IN IMAGE SPACE TRANSFORMS TO A POINT IN HOUGH SPACE
The basics of Hough Transform is to find aligned points in images that create lines
VERY IMPORTANT: https://www.youtube.com/watch?v=4zHbI -fFIlI
Teta and Ro parameters will define a line equation in terms of angle and radius
The point where the curves intersect gives a distance and angle, and they indicate the line which
intersects the point being tested
Karen Louise De Sousa Pesse
82
Pattern Indexation
Assuming the pattern is from a known structure, the first step is to identify the (hkl) indices of the
strongest diffracting planes in the crystal;
A look up table is constructed from th ese planes
The bands identified by Hough transform are processed to extract geometrical relationship between
bands and then compared to the table to identify potential (hkl) indices for the detected bands;
To correlate the bands with particular planes in the crystal lattice, and to identify the crystal
orientation, the angles between the bands are calculated and compared to theoretical values; once
the bands have been found, next step is to determine the orientation of the crystal lattice from the
geometrical arrangement of the bands;
Often the first step in the EBSD process after pattern collection is indexing: this allows
identification of crystal orientation at the
Resume:
Influences on EBSD:
beam current
accerlerating voltage
spotsize and tilt angle
features that can be seen are crystal latice
Latice parameter
orientation
Sources:
a.
Lecture 7 ”EBSD” slides 1-26
b.
V. Randle and O. Engler “Introduction to Texture Analysis-Macrotexture, Microtexture and Orientation Mapping” Chapter6 pages
127-151 and 157-176. –Recommended. (file Introduction to Texture Analysis.pdf)
c.
Flewitt:”Physical methods for materials characterization-Second Edition”, Chapter 6. Parts 6.4.1., 6.4.2, 6.4.3 and 6.4.5 (file
IP556_CH06.pdf)
d.
Bob Hafner,” Introductory Transmission Electron Microscopy Primer” file “tem_primer.pdf”-Recommended
e.
Practical classes report
Karen Louise De Sousa Pesse
83
7.2 Question 19
Evolution of electron back-scatter diffraction (EBSD. Orientation Image Analysis. Special
resolution and angular resolution of the EBSD. What is IQ, (BC) CI (MAD)? Experiment design
philosophy. What kind of information can be obtained from an EBSD measurement? (Examples).
Sample preparation for the EBSD measurement. Compare the EBSD with the XRD method for
texture characterization.
7.2.1 Evolution of electron back-scatter diffraction EBSD
30-50s: first observation of Kikuchi patterns
70s – Electron backscattered patterns recorded in the SEM
80s – Computer routines for interactive EBSD pattern evaluation
80s – Automated EBSD pattern analysis
90s – Orientation contrasting map
7.2.2 Orientation Image Analysis.
ORIENTATION IMAGING MICROSCOPY (OIM) – automation technique of the orientation
measurement process using EBSD; in OIM the electro n beam is scanned over the specimen surface in
a regular grid, at each pixel in the grid and EBSD pattern is captured and automatically indexed; the
following data are calculated:
Crystallographic Orientation data
IQ Image Quality: a quality factor definin g the sharpness of the diffraction pattern
CI confidence index: a patented parameter indicating the degree of confidence that the orientation
calculation is correct
Material Phase
Specimen data collection coordinates (x, y)
