Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
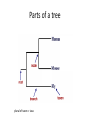
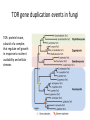
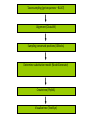
Phylogenetics workshop: Protein sequence phylogeny Darren Soanes Parts of a tree plural of taxon = taxa Phylogenetic tree: evolutionary family tree Nodes in the tree represent speciation events, where an ancestral lineage gives rise to daughter lineages. Relationships in trees Rooting a tree outgroup Root - most recent common ancestor of all the taxa in a tree outgroup — a taxon outside the group of interest. All the members of the group of interest are more closely related to each other than they are to the outgroup. Used to root the tree. Outgroup Rooted and unrooted trees rooted tree unrooted tree Cladogram Phylogram Evolution of Amino Acid Sequences • Amino acid sequences change due to mutations in DNA sequence. • Amino acid sequences evolve more slowly than DNA sequences. • Evolutionary selection occurs on protein sequences. • Gene trees created using protein sequences. DNA mutations (1) • Synonymous substitution – change in DNA sequence that does not affect the amino acid sequence, often in the third position of a codon, e.g. CCG (Pro)→CCA (Pro). • Non-synonymous substitution - change in DNA sequence that does affect the amino acid sequence, often in the first or second position of a codon, e.g. CCG (Pro)→CAG (Gln). Genetic Code DNA mutations (2) • Non-synonymous substitution also called missense mutation. • Nonsense mutation – where a a stop codon is introduced into the middle of a sequence, e.g. TGG (Trp)→ TAG (Stop) • Insertion / deletion (indel), causes a frame shift if not a multiple of three bases. • Nonsense and frame-shift mutations usually produce non-functional proteins. Amino acid substitution matrices (1) • Substitutions between amino acids that are similar in properties are more common. • Cysteine, glycine and tryptophan rarely change. • Substitution matrices measure the likelihood that one amino acid is likely to change to another. Families of amino acids Amino acid substitution matrices (2) • Amino acid substitution matrices are empirically derived by alignment of sets of closely related protein sequences. • Examples include Dayhoff, BLOSUM (used in BLAST searches), WAG, JTT, LG. • Different matrices suitable for looking at proteins encoded by mitochondrial genome e.g. MtREV. BLOSUM 62 Matrix Rates of amino acid change • Rate of substitution varies at different positions in an amino acid sequence. • A proportion of sequences are likely to be invariant, generally have an essential role in the function of a protein. • A gamma distribution models the variation of rates at different sites. • Sites are sorted into gamma rate categories. Structure of thrombin showing catalytic triad (conserved in serine proteases) Phylogenetic analysis • Phylogenetic analysis programs take an alignment of protein sequences and attempt to produce a phylogenetic tree showing evolutionary relationships between the sequences. • User can select amino acid substitution matrix and number of gamma rate categories, the program will estimate the proportion of invariant sites. • Programs use these parameters and protein alignment to estimate evolutionary distance between sequences. • They calculate topology and branch length of final tree. Distance Methods • Evolutionary distance calculated for all pairs of taxa. • UPGMA - assumes rate of substitution is constant. • Least squares – allows different rates of substitution in different branches. • Minimum evolution (ME)– topology chosen where the sum of branch lengths is the smallest. Can take a long time to compute, neighbour joining (NJ) method is simplified version of ME – much quicker. Maximum parsimony • For each topology the smallest number of amino acid substitutions are calculated that could explain the evolutionary process. • The topology that requires the smallest number of substitutions is chosen as the best one. Maximum likelihood (ML) • For each topology the likelihood is calculated that the known sequences could have evolved on that tree (branch lengths