Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Discovery and development of integrase inhibitors wikipedia , lookup
Orphan drug wikipedia , lookup
Compounding wikipedia , lookup
Discovery and development of cephalosporins wikipedia , lookup
Psychopharmacology wikipedia , lookup
Pharmaceutical industry wikipedia , lookup
Drug design wikipedia , lookup
Prescription drug prices in the United States wikipedia , lookup
Prescription costs wikipedia , lookup
Drug discovery wikipedia , lookup
Pharmacogenomics wikipedia , lookup
Pharmacognosy wikipedia , lookup
Theralizumab wikipedia , lookup
Plateau principle wikipedia , lookup
Neuropharmacology wikipedia , lookup
Neuropsychopharmacology wikipedia , lookup
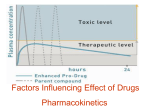
Pharmacokinetics ผศ.มนุพศั โลหิตนาวี [email protected] [email protected] Outline Introduction Physicochemical properties Absorption, Bioavialability, routes of admistration Distribution Biotransformation (Metabolism) Excretion Clinical pharmacokinetics Components of pharmacokinetics Input, dosing by using routes of administration Pharmacokinetic processes (figure 1, drawing) – Absorption – Distribution – Biotransformation (Metabolism) – Excretion Cell membrane barrier of drug permeation (drawing), with semipermeable property factors affecting drug across cell membrane – cell membrane properties – physicochemical properties of drugs Cell membrane physicochemical properties of drugs –size and shape –solubility –degree of ionization –lipid solubility Cell membrane Characteristics of Cell membrane – Lipid bilayer: mobile horizontally, flexible, high electrical resistance and impermeable to high polar compounds – protein molecules function as receptors or ion channels or sites of drug actions. Diffusion across the cell membrane Passive transport (drawing) – higher conc to lower conc area – energy independent – at steady state both sides have equal conc.(non electrolye cpds) – electrolyte: conc. of each side depends on pH (fig 2) – weak acid and weak base Diffusion across the cell membrane Carrier-mediated membrane transport (drawing) – lower conc to higher concentration area (agianst concentration gradient) – structure specific – rapid rate of diffusion – Active and Facillitated transport Diffusion across the cell membrane Active transport – energy dependent – structure specific, inhibited by structure-related cpds, saturable Facillitated transport – energy independent – structure specific, inhibited by structure-related cpds, saturable Drawing almost Saturable process all protein-mediated process in our body can occur this process saturation not only transport system but also others such as enzymatic reaction, drug-ligand binding and so on. because functional protein molecules are limited. Parameters Drug absorption in drug absorption – Rate constant of drug absorption (Ka) – Bioavialability (F) Anatomical aspects affecting absorption parameters (Drawing) – GI tract (metabolzing organ and barrier of drug movement) – Liver (portal and hepatic vien, excretion via biliary excretion) – cumulative degradation so called “First pass effect” Drug absorption Factors affecting drug absorption (Drawing) – Physicochemical properties of drugs – pH at site of absorption – Concentration at the site of administration – Anatomical and physiological factors Blood flow Surface area Routes of administration Enteral and parenteral routes Pros and cons between Enteral and parenteral Enteral administration Pros – most economical, – most convenient Cons –high polar cpds could not be absorbed –GI irritating agents –enzymatic degradaion or pH effect –Food or drug interaction (concomitant used) –cooperation of the patients is needed –first pass effect due to GI mucosa Parenteral administration Pros – Rapidly attained concentration – Predictable conc by the calculable dose – Urgent situation Cons –Aseptic technic must be employed –Pain –limited self adminstration –More expensive Enteral administration Common use of enteral administration –Oral administration –Sublingual administration –Rectal administration Enteral administration Concentrion-time course of oral administration (Drawing) Rapid increase in plasma conc until reaching highest conc and subsequent decrease in plasma conc Drawing (concept of MTC and MEC) – Absorption phase – Elimination phase Enteral administration Prompt release: the most common dosage form Special preparation: Enteric-coat, SR SR, Controlled release: Purposes and limitation Enteral administration Sublingual administration – Buccal absorption – Superior vana cava directly: no first pass effect – Nitroglycerin (NTG): highly extracted by the liver, high lipid solubility and high potency (little amount of absorbed molecules be able to show its pharmacological effects and relieve chest pain). Enteral administration Rectal adminstration – unconscious patients, pediatric patients – 50% pass through the liver and 50% bypass to the inferior vena cava – lower first pass effect than oral ingestion – inconsistency of absorption pattern – incomplete absorption – Irritating cpds Parenteral administration Common use of parenteral administration – Intravenous – Subcutaneous – Intramuscular Simple diffusion Rate depends on surface of the capillary, solubility in interstitial fluid High MW: Lymphatic pathway Parenteral administration Intravenous – precise dose and dosing interval – No absorption (F=1), all molecules reach blood circulation – Pros: Calculable, promptly reach desired conc., Irritating cpds have less effects than other routes – Cons: unretreatable, toxic conc, lipid solvent cannot be given by this route (hemolysis), closely monitored Parenteral administration Subcutaneous – suitable for non-irritating cpds – Rate is usually slow and constant causing prolonged pharmacological actions. Parenteral administration Intramuscular – more rapid than subcutaneous – rate depends on blood supply to the site of injection – rate can be increased by increasing blood flow (example) Pulmonary absorption gaseous or volatile substances and aerosol can reach the absorptive site of the lung. Highly available area of absorption Pros: rapid, no first pass effect, directly reach desired site of action (asthma, COPD) Cons: dose adjustment, complicated method of admin, irritating cpds. Bioequivalence Pharmaceutical equivalence (drawing) Bioequivalence: PharEqui+ rate+ bioavialable drugs Factors: – Physical property of the active ingredient: crystal form, particle size – Additive in theformulation: disintegrants – Procedure in drug production: force An example of a generic product that could pass a bioequivalence test: Simvastatin (Parent form, n=18) Plasma concentration (ng/ml) 8.00 6.00 A 4.00 B 2.00 0.00 0 4 8 12 Time (hr) 16 20 24 An example of a generic product that could pass a bioequivalence test: Ondansetron (n=14) Serum ondansetron concentration (ng/mL) 60 A B 40 20 0 0 6 12 Time (hr) 18 24 An example of a generic product that could pass a bioequivalence test: Clarithromycin (n=24) Plasma clarithromycin concentration (ng/mL) 2500 2000 Klacid (A) Claron (B) 1500 1000 500 0 0 4 8 12 Time (h) 16 20 24 Distribution Drawing distribution site: well-perfused organs, poor-perfused organs, plasma proteins Well-perfused: heart, liver, kidney, brain Poor-perfused: muscle, visceral organs, skin, fat Distribution Plasma proteins – Albumin: Weak acids – alpha-acid glycoprotein: Weak bases Effects of plasma protein binding – Free fraction: active, excreted, metabolized – the more binding, the less active drug – the more binding, the less excreted and metabolized: “longer half-life” Distribution Effects tissues of well distribution into the – deep tissue as a drug reservoir – sustain released drug from the reservoir and redistributed to the site of its action – prolong pharmacologic actions Distribution CNS and CSF Blood-Brain Barrier (BBB) – unique anatomical pattern of the vessels supplying the brain – only highly lipid soluble compounds can move across to the brain – infection of the meninges or brain: higher permeability of penicillins to the brain. Distribution Placental transfer Simple diffusion Lipid soluble drug, non-ionized species first 3 mo. of pregnancy is very critical: “Organogensis” Biotransformation Why biotranformed? (Figure 5) – Normally, drugs have high lipid solubility therefore they will be reabsorbed when the filtrate reaching renal tubule by using tubular reabsorption process of the kidney. – Biotransformation changes the parent drug to metabolites which always have less lipid solubility (more hydrophilicity) property therefore they could be excreted from the body Biotransformation Biotransformation – to more polar cpds – to less active cpds – could be more potent (M-6-G) or more toxic (methanol to formaldehyde) Biotransformation Phase I and II Biotransformation – Phase I : Functionization, Functional group – Phase II: Biosynthetic, Molecule Biotransformation Phase I Reactions (Table 2) – Oxidation – Reduction – Hydrolysis Biotransformation Phase II Reactions (Table 3) – Glucuronidation – Acetylation – Gluthathione conjugation – Sulfate conjugation – Methylation Biotransformation Metabolite from conjugation reaction – Possibly excreted into bile acid to GI – Normal flora could metabolize the conjugate to the parent form and subsequently reabsorbed into the blood circulation. This pheonomenon is socalled “Enterohepatic circulation” which can prolong drug half-life. Biotransformation Site of biotransformation – Mostly taken place in the liver – Other drug metabolizing organs: kidney, GI, skin, lung – Hepatocyte (Drawing) Biotransformation The Liver: Site of biotransformation: – mostly enzymatic reaction by using the endoplasmic reticulum-dwelling enzymes.