Karen Louise De Sousa Pesse
84
7.2.3 Spatial resolution and angular resolution of the EBSD.
7.2.3.1 Spatial Resolution:
Def.: ability of the imaging modality to differentiate two objects; measure of how closely lines can
be resolved; ability to see fine detail
The spatial resolution of the technique is governed by the SEM elec tron optics as in conventional
backscattered electron imaging. For high resolution imaging on Nano grains, high performance FESEMs are required, along with a small sample and short working distances;
Minimum grain diameter for EBSD imaging is 10nm
The Spatial Resolution is primarily determined by
o
SEM
o
Geometry of the sample/lens/EBSD detector relationship (the specimen/microscope geometry:
specimen → screen distance; specimen tilt; specimen height)
o
Material (↑ Z - ↑ backscattered signal - ↑ pattern clarity)
o
Accelerating Voltage (↑ voltage = ↑ brighter diffraction pattern, ↓ interference from EM fields,
electron beam penetrates further and surface contamination/damage effects are minimized)
o
Probe (beam) Current (narrow beam, ↑ current density)
o
Pattern Clarity
7.2.3.2 Angular Resolution of Ebsd
o
AR of an individual EBSD pattern is usually 1°
o
Important to determine misorientation between two crystal lattices (grains) – the accuracy is
determined by measuring misorientation between adjacent sampling points in a single cry stal;
o
The accuracy, or angular resolution of EBSD related directly to the precision with which the
diffraction pattern can be indexed;
7.2.4 What is IQ, (BC) CI (MAD)
7.2.4.1 The image quality parameter IQ
describes the quality of an electron backscattered diffractio n pattern;
IQ = sum of detected peaks in the Hough Transform
The IQ is dependent on the material and condition, and is a function of the technique and
parameters used to index the pattern as well as other factors such as video processing;
The greatest effect on the quality of diffraction patterns is the perfection of the crystal lattice in
the diffracting volume;
Distortions of the crystal lattice lower quality of image;
IQ reveals grain boundaries and precipitates (black dots)
Gradients in IQ reveal disloc ation structures
Karen Louise De Sousa Pesse
85
In the Oxford HKL software, the equivalent of the IQ is the Band Contrast Parameter (BC) and the
Confidence Index or Indexation Reliability is quantified by the Minimum Angular Deviation (MAD);
MAD shows the difference between the measured value of the angles and the theoretical ones for
the given crystal lattice;
Karen Louise De Sousa Pesse
86
7.2.4.2 CI Confidence Index
Parameter that indicates the degree of confidence that the orientation calculation is correct
For a given diffraction pattern, several possible orientations may be found (that satisfy the
diffraction bands detected by the image analysis routine);
The orientation is then “voted”: ex.: if you have 5 colors (bands) that can form different triplets (10
combinations), you can have several solutions. Each solutio n assigns an hkl to a band triplet; If a
solution yields inter-planar angles within tolerance, a vote or an “x” is marked in the solution
column;
The solution chosen will be the one with more votes
Once the solution is chosen, it is compared to the Hough a nd the angular deviation is calculated as
the fit
Band triplets
Solution
#
n votes
S1 (solution w/most votes)
nd
S2 (solution w/ 2 most votes)
CI
Karen Louise De Sousa Pesse
n votes of S1 - n votes of S2 10 4
0.6
number of band triplets
10
87
7.2.5 Experiment design philosophy - What kind of information can be obtained from an EBSD measurement?
(Examples)
A great advantage of the EBSD measurement is that the output data can be easy represented as text
files which could be further exchanged, post -processed or exported to their programs for texture
calculations, modelling; etc. A table can be obtained with:
Information for the scanned phase
X and y coordinates of the pixel from where the diffraction pattern was acquired
Orientation of the crystal in the point with coordinates x and y given by three Euler angles (phi1,
phI, phi2)
Image Quality factors (BC, BS, IQ) and Confidence Index (CI, MAD)
7.2.6 Sample preparation for the EBSD measurement
Sectioning/cutting
Mounting (convenience in handling specimens of difficult shapes or sizes/protect and preserve
extreme edges or surface defects during preparation)
Grinding (different grits of sandpaper for 15 -20s)
Mechanical Polishing (diamond/alpha alumina solution 5-10 min)
7.2.6.1 Additional steps:
i.
Vibratory polishing,
ii.
Electro polishing (best quality of EBSD, but several bad aspects like surface relief, rounds sample
edges, sensitive to polish parameters)
iii.
Chemical etching (simpler technique, but surface relief is cre ated and any topography is enhanced)
iv.
Ion etching
v.
Ion Beam Milling (works on almost every type of material, but only small surfaces can be polished
and takes long time to prepare; ion beam interacts with material and unstable phases can
transform);
vi.
Coating
7.2.6.2 Sample storage
i.
EBSD is a surface technique – information is obtained at 10 -50nm from the surface
ii.
Any surface damage influences the result
iii.
EBSD is a SEM technique – samples must be conductive
iv.
Non-conductive samples require special measures
v.
Selection of correct phase (crystal structure) is necessary
Karen Louise De Sousa Pesse
88
7.2.7 Compare the EBSD with the XRD method for texture characterization.
The characterization of phase fractions with EBSD and XRD shows differences, due to interacting
volumes and the spatial resolution of differe nt methods.
