Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Biochemical cascade wikipedia , lookup
Signal transduction wikipedia , lookup
Biochemistry wikipedia , lookup
Point mutation wikipedia , lookup
Gene expression wikipedia , lookup
Paracrine signalling wikipedia , lookup
Ribosomally synthesized and post-translationally modified peptides wikipedia , lookup
Magnesium transporter wikipedia , lookup
Expression vector wikipedia , lookup
Ancestral sequence reconstruction wikipedia , lookup
G protein–coupled receptor wikipedia , lookup
Bimolecular fluorescence complementation wikipedia , lookup
Structural alignment wikipedia , lookup
Interactome wikipedia , lookup
Metalloprotein wikipedia , lookup
Protein purification wikipedia , lookup
Homology modeling wikipedia , lookup
Western blot wikipedia , lookup
Two-hybrid screening wikipedia , lookup
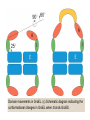
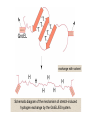
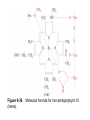
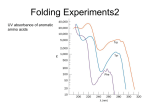
Reductive denaturation and oxidative renaturation of RNase A Plausible mechanism for the thiol- or enzyme-catalyzed disulfide interchange reaction in a protein protein disulfide isomerase C-chain needed to direct proper disulfide bond formation Primary structure of porcine proinsulin Determinants of Protein Folding A. helices/sheets predominate in proteins because they fill space efficiently B. protein folding is directed mainly by internal residues (protein folding is driven by hydrophobic forces) C. protein structures are organized hierarchically Hierarchical organization of globular proteins (subdomains) Determinants of Protein Folding (cont.) D. protein structures are highly adaptable E. secondary structure can be context-dependent “chameleon” sequence: AWTVEKAFKTF (unfolded free in solution) NMR structure of protein GB1 Determinants of Protein Folding (cont.) F. dependence of protein fold on primary sequence created by changing 50% of the 56 residues in GB1 not all residues have equally important roles in specifying a specific fold X-ray structure of Rop protein, a homodimer of aa motifs that associate to form a 4-helix bundle The Levinthal Paradox 2n backbone torsions, n-residue protein: ~10n structures time to explore all structures: t = 10n/1013 s-1 for a 100-residue protein: t = 1087 s Conclusion: proteins fold via an ordered pathway or set of pathways Experimental Methods to Monitor Protein Folding (UV/ VIS/ fluorescence /CD) A stopped-flow device: 40 ms dead-times cold denatured proteins / T-jump molar extinction coefficient UV absorbance spectra of the three aromatic amino acids, phenylalanine, tryptophan, and tyrosine eL and eR differ (circularly polarized light); measure ∆e Circular dichroism (CD) spectra of polypeptides Pulsed H/D Exchange X-H + D2O X-D + HOD Used to follow the time course of protein folding by 2D NMR a. b. c. d. e. Denatured protein in D2O Dilute with H2O and allow to fold for time tf Increase pH to initiate D-H exchange (10-40 ms) Lower pH; allow to completely fold Determine which amide protons are protonated and deuterated Landscape Theory of Protein Folding Polypeptides fold via a series of conformational adjustments that reduce their free energy and entropy until the native state is reached. There is no single pathway or closely related set of pathways that a polypeptide must follow in folding to its native state. The sequence information specifying a particular fold is both distributed throughout the polypeptide chain and highly overdetermined. Folding funnels: An idealized funnel landscape Folding funnels: The Levinthal “golf course” landscape Folding funnels: Classic folding landscape Closer mimic of an actual folding pathway Folding funnels: Rugged energy surface Folds via an ordered pathway: involves well defined intermediates Polypeptide backbone and disulfide bonds of native BPTI (58 residues, three disulfide bonds) Renaturation of BPTI: protein primary structures evolved to specify efficient folding pathways as well as stable native conformations Folding accessory proteins A. Protein disulfide isomerases (PDI) B. Peptidyl prolyl cis-trans isomerases C. Molecular chaperones A folding accessory protein Reactions catalyzed by