(Phase I), Cytosolic enzymes are mostly involved in the phase II Rxm. – Method of study phase I Rxm Breaking liver cells Centrifugation very rapidly microsomes and microsomal enzymes Biotransformation Cytochrome (figure 6) P450 monooxygenase system – microsomal enzymes – Oxidation reaction using reducing agent (NADPH), O2 – System requirement Flavoprotein (NADPH-cytochrome P450 reductase, FMN+FAD) fuctions as an electron donor to cytochrome c. Cytochrome P450 (CYP450) Biotransformation Steps in oxidative reactions (figure 6) – Step 1:Parent + CYP450 – Step 2:Complex accepts electron from the oxidized flavoprotein – Step 3:Donored electron and oxygen forming a complex – Step 4: H2O and Metabolite formation Biotransformation CYP450 is a superfamily enzyme, many forms of them have been discovered (12 families). Important CYP450 families in drug metabolism (Fig. 7) – CYP1 (1A2) – CYP2 (2E1, 2C, 2D6) – CYP3 Biotransformation Factors affecting biotransformation – concurrent use of drugs: Induction and inhibition – genetic polymorphism – pollutant exposure from environment or industry – pathological status – age Biotransformation Enzyme induction – Drugs, industrial or environmental pollutants – increase metabolic rate of certain drugs leading to faster elimination of that drugs. – “autoinduction” – Table 4 Biotransformation Enzyme induction – important inducers: antiepileptic agents, glucocorticoids for CYP3A4 isoniazid, acetone, chronic use of alcohol for CYP2E1 Biotransformation Enzyme inhibition: (drawing) – Competitive binding and reversible: Cimetidine, ketoconazole, macrolide metabolites – Suicidal inactivators: Secobarbital, norethindrone, ethinyl estradiol – Clinical significance: erythormycin and terfenadine or astemizole causing cardiac arrhythmia. Genetic Biotransformation polymorphism – Gene directs cellular functions through its products, protein. – Almost all enzymes are proteins so they have been directed by gene as well. – Drug-metabolizing enzymes: Isoniazid: causing more neuropathy in caucaasians leading to identification of the first characterized pharmacogenetics. due to the rate of N-acetylation: Slow and fast acetylators Biotransformation Pathologic conditions – Hepatitis – Cirrhosis due to chronic alcohol intake – Hypertensive pts recieving propranolol which lowers blood supply to the liver may lead to less biotransformation of the high extraction drugs such as lidocaine, propranolol, verapamil, amitryptyline Excretion Parent and metabolite Hydrophilic compounds can be easily excreted. Routes of drug excretion – – – – Kidney Biliary excretion Milk Pulmonary Renal Excretion excretion: Normal physiology – Glomerular filtration: Free fraction, filtration rate – Active tubular secretion: Energy dependent, carrier-mediated, saturable Acids: penicillins and glucuronide conjugate (uric excretion) Bases:choline, histamine and endogenous bases – Passive tubular reabsorption non-ionized species back diffuse into blood circulation Excretion Clinical application of urine pH modification Drug toxicity – Weak base: Acidic urine pH enhances drug excretion by increasing numbers of inoized species by using ammonium chloride. – Weak cid: Basic urine pH enhances drug excretion by increasing numbers of inoized species by using sodium bicarbonate. Excretion Cationic, anionic and glucuronide conjugates can be excreted into bile acid and show enterohepatic cycle. Clinical pharmacokinetics Assumption: correlation between blood concentration and effects MEC and MTC (figure 8) Therapeutic range Clinical pharmacokinetics Order of reaction – zero order pharmacokinetics (Drawing): ethanol, high dose phenytoin and aspirin – first order pharmacokinetics: most drugs show first order pharmacokinetic fashion. Clinical pharmacokinetics Data: relationship between concentration and time (Drawing) Compartmental model to explain above relationship (fig. 9) Dosing and route of administration: IV bolus, IV infusion and oral ingestion Clinical pharmacokinetics Using first order: – IV bolus: concentration-time curve profile (fig 10) – explain equation number 1 – which leads to these pharmacokinetic parameters: clearance, volume of distribution, half-life, Css, onset, duration, F Clinical pharmacokinetics Clearance Vd Half-life and Elimination constant Onset Duration Steady state concentration Absolute bioavialability