Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
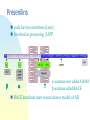
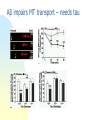
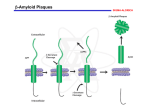
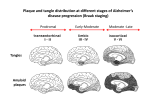
Alzheimer’s disease Aim Alzheimer’s symptoms neurodegeneration genetic basis of early onset AD amyloid hypothesis treatment Dementia – economic costs Dementia increases with age at 65, 11% of USA had dementia 70% of dementia is Alzheimer’s 15% from strokes at 85, 47% affected Early onset Alzheimer’s inherited <1% of cases ~5 years from MCI to diagnosis by physician survival depends on age @70 ~ 8 years @90 ~ 3 years Alois Alzheimer On November 3, 1906, Alois Alzheimer gave a lecture to the Meeting of the Psychiatrists of South West Germany, presenting the neuropathological and clinical description of the features of one of his cases, Auguste D., who had died of a dementing illness at the age of 55, Alzheimer’s Symptoms ?preceded by MCI (mild cognitive impairment) Forgetfulness untidiness confusion less movement storage of new memory reduced finally loss of bodily function First: what happens to the brain in AD ? Neuroanatomy cortex very reduced normal Alzheimer Neuroanatomy cortex reduced - note gaps between folds NL : normal MCI: mild cognitive impairment PET scan hippocampus cortex loss of energy metabolism: hippocampal hypometabolism predicts cognitive decline from normal aging Cellular changes AD brains feature plaques (Ab = b-amyloid) tangles (tau) Next: tau Neurofibrillary tangles micrograph drawing by Alois Alzheimer Development of tau tau hypothesis tau and microtubules T : taxol binding Although tau gets in way of cargo transport, tau is required for MT integrity. Normal equilibrium of unbound tau-P and tau (bound) Phosphorylation of tau tau-P mutations lead to neurodegeneration these mutations more readily phosphorylated kinases: glycogen synthase kinase 3 (GSK3), cyclin-dependent kinase 5 (CDK5) microtubule-affinity-regulating kinase (MARK) Amyloid hypothesis Down’s syndrome leads to AD by 40 linked to chromosome 21 Positional cloning identified: mutations in bAPP (amyloid precursor protein) 670 / 692 / 716 & 717 amyloid-b (Ab) peptide 40-42 amino acids amyloid b toxic to cultures Presenilins Familial early onset dominant AD linked to mutations on chromosomes 14 & 1 presenilin I : mutations lead to onset at age 28 presenilin II : second homologous gene mutations are in regions conserved between PSI and PSII associated with AD lead to increased Ab production Presenilins code for two secretases b and g involved in processing bAPP b a g a secretase now called ADAM b secretase called BACE BACE knockout mice rescue mouse model of AD Proteolysis of APP Normal AD Where does BACE act ? Promote a cleavage treat with BACE1 inhibitor localised to membrane flies expressing APP / presenilins (%eclosing) mice with inhibitor, membrane localised inhibitor (Ab level) therapy ???? Proteolysis of Ab In non-familial AD, plaques caused not by production of Ab but by failure to degrade it Little evidence for increased production of Ab peptide maybe normally degraded quickly half life 1-2 hr in mice 8hr in human plaques resistant to degradation enzymes: neprilysin & insulin-degrading-enzyme Neprilysin Neprilysin knockout mice have more Ab42 Summary so far AD is disease of older people early onset linked to Ab plaques presenilins linked to tau tangles Major problem : how does faulty b-amyloid lead to tangles of tau? Aβ impairs MT transport – needs tau Do tau and Ab form complexes? form soluble complex which then precipitates? GSK-3 phosphorylates tau in complex Ab is extracellular? tau Ab in neurons merge lower magn. merge Aβ oligomers induce missorting of Tau control Aβ Oligomers yellow colour indicates tau in dendrites Summary so far AD is disease of older people early onset linked to Ab plaques presenilins linked to tau tangles tau and Ab ???? Next: another genetic risk factor! Apolipoprotein E Another family gene for late onset of AD produces Apolipoprotein E Apolipoprotein E - cont 299 aa protein secreted by astrocytes and microglia Interacts with receptors in the lowdensity lipoprotein receptor family LRP1 expressed in neurons LDLR in astrocytes normal role of ApoE is in cholesterol transport may aid in clearance of b-amyloid from brain to blood HSPG: heparin sulfate proteoglycan Oxidative stress main function of b-amyloid may be to protect cells from reactive Oxygen radicals damage to mitochondria leads to *OH shortage of energy (or oxygen) increases likelihood of AD through high [Ca] metal ions might affect build up of b-amyloid Environmental factors Cold sores 'an Alzheimer's risk' Therapy ?? cholinergic therapy NMDA block (Memantine) secretase blockers relief of oxidative stress Apolipoprotein therapy stem cells for replacement vaccination ginko biloba see http://www.cnsspectrums.com/aspx/articledetail.aspx?articleid=972 for review Vaccination trial halted (2002) meningoencephalitis follow up (2008) showed Ab clearance, but no cognitive effect new vaccine(s) 2010 ? 29th July 2008 “drug works by dissolving the tangle of tau fibres” Cholinergic hypothesis cholinergic neurones in basal forebrain project to cortex and hippocampus muscarinic antagonist, (M1), pirenzipine, causes memory loss in hippocampus agonists, e.g. physostigmine, improve memory But other systems interact Cholinergic therapy Cholinesterase inhibitors – delay symptoms Tacrine: allosteric – 1993 (toxic in liver) Donepezil (aricept); mixed binding Rivastigmine: low interaction with other drugs preferentially blocks form of ACh-esterase found in brain delays decrease in MCI ~ 2 years Tacrine Rivastigmine Donepezil Try Cholinergic agonist M2 on basal ganglia and intestine Depletion of M1 receptors? M1 and M3 receptors in hippocampus Drug trials discontinued Other therapies ? bapineuzumab, a monoclonal anti-amyloid antibody (Phase III) tarenflurbil (modulates gamma secretase activity) (terminated in Phase III) dimebon (antihistamine) – phase II very +, phase III no effect Summary of AD Full mechanism not known amyloid hypothesis well – established role of tau also established role for glia and neurons No one effective treatment ! cholinotherapy promising ? MS – PD – AD – what have we learnt? Genetics provided major insight Despite short lifespan, animal models of neurodegeneration remarkably successful Range of therapies under development many disappointments some successes Still no major understanding of the cause(s) Happy Christmas & New Year