The minimum detectable sample area in EBSD is 0.1µm of diameter while in XRD is 10 -100µm
diameter;
The angular accuracy of EBSD is 0.5 -1.0°; XRD is 2°;
Applications of XRD are mostly for Grains; EBSD reaches Subgrains;
Sources:
Lecture 7 ”EBSD” slides 1-26
V. Randle and O. Engler “Introduction to Texture Analysis -Macrotexture, Microtexture and Orientation
Mapping” Chapter 7; pages 157 -176. –Recommended (file Introduction to Texture Analysis.pdf)
Karen Louise De Sousa Pesse
89
8 3D Microstructure Characterization (3D-EBSD)
8.1 Question 20
Give an overview of the special techniques that are used to do a 3D microstructure
characterization. Explain details on 3D-EBSD with focused Ion beam and 3D-Xray diffraction.
WHY 3D: THE MATERIALS MICROSTRUCTURE IS OF 3D NATURE. THE STATISTICAL 3D METHODS GIVE AN
ACCURATE BUT NOT A COMPLETE DESCRIPTION OF THE MICROSTRUCTURE.
8.1.1 Overview of special techniques
Method
Multiple sectioning
+OM or SEM or EBSD
Most simple
Destructive method
Multiple cutting or polishing
Atom probe
microscopy
Destructive method
+ Chemical composition
No crystallographic structure
X-Ray tomography
Non-destructive method
Uses x-rays to create cross-sections
due to the difference in absorption
Used for non-metals: porosity of
biological objects
3D XRD
Non-destructive method
Texture + grain shape reconstruction
With variation of time and
temperature (in situ: -4D)
3D EBSD with FIB
Destructive method
Sectioning by FIB
Observation by EBSD
8.1.2 3D-EBSD with focused ion beam
The geometry of the sample holder for 3D EBSD looks like the figure at
angles handle the geometrical condition. The perfect alignment of the ion
the electron beam is a critical point in this technique.
Karen Louise De Sousa Pesse
right. The
beam and
90
First the Ga + beam glides over the sample and removes a thin layer. Then the Ga + beam is stopped and the
sample holder rotates. Now the e - beam reacts with the sample and the reflected electrons can be
collected by the EBSD.
In each step you go deeper in the specimen by removing a thin layer. Then you put the EBSD patterns
together and you get a 3D image.
Karen Louise De Sousa Pesse
91
8.1.3 3D-Xray diffraction
High energy, short wavelength penetrate the sample
The primary beam (yellow) is stopped; the diffracted beam (red) gives the x -rays.
The sample can be heated up for some experiments.
Sources:
Lecture 12 ”3D microstructure characterization by means of combined FIB -EBSD method” slides 1-26
Karen Louise De Sousa Pesse
92
9 AFM/APM
9.1 Question 21
What observation techniques stay behind the abbreviations AFM and APM? Discuss the
operational principle, requirements for the sample preparation and the application field.
9.1.1 APM
APM stands for atomic probe field ion microscopy. APM combines two techniques
Field ion microscope: first microscope with atomic resolution
Atom probe: time-of-flight mass spectrometer: identification of atoms
9.1.2 Field ion microscope
In this microscope a sharp (<50nm tip radius) is placed in an ultrahigh vacuum chamber, which is filled with
an imaging gas such as argon, neon, hydrogen or helium. To avoid thermal emission, the tip is cooled to
cryogenic temperatures (20-100K). A high positive voltage of 5 -20kV is applied to the tip, therefore the
sample is placed on an electrically insulated stage. Imaging gas atoms adsorbed on the positive charged tip
are ionized by the strong electric field in de neighborhood of the t ip. So the gas atoms are becoming
positively charged and then being repelled from the tip. The ions are repelled in a direction perpendicular
to the surface due to the curvature of the surface. A detector (fluorescent screen) is placed so as to collect
these repelled ions.
When the voltage increase, we could have field evaporation. This means that an atom contains enough
energy to evaporate from the surface. With this effect we are not restricted to the investigation of surface
atoms because the next layer is available: 3D imaging is possible.
9.1.3 Atom probe
A pulsed high voltage source (typically 0 -7 kV) is generated and applied to the specimen. The application of
the pulsed voltage to the sample allows for individual ions at the sample surface to have their electric
field, and hence atomic bonding, temporarily disrupted. This results in ejection of an ionized atom from
the sample surface at a known time. The delay between application of the pulse and detection of the ion
allows for the computation of a mass-to-charge ratio (time of flight).
