Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
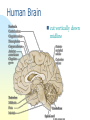
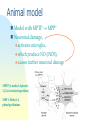
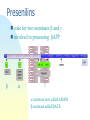
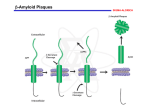
725 - Molecular neurobiology of disease Parkinson’s disease Schizophrenia Alzheimer’s disease Reference List Approaches epidemiology genetic chromosome gene / protein pharmacology anatomical post-mortem MRI/PET animal models Human Brain cut vertically down midline Parkinson’s disease Loss of dopaminergic neurons normal: 4% per decade Parkinson’s: 70-80% loss normal substantia nigra Parkinson’s Symptoms Hard to initiate movement Interaction of substantia nigra with cortex see 746 lecture 6 Therapy L-DOPA cross blood-brain barrier dopamine agonists MAO-B inhibitors (selegiline = deprenyl) cell replacement fetal midbrain transplants pigs carotid body stem cells deep brain [=thalamus] stimulation Animal model Model with MPTP MPP+ Neuronal damage, activates microglia, which produce NO (iNOS), causes further neuronal damage MPTP (1-methyl-4-phenyl 1,2,3,6-tetrahydropyridine) MPP 1-Methyl-4phenylpyridinium Causation Inherited disorder *a-synuclein (folds SNAREs) Parkin (E3 ubiquitin ligase) DJ-1 (stress response chaperone) PINK-1 (mitochondrial protein kinase) *LRRK2 (another ?mitochondrial kinase) It is not clear why mutations in a-synuclein, or parkin or [] genes cause nigral dopaminergic cell death in familial PD [Le W & Appel SH (2004)] *dominant – others are recessive Causation Environmental factors too Rotenone fish poison blocks mitochondrial function upregulates a-synuclein oxidises DJ-1 Paraquat One model inhibitors of parkin Another model Summary Parkinson’s has well-defined deficit – loss of dopaminergic cells well-described pathology & behaviour variety of therapies no cure no known cause Schizophrenia Positive (hallucinations) & negative symptoms (asociality) possibly several illnesses seasonal highly inherited Developmental disease genetic cause : DISC1 or a chromosome translocation caused by failure of neurons to migrate ? red shows areas less in Sc Dopamine hypothesis positive symptoms respond to treatment negative symptoms do not respond to treatment DA antagonists Chlorpromazine side effects, e.g. Parkinsonism, constipation Haloperidol D2 (+D3, D4 +5-HT2A) blocker Newer drugs e.g. clozapine dopamine D2 receptors and 5-HT action D2 receptor block is key point e.g. mouse model -ve symptoms from DA in prefrontal cortex 5-HT action helps -ve symptoms NMDA (glutamate) receptors blocked by phencyclidine, relieves many symptoms Depression 5-HT (=serotonin) main treatment is with uptake inhibitors SSRI eg Prozac Noradrenaline also selective reuptake inhibitors PFC: pre-frontal cortex Summary so far ethical issues “impede” research animal models hard to interpret key concept: neural diseases identified with cellular / molecular deficit disease related to change in specific neurotransmitter complexity of CNS leads to side effects Dementia Reduction of brain volume and cells with age Dementia increases with age at 65, 11% of USA had dementia 70% of dementia is Alzheimer’s 15% from strokes at 85, 47% affected Early onset Alzheimer’s inherited <1% of cases Alois Alzheimer On November 3, 1906, Alois Alzheimer gave a lecture to the Meeting of the Psychiatrists of South West Germany, presenting the neuropathological and clinical description of the features of one of his cases, Auguste D., who had died of a dementing illness at the age of 55, Alzheimer’s Symptoms Forgetfulness untidiness confusion less movement storage of new memory reduced finally loss of bodily function Neuroanatomy cortex very reduced normal Alzheimer Neuroanatomy cortex reduced - note gaps between folds Neurodegeneration brains feature plaques (Ab = b-amyloid) tangles (tau) Neurofibrillary tangles micrograph drawing by Alois Alzheimer Development of tau Amyloid hypothesis Down’s syndrome leads to AD by 40 linked to chromosome 21 Positional cloning identified: amyloid-b (Ab) peptide 40-42 amino acids families 670 with mutations in bAPP / 692 / 716 & 717 amyloid b toxic to cultures Presenilins Familial early onset dominant AD linked to mutations on chromosomes 14 & 1 presenilin I : mutations lead to onset at age 28 presenilin II : second homologous gene mutations are in regions conserved between PSI and PSII associated with AD lead to increased Ab production Presenilins code for two secretases b and g involved in processing bAPP b a g a secretase now called ADAM b secretase called BACE Proteolysis of APP Normal amyloidogenic APP Proteolysis of Ab In non-familial AD, plaques caused not by production of Ab but by failure to degrade it Little evidence for increased production of Ab peptide maybe normally degraded quickly half life 1-2 hr tangles resistant to degradation enzymes: neprilysin & insulin-degrading-enzyme Neprilysin Neprilysin knockout mice have more Ab42 Major problem how does faulty b-amyloid lead to tangles of tau? tau is hyperphosphorylated GSK-3 glycogen synthase kinase More direct interaction? tau and Ab form complexes GSK-3 phosphorylates tau in complex tau Ab in neurons Ab is extracellular tau v Ab AD has both tau and Ab other diseases have just tangles of tau Apolipoprotein E Another family gene for late onset of AD produces Apolipoprotein E Apolipoprotein E - cont receptor (LRP) expressed in astrocytes normal role is in cholesterol transport may aid in clearance of b-amyloid from brain to blood mutations disrupt clearance Oxidative stress main function of b-amyloid may be to protect cells from reactive Oxygen radicals damage to mitochondria leads to *OH shortage of energy (or oxygen) increases likelihood of AD through high [Ca] metal ions might affect build up of b-amyloid Therapy ?? cholinergic therapy secretase blockers relief of oxidative stress Apolipoprotein therapy stem cells for replacement vaccination ginko biloba Cholinergic hypothesis cholinergic neurones in basal forebrain project to cortex and hippocampus muscarinic antagonist, (M1), pirenzipine, causes memory loss in hippocampus agonists, e.g. physostigmine, improve memory But other systems interact Cholinergic therapy Cholinesterase inhibitors – delay symptoms Tacrine: allosteric – 1993 (toxic in liver) Donepezil; mixed binding Try Cholinergic agonist M2 on basal ganglia and intestine Depletion of M1 receptors? M1 and M3 receptors in hippocampus Drug trials discontinued Summary of AD Full mechanism not known amyloid hypothesis well – established role of tau also established role for glia and neurons No one effective treatment cholinotherapy promising ? Happy Christmas & New Year!