Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
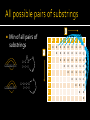
Doug Raiford Lesson 7 RNA World Hypothesis RNA world evolved into the DNA and protein world DNA advantage: greater chemical stability Protein advantage: more flexible and efficient enzymes (biomolecules that catalyze) ▪ 20 amino acids vs. 4 nucleotides ▪ Chemically, more diverse Remnants remain in ribosomes, nucleases, polymerases, and splicing molecules Primary: sequence Secondary: double stranded regions >tRNA. Carries amino acid for Isolucine AGGCUUGUAGCUCAGGUGGUUAGAGCGCACCCCUGAUAAGGGUGAGGUCGGUGGUUCA AGUCCACUCAGGCCUACCA Reverse complements Tertiary: threedimensional structure T arm CCA Tail Acceptor Step D arm Anticodon arm Anticodon How find regions of reverse complementation? What do we have? Sequence A’s like pairing with U’s and G’s like pairing with C’s Stronger bond (3 hydrogen bonds) between G’s and C’s Should result in lowest free energy (max enthalpy) tRNA Transports amino acid to the ribosome T arm CCA Tail Acceptor Step D arm Anticodon arm Anticodon Visualization Good at finding longer basepairings (stacked base-pairs) Need to find the conformation that provides the minimal total free energy RNA often has many alternate conformations at different temperatures Stacked base-pairs add stability Loops/bulges introduce positive free energy and are destabilizing First nucleotide basepairs with last Recurse on rest Recurrence relations First nucleotide basepairs with some other on every (ri , rj ) E ( Si 1, j Recurse ) 1 E ( S ) min i, j possible set of (other than last) min E ( S ) E ( S ) for i k j i , k 1 k, j two strings nucleotide (including none) j G G i As luck would have it… Zuker came up with a dynamic programming solution G G A A A U C C G G A A A U C C 0 0 0 0 0 0 0 0 0 j G Start with zeros on diagonal Populate diagonally i G 0 G 0 G A A A U C C G G A A A U C C 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 Will look at last value to illustrate Match first and last character, recurse on rest (ri , rj ) E ( Si 1, j 1 ) 1 (2) j i G G G A A A U G 0 0 0 0 0 0 -1 -2 -3 G 0 0 0 0 0 0 -1 -2 -3 0 0 0 0 0 -1 -2 -2 0 0 0 0 -1 -1 -1 0 0 0 -1 -1 -1 0 0 -1 -1 -1 0 0 0 0 0 0 0 0 0 G A A A U α A C U G C A 0 0 -1 0 C C 0 0 0 -1 U -1 0 0 0 G 0 -1 0 0 C C j Min of all pairs of substrings G i GGGAAAUCC G G-G A C-C-U A GGGAAAUCC G A A G-G-G-A C-C-U G A A A U A C C G G G A A A U 0 0 0 0 0 0 C -1 -2 -3 0 0 0 0 0 -1 -2 -3 0 0 0 0 -1 -2 -2 0 0 0 -1 -1 -1 0 0 -1 -1 -1 0 -1 -1 -1 0 C 0 0 0 0 0 n2 plus 2n for each visited cell So O(n3) Populate matrix plus traverse row/column for each cell Any prediction method must account for these Now O(n4) Interior loops most expensive Can exploit the fact that along diagonals, loops have same size Can calculate once Limits search space Back to O(n3) E ( Si 1, j ) E (S ) i , j 1 E ( Si , j ) min min E ( Si ,k ) E ( S k 1, j )for i k j E ( Li , j ) (ri , rj ) ( j i 1), if Li , j is a hairpin loop (r , r ) E ( S i 1, j 1 ), if Li , j is a helical region i j (ri , rj ) (k ) E (Sik 1, j 1 ), if Li, j is a bulge on i E ( Li , j ) min k 1 min (ri , rj ) (k ) E ( Si 1, j k 1 ), if Li , j is a bulge on j k 1 min (ri , rj ) (k1 k 2 ) E ( Si 1 k1 , j 1 k 2 ) , if Li , j is an interior loop k 1 (k ) destabiliz ing free energy of a hairpin loop with size k stabilizin g free energy of adjacent base pairs (k ) destabiliz ing free energy of a bulge of size k (k ) destabiliz ing free energy of an interior loop of size k Zuker’s site T arm CCA Tail Acceptor Step D arm Anticodon arm Anticodon Codon: uua Anti-codon: aat tRNA for Leucine in E. coli, a prototypical organism 1 gccgaggtgg tggaattggt agacacgcta ccttgaggtg gtagtgccca atagggctta 61 cgggttcaag tcccgtcctc ggtacca Just like proteins: conformation What if a T-A base-pair mutate to an G-C Still same function What would this do to a search or sequence alignment? GCAGGACCAUAUA ||||||||||||| CGUCCUGGUAUAU GCAGGACCAGAUA ||||||||||||| CGUCCUGGUCUAU Phenomenon known as covariance (not to be confused with statistical covariance) GCAGGACCAUAUA ||||||||||||| CGUCCUGGUAUAU GCAGGACCAGAUA ||||||||||||| CGUCCUGGUCUAU How might we locate covariant pairs? MSA then compare all pairwise combinations of columns High degree of agreement in two columns (G’s match with C’s, A’s match with U’s) an indication of base-pairing χ2 test Compare to expected number of parings given sequence composition Pairing depicted with nested parentheses AAGACUUCGGUCUGGCGACAUUC ((( ))) (( ( ))) Mountain plots A mountain plot represents a secondary structure in a plot of height versus position, where the height m(k) is given by the number of base pairs enclosing the base at position k. I.e. loops correspond to plateaus (hairpin loops are peaks), helices to slopes. Circle plot Data structure capable of capturing secondary structure Ordered Binary Tree Productions S → aSu | uSa | cSg | gSc S → aS | cS | gS | uS S → Sa | Sc | Sg | Su S → SS S →⍉ Derivation S → aS S → aSc S → aScc S → acSgcc S → acgScgcc S → acggSccgcc S → acgggScccgcc S → acggggSccccgcc S → acgggguSccccgcc S → acgggguuSccccgcc S → acgggguucSccccgcc S → acgggguucgSccccgcc S → acgggguucgaSccccgcc S → acgggguucgaaSccccgcc S → acgggguucgaauSccccgcc S → acgggguucgaauccccgcc Parse tree a←S | S→c | S→c | c←S→g | g←S→c | g←S→c | g←S→c | g←S→c S→u | | u←S S→a \ / u←S S→a \ / c←S—S→g Conformation of RNA dictates function Determining secondary structure can help determine tertiary structure Dynamic programming approach to identifying minimum energy conformations Zuker MFOLD View using dot plots, nested parens, mountain or circular plots Covariance: base-pairs mutate but still form pairs, exploit to find pairings