protein disulfide isomerase (PDI). (a) Reduced PDI catalyzes the rearrangement of the non-native disulfide bonds. Reactions catalyzed by protein disulfide isomerase (PDI). (b) The oxidized PDI-dependent synthesis of disulfide bonds in proteins. homodimer; eukaryotic; each subunit consists of four domains Cys 36 and 39 exposed NMR structure of the a domain of human protein disulfide isomerase (PDI-a) in its oxidized form. (a) The polypeptide backbone is shown in ribbon form. Cys 36 is located in a hydrophobic patch Oxidized PDI-a is less stable than reduced PDI-a NMR structure of the a domain of human protein disulfide isomerase (PDI-a) in its oxidized form. (b) The molecular structure as viewed from the bottom. Peptidyl Prolyl Cis-Trans Isomerases (PPIs) Xaa-Pro peptide bonds: ~10% cis PPIs catalyze the otherwise slow interconversion of Xaa-Pro peptide bonds between their cis and trans conformations; accelerate the folding of Pro-containing polypeptides. Two families: cyclophilins and FKBP12 (based on known inhibitors) Unfolded proteins in vivo have a great tendency to form intramolecular and intermolecular aggregates. Molecular chaperones prevent/reverse improper associations, especially in multidomain and multisubunit proteins. Function by binding solvent-exposed hydrophobic surfaces reversibly to promote proper folding Many chaperones are ATPases. Classes of Chaperones A. Heat shock proteins 70: 70 kD monomeric proteins B. Chaperonins: form large multisubunit cage-like assemblies C. Hsp90: involved in signal transduction; very abundant in eukaryotes D. Nucleoplasmins: acidic nuclear proteins involved in nucleosome assembly Chaperonins GroEL/ES system 14 identical ~60 kD subunits in two rings; creates a central cavity Electron micrograph-derived 3D image of the Hsp60 (GroEL) chaperonin from the photosynthetic bacterium Rhodobacter sphaeroides. X-ray structure of GroEL. (a) Side view perpendicular to the 7-fold axis. X-ray structure of GroEL. (b) Top view along the 7-fold axis. X-ray structure of GroES as viewed along its 7-fold axis. cis ring trans ring X-ray structure of the GroEL-GroES-(ADP)7 complex. X-ray structure of the GroEL-GroES-(ADP)7 complex. bound ADP shown in cis ring X-ray structure of the GroEL-GroES-(ADP)7 complex. apical intermediate equatorial Domain movements in GroEL. (a) Ribbon diagram of a single subunit of GroEL in the X-ray structure of GroEL alone. Domain movements in GroEL. (b) A GroEL subunit in the X-ray structure of GroEL-GroES-(ADP)7. Domain movements in GroEL. (c) Schematic diagram indicating the conformational changes in GroEL when it binds GroES. Apical domain of GroEL in complex with a tight-binding 12-residue polypeptide (SWMTTPWGFLHP). (a) (b) Movements of the polypeptide-binding helices of GroEL. Reaction cycle of the GroEL/ES chaperonin system in protein folding. Models for GroEL/ES Action A. Anfinsen cage model: folding within complex B. Interative annealing: reversible release of partially folded intermediates Refolding of RiBisCO; requires assistance to reach native state black: no components red: released only after native state is reached green: released after one turnover Rate of hydrogen-tritium exchange of tritiated RuBisCO. exchange with solvent Schematic diagram of the mechanism of stretch-induced hydrogen exchange by the GroEL/ES system. Protein Structure Prediction Secondary structure a) Chou-Fasman method Frequency at which a given aa occurs in an a helix in a set of protein structures = fa = na/n, where na = number of amino acid residues of the given type that occur in a helices, and n = total number of residues of this type in the protein set Propensity of a particular aa residue to occur in an a helix = Pa = fa/<fa>, where <fa> is the average value of fa for all 20 residues Pa > 1: residue occurs with greater than average frequency in an a helix Also applies to b-structure H = strong former h = former I = weak former i = indifferent b = breaker B = strong breaker Propensities and classifications of amino acid residues for a helical and b sheet conformations. B. Reverse turns: Rose method Occur on the surface of a protein; locations of minimal hydropathy (exclude helical regions) Rationale for Observed Propensities