and substitution rate parameters optimised). • Topology with the best likelihood score is chosen. • Takes a long time to compute ML of every possible tree. • Heuristic methods such as quartet puzzling reduce the number of candidate trees. • Programs that use ML methods: PhyML, RAxML, TreePuzzle (uses quartet puzzling). Bootstrapping • Tests the reliability of a tree. • Initial protein alignment is randomised (by sampling columns at random). • Tree construction repeated for each randomised alignment. • For each group of taxa in the original tree it is determined what percentage of the randomised trees contain the same group. • Alternative: Approximate likelihood-ratio test Bayesian methods • A sample is taken of a large number of trees with high ML. • Posterior probabilities calculated for different events of interest. • Markov Chain Monte Carlo method used to generate samples of trees. • Mr Bayes uses these methods. Taxon sampling • Take initial protein sequence. • Decide which range of species you are interested in. • Use BLAST to find homologous sequences in databases, either NCBI database or individual genome databases. FASTA formatted file • • >YJL052W_Saccharomyces_cerevisiae MIRIAINGFGRIGRLVLRLALQRKDIEVVAVNDPFISNDYAAYMVKYDSTHGRYKGTVSH DDKHIIIDGVKIATYQERDPANLPWGSLKIDVAVDSTGVFKELDTAQKHIDAGAKKVVIT APSSSAPMFVVGVNHTKYTPDKKIVSNASCTTNCLAPLAKVINDAFGIEEGLMTTVHSMT ATQKTVDGPSHKDWRGGRTASGNIIPSSTGAAKAVGKVLPELQGKLTGMAFRVPTVDVSV VDLTVKLEKEATYDQIKKAVKAAAEGPMKGVLGYTEDAVVSSDFLGDTHASIFDASAGIQ LSPKFVKLISWYDNEYGYSARVVDLIEYVAKA* >YJR009C_Saccharomyces_cerevisiae MVRVAINGFGRIGRLVMRIALQRKNVEVVALNDPFISNDYSAYMFKYDSTHGRYAGEVSH DDKHIIVDGHKIATFQERDPANLPWASLNIDIAIDSTGVFKELDTAQKHIDAGAKKVVIT APSSTAPMFVMGVNEEKYTSDLKIVSNASCTTNCLAPLAKVINDAFGIEEGLMTTVHSMT ATQKTVDGPSHKDWRGGRTASGNIIPSSTGAAKAVGKVLPELQGKLTGMAFRVPTVDVSV VDLTVKLNKETTYDEIKKVVKAAAEGKLKGVLGYTEDAVVSSDFLGDSNSSIFDAAAGIQ LSPKFVKLVSWYDNEYGYSTRVVDLVEHVAKA* Multiple sequence alignment • Take FASTA file of sequences you are interested in. • Align sequences using ClustalW, Muscle, TCoffee. Sampling of conserved blocks • To get reliable trees non-aligned and poorly conserved areas of sequence need to be removed. • Gblocks samples highly conserved blocks of sequence. Sequence alignment and sampling conserved block Which substitution model should I use? • ModelGenerator takes your sequence alignment and calculates the best amino acid substitution model to use. Creating tree • Take alignment produced by Gblocks and use program of choice to generate a tree (using substitution model suggest by ModelGenerator and specifying number of gamma rate categories, 4 is sufficient). • File format problems, different programs use different file formats – use Readseq to convert between file formats. • Use tree viewing program to look at graphical representation of tree (TreeView, TreeDyn). TOR gene duplication events in fungi TOR: protein kinase, subunit of a complex that regulate cell growth in response to nutrient availability and cellular stresses Workshop task Looking at evolution of genes encoding two types of phosphoglycerate mutase in fungi. Two types of phosphoglycerate mutase (PGM) • Both catalyse the same overall reaction: – 3-phosphoglycerate → 2-phosphoglycerate • cofactor-dependent PGM (dPGM) uses 2,3bisphosphoglycerate (2,3BPG) as a cofactor: • 3PG + P-Enzyme → 2,3BPG + Enzyme → 2PG + P-Enzyme • cofactor-independent PGM (iPGM) has two bound Mn(II) ions at its active site. • 3PG + Enzyme → PG + P-Enzyme → 2PG + Enzyme Two types of phosphoglycerate mutase (PGM) • dPGM found in yeasts and vertebrates • iPGM found in filamentous fungi, plants and some invertebrates • Both can be found in bacteria. • No sequence similarity between the two forms of the enzyme. Structure of iPGM Structure of dPGM Task • Use BLAST search to find PGM protein sequences in a sample of fungal species. • Use these to create phylogenetic trees showing the evolution of genes encoding these enzymes. Taxon sampling (get sequences – BLAST) Alignment (ClustalW) Sampling conserved positions (GBlocks) Determine substitution model (ModelGenerator) Create tree (PhyML) Visualise tree (TreeDyn)