Karen Louise De Sousa Pesse
93
9.1.4 Sample preparation
We need to have a very thin tip. We can do this with mechanical grinding (left image) of a rod or wire or
with electro polishing (right image).
9.1.5 Applications
You can determine the location and distribution
of elements in alloys. You can also identify the
elements segregated to dislocations. This
technique is the only one who combine
chemistry and morphology in 3D.
Karen Louise De Sousa Pesse
94
9.1.6 AFM
AFM stands for atomic force microscopy.
AFM consist of a cantilever with a sharp tip (silica crystal) at its end that is used to scan the specimen
surface. Forces between the tip and the surface leads to deflections.
These deflections caused by the surface topology are measured by a laser light. A laser l ight is reflected off
the back of a cantilever and the light is collected by a 4 quadrant photodiode. The output signal is
proportional to the deflection. The detection limit is approximately 0.1 A.
There are two primary modes of operation (imaging mode s). The static mode (contact mode AFM), where
the force between the tip and the surface and the deflection is kept constant. The dynamic mode (non contact mode AFM and tapping mode AFM), where the cantilever oscillate around resonance frequency and
the change in oscillation, after reaction with the surface, gives information about the sample.
9.1.7 Contact mode AFM
In contact mode, the force between the tip and the surface is kept constant during scanning by maintaining
a constant deflection. The contact mode us es the repulsive force between the tip and the sample and uses
a DC-detection method. Because the distance tip -sample changes, the applied voltage can determine the
height of sample.
The disadvantage of this mode is that you can cause damage to the surf ace.
Karen Louise De Sousa Pesse
95
9.1.8 Non-contact mode AFM
The non-contact mode uses attractive forces between the tip and the sample and uses an AC -detection
method. The tip oscillates above the sample with a constant oscillation frequency or amplitude. The tip sample distance is changing and the applied voltage is a measure for the height of the sample.
The non-contact AFM uses weaker forces so an AC -detection method is
needed. This mode also requires vacuum due to adsorbents.
9.1.9 Tapping mode AFM
In the tapping mode the tip oscillates above the sample with a constant
amplitude. The amplitude changes when surface relief. The tip -sample
distance is changing and the applied voltage is a measure for the height of the sample.
With this mode, the surface isn’t damaged and there is no influ ence
by adsorbent layer. It can be implemented in air. Even a very soft and
fragile sample can be imaged with this mode.
9.1.10 Advantages of AFM
The sample doesn’t have to be conductive and there is no surface preparation necessary. It is a 3D -imaging
technique to investigate the surface of the specimen with a very good resolution: lateral resolution 5nm,
vertical resolution 0.01 nm. There is no vacuum needed (except for non -contact mode) so air and liquid
environment are possible.
9.1.11 Disadvantage of AFM
You have only a small imaging area: 150µm by 150µm with µm height (if you increase x and y, you decrease
z) (↔ SEM: mm by mm with mm depth of field). The imaging of the sample (several minutes) is slower than
with SEM (nearly real-time). Drift in the image is also possible. It is required to have a good choice of the
tip, otherwise you can get artifacts (see picture below).
Sources:
Lecture 13 ”Microstructure characterization by means of combined Atomic Force Microscopy, Atom Probe
Microscopy and Laser Confocal M icroscopy”
Karen Louise De Sousa Pesse
96
Practical classes
EBSD, XRD, LOM/QM, SP, TEM, SEM
GROUP 4
9.2 Extra on EBSD
EBSD – Analysis technique based on a scanning electron microscope (SEM)
Measures the local crystal orientation with sub-micron spatial resolution
Can also be used for phase distribution analysis
When the electron beam hits the sample, a cloud of backscattered electrons is generated
A phosphor screen placed within this cloud transform each arriving electron into a photon, thus transforming
the backscattered signal into a visible light signal;
The diffracted part of the backscattered electrons is highly anisotropic and when interacting with the phosphor
it will create a KIKUCHI pattern
The kikuchi pattern is captured by a high speed and high sensitivity CCD camera placed behind the phosphor
screen and transferred to a computer where it is being analyzed;
Hundreds of patterns per second can be acquired and analyzed due to state of the art hardware and software
programming technology;
EBSD data is displayed and interpreted in real time; the phase distribution map reveals the presence of hard
and brittle intermetallic phases; the orientation distribution map displays the local crystal orientations;
https://www.youtube.com/watch?v=Ny_lTzPnynY
Karen Louise De Sousa Pesse
97
10 Sources
1.