For a-helix: appears related to the amount of side-chain hydrophobic surface buried in the protein For Pro: low a propensity caused by strain For Gly: low a propensity caused by reduced entropy and lack of hydrophobic stabilization For Ala: high a propensity caused by lack of a g substituent; reduced entropic cost; minimal hydrophobic stabilization Computer-based Secondary Structure Algorithms Combine three or more methods: accurate to ~75% Jpred: public domain software The moderate accuracy is caused by failure to take tertiary interactions into account (tertiary structure influences secondary structure). Secondary structure prediction in adenylate kinase ( N-terminal 24 residues) Tertiary Structure Prediction a. comparative or homology modeling b. fold recognition or threading c. ab initio methods Protein Design Structures of the second zinc finger motif of Zif268 (DNAbinding protein): X-ray structure. sequence has only 6 of the 28 residues identical to Zif268 (5 are similar) group of Phe residues replaces zinc finger Structure of de novo designed peptide, FSD-1: NMR structure (a bba motif; 28 residues) Comparison of the structures of the second zinc finger motif of Zif268 and FSD-1: best-fit superpositions of their backbones. Protein Dynamics Proteins undergo structural motions that have functional significance. Conformational fluctuations (breathing motions) in the oxygen binding protein, myoglobin. Classes of Motions 1. atomic fluctuations (10-15-10-11 s; 0.01 - 1Å displacements) 2. collective motions (10-12-10-3 s; 0.01 - 5 Å displacements) 3. triggered conformational changes (10-9 -103 s; 0.5 - 10 Å displacements) Techniques: crystallography, NMR, MD blue = least mobile red = most mobile The mobility of the GroEL subunit in the X-ray structure of GroEL alone. blue = least mobile red = most mobile The mobility of the GroEL subunit in the X-ray structure of the GroEL-GroES-(ADP)7 complex. The internal motions of myoglobin as determined by a molecular dynamics simulation: the Ca backbone and the heme group. The internal motions of myoglobin as determined by a molecular dynamics (MD) simulation: an a helix. Detecting infrequent motions (time scale of seconds) Exchange rate of a particular proton correlates with the conformational mobility of its surroundings The hydrogen-tritium “exchange-out” curve for hemoglobin that has been pre-equilibrated with tritiated water. Conformational Diseases: Amyloid and Prions Alzheimer’s disease; transmissible spongiform encephalopathies (TSEs); amyloidoses Common characteristic: formation of amyloid fibrils The involved proteins assume two different stable conformations (native and amyloid) Amyloid fibrils: an electron micrograph of amyloid fibrils of the protein PrP 27-30. a b Fibrils consist mainly of b-sheets whose b-strands are perpendicular to the fibril axis. Amyloid fibrils (PrP 27-30): Model (a) and isolated (b) b sheet. Amyloidogenic proteins are mutant forms of normally occuring proteins Lysozyme mutants occur in familial visceral amyloidosis Superposition of wild-type human lysozyme and its D67H mutant. Prion Diseases Evidence that the scrapie agent is a protein: scrapie agent is inactivated by treatment with diethylpyrocarbonate, which reacts with His sidechains. Evidence that the scrapie agent is a protein: scrapie agent is unaffected by treatment with hydroxylamine, which reacts with cytosine residues. Evidence that the scrapie agent is a protein: hydroxylamine rescues diethylpyrocarbonate-inactivated scrapie reagent. Prion protein conformations: NMR structure of human prion protein (PrPC). Note the disordered N-terminal tail residues (dots). PrP may be a cell-surface signal receptor. Prion hypothesis: PrPSc induces the conversion of PrPC to PrpSc Conversion may be mediated by a molecular chaperone. Prion protein conformations: a plausible model for the structure of PrPSc (very insoluble) END Figure 9-36 (heme). Molecular formula for iron-protoporphyrin IX Figure 9-37 Primary structures of some representative ctype cytochromes. Figure 9-38 Three-dimensional structures of the c-type cytochromes whose primary structures are displayed in Fig. 9-37. Figure 9-39 rhodanese. The two-structurally similar domains of