Lecture 1, slides 14 to 28;
2.
Flewitt ”Physical methods for materials characterization -Second Edition”, Chapter 1.9 -Microstructure (only)(file
IP556_CH01.pdf);
3.
Lecture 1, slides 29 to 35;
4.
Flewitt ”Physical methods f or materials characterization -Second Edition”, Chapter 1.9 -Microstructure (only)(file
IP556_CH01.pdf);
5.
Lecture 4, slides 1 to 30;,
6.
Flewitt ”Physical methods for materials characterization -Second Edition”, Chapter 2.Parts 2.1, 2.2, and 2.3 Without
2.2.4. “Protons” (file IP556_CH02.pdf); I strongly recommend you to go through the rest of the Chapter 2 until Chapter
2.8
7.
B.C.De Cooman “Materiaalkundige observatietechniken”Chapters 1.6.1.”Materiaalen en denciteiten” en chapter
1.6.2.”Basisformule van de micr okarakterisatie”
8.
Lecture 2. Slides 1-26;
9.
Practical classes notes and discussions;
10. Lecture 2, slides 27 to 56;,
11. Flewitt ”Physical methods for materials characterization -Second Edition”, Chapter 5. Parts 5.1, and 5.4
12. Practical classes report
13. Lecture 2, slides 34 to 39,
14. Flewitt:”Physical methods for materials characterization -Second Edition”, Chapter 5. Parts 5.2 and 5.3,
15. Practical classes report
16. Lecture 2, slides 57 to 72
17. Practical classes report
18. Lecture 9, slides 1 to 23,
19. Flewitt:”Physical methods f or materials characterization -Second Edition”, Chapter 4. Parts 4.31, 4.3.2, 4.3.4 and 4.3.3
(4.3.3 only for information), (file IP556_CH04.pdf)
20. Website: http://www.matter.org.uk/diffraction/geometry/3D_reciprocal_lattices.htm
21. Practical classes report
22. B.C.De Cooman “Materiaalkundige observatietechniken”; Chapter 3.1, 3.2(Interactie tussen X -stralen en vaste stof);
3.3.1, 3.3.2 (Productie van X -stralen). In file “Chapter 16.pdf”
23. Lecture 9, slides 24 to 46,
24. Flewitt:”Physical methods for materials characterization -Second Edition”, Chapter 4. Parts 4.3.5, 4.3.6 (file
IP556_CH04.pdf)
25. B.C.De Cooman “Materiaalkundige observatietechniken”; In file “Chapter 16.pdf” Chapter 3.1.1,until 3.1.2
26. Practical classes report
27. Lecture 9, ”X-ray diffraction” slides 47 to 51,
28. Flewitt:”Physical methods for materials characterization -Second Edition”, Chapter 4. Parts 4.3.8, (file IP556_CH04.pdf)
29. B.C.De Cooman “Materiaalkundige observatietech niken” File: “Chapter 18.pdf” Chapters 3.6.1.”Meting van interne
spanningen”
30. Practical classes report
Karen Louise De Sousa Pesse
98
31. Lecture 8 “Introduction to quantitative texture analysis”, slides 1 to 35,
32. B.C.De Cooman “Materiaalkundige observatietechniken” File “Chapter 18.pdf” Chapter 3.6.2.”Textuuranalyse”
33. V. Randle and O. Engler “Introduction to Texture Analysis -Macrotexture, Microtexture and Orientation Mapping”
Chapters 2.1 to 2.6. page 13 -36.-Recommended. (file Introduction to Texture Analysis.pdf)
34. Practical classes report
35. Lecture 9 ”X-ray diffraction” slides 52 -60
36. Lecture 8 ”Introduction to quantitative texture analysis”, slides 36 to 52
37. V. Randle and O. Engler “Introduction to Texture Analysis -Macrotexture, Microtexture and Orientation Mapping”
Chapter 3. Pages 41-54. and 61 -88. –Recommended. (file Introduction to Texture Analysis.pdf)
38. Flewitt:”Physical methods for materials characterization -Second Edition”, Chapter 4. Parts 4.3.6, last part of the
chapter, (file IP556_CH04.pdf)
39. B.C.De Cooman “Materiaalkundige observati etechniken” File: Chapter 19.pdf” Chapter 3.6.2.3”Practische aspekten
van de textuurmeting”
40. Practical classes report
41. Lecture 6 ”Introduction to SEM” slides 1 -32
42. Flewitt:”Physical methods for materials characterization -Second Edition”, Chapter 6. Parts 6. 1,6. 2 and 6.3. (file
IP556_CH06.pdf)
43. Bob Hafner ”Scanning Electron Microscopy Primer” –strictly recommended ! File “sem_primer.pdf”
44. B.C.De Cooman “Materiaalkundige observatietechniken” Files: “ Chapter 8.pdf” “Chapter 9.pdf”, “Chapter 11.pdf”and
Chapter 12.pdf Chapters 2.3.1,(Elektronen bron), 2.3.2 (Elektronen optika), 2.3.3(Elektronen detektoren), 2.5
(Elementen van de elektronen microscopen)
45. Practical classes report
46. Lecture 6 ”Introduction to SEM” slides 1 -32
47. Flewitt:”Physical methods for materials charac terization-Second Edition”, Chapter 6. Parts 6.1,6. 2 and 6.3. (file
IP556_CH06.pdf)
48. Bob Hafner ” Energy Dispersive Spectroscopy on the SEM:A Primer ! File “eds_on_sem_primer.pdf” Recommended!
49. B.C. De Cooman “Materiaalkundige observatietechniken” Files: “ Chapter 11.pdf”, “Chapter 12.pdf” Chapters 2.4. (Xstralen spectrometers) en Chapter 2.5.5. (Microanalize m.b.v. characteristieke X -stralen)
50. Practical classes report
51. Lecture 11 ”Introduction to TEM” slides 21 -42
52. Flewitt:”Physical methods for materials char acterization-Second Edition”, Chapter 6. Parts 6.4.4. (file IP556_CH06.pdf)
53. Practical classes report
54. Lecture 11 ”Introduction to TEM” slides 1 -21
55. Bob Hafner,” Introductory Transmission Electron Microscopy Primer” file “tem_primer.pdf” -Recommended
56. Flewitt: ”Physical methods for materials characterization -Second Edition”, Chapter 6. Parts 6.4.1., 6.4.2, 6.4.3 and 6.4.5
(file IP556_CH06.pdf)
57. Practical classes report
58. Lecture 11 ”Introduction to TEM” slides 1 -21
59. Flewitt:”Physical methods for materials character ization-Second Edition”, Chapter 6. Parts 6.4.1., 6.4.2, 6.4.3 and 6.4.5
(file IP556_CH06.pdf)
60. Bob Hafner,” Introductory Transmission Electron Microscopy Primer” file “tem_primer.pdf” -Recommended
61. Practical classes report
Karen Louise De Sousa Pesse
99
62. Lecture 7 ”EBSD” slides 1 -26
63. V. Randle and O. Engler “Introduction to Texture Analysis -Macrotexture, Microtexture and Orientation Mapping”
Chapter6 pages 127-151 and 157-176. –Recommended. (file Introduction to Texture Analysis.pdf)
64. Flewitt:”Physical methods for materials characterization -Second Edition”, Chapter 6. Parts 6.4.1., 6.4.2, 6.4.3 and 6.4.5
(file IP556_CH06.pdf)
65. Bob Hafner,” Introductory Transmission Electron Microscopy Primer” file “tem_primer.pdf” -Recommended
66. Practical classes report
67. Lecture 7 ”EBSD” slides 1 -26
68. V. Randle and O. Engler “Introduction to Texture Analysis -Macrotexture, Microtexture and Orientation Mapping”
Chapter 7; pages 157-176. –Recommended (file Introduction to Texture Analysis.pdf)
69. Practical classes report
70. Lecture 12 ”3D microstructure characterization by means of combined FIB -EBSD method” slides 1 -26
71. Lecture 13 ”Microstructure characterization by means of combined Atomic Force Microscopy, Atom Probe Microscopy
and Laser Confocal Microscopy”
Karen Louise De Sousa Pesse
100