Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Coronary artery disease wikipedia , lookup
Heart failure wikipedia , lookup
Cardiac contractility modulation wikipedia , lookup
Mitral insufficiency wikipedia , lookup
Myocardial infarction wikipedia , lookup
Hypertrophic cardiomyopathy wikipedia , lookup
Quantium Medical Cardiac Output wikipedia , lookup
Heart arrhythmia wikipedia , lookup
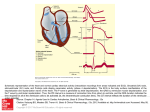
Electrocardiography wikipedia , lookup
Ventricular fibrillation wikipedia , lookup
Arrhythmogenic right ventricular dysplasia wikipedia , lookup
Depolarization wave and mechanics in the paced heart : model and experiment Kerckhoffs, R.C.P. DOI: 10.6100/IR564613 Published: 01/01/2003 Document Version Publisher’s PDF, also known as Version of Record (includes final page, issue and volume numbers) Please check the document version of this publication: • A submitted manuscript is the author’s version of the article upon submission and before peer-review. There can be important differences between the submitted version and the official published version of record. People interested in the research are advised to contact the author for the final version of the publication, or visit the DOI to the publisher’s website. • The final author version and the galley proof are versions of the publication after peer review. • The final published version features the final layout of the paper including the volume, issue and page numbers. Link to publication Citation for published version (APA): Kerckhoffs, R. C. P. (2003). Depolarization wave and mechanics in the paced heart : model and experiment Eindhoven: Technische Universiteit Eindhoven DOI: 10.6100/IR564613 General rights Copyright and moral rights for the publications made accessible in the public portal are retained by the authors and/or other copyright owners and it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights. • Users may download and print one copy of any publication from the public portal for the purpose of private study or research. • You may not further distribute the material or use it for any profit-making activity or commercial gain • You may freely distribute the URL identifying the publication in the public portal ? Take down policy If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim. Download date: 15. May. 2017 Depolarization wave and mechanics in the paced heart Model and Experiment CIP-DATA LIBRARY TECHNISCHE UNIVERSITEIT EINDHOVEN Kerckhoffs, Roy C.P. Depolarization wave and mechanics in the paced heart: model and experiment / by Roy C.P. Kerckhoffs. – Eindhoven : Technische Universiteit Eindhoven, 2003. Proefschrift. – ISBN 90-386-3004-2 NUR 954 Subject headings: cardiac mechanics / cardiac electromechanics / cardiac electromechanical delay / ventricular pacing / interventricular asynchrony / excitation-contraction coupling / depolarization ; propagation / hemodynamics ; pressure volume loop / cardiac cycle / eikonal-diffusion equation / 3-D simulations / mathematical modeling / non-invasive geometrical measurements c 2003 by R.C.P. Kerckhoffs Copyright All rights reserved. No part of this book may be reproduced, stored in a database or retrieval system, or published, in any form or in any way, electronically, mechanically, by print, photoprint, microfilm or any other means without prior written permission of the author. Printed by Universiteitsdrukkerij TU Eindhoven, Eindhoven, The Netherlands. This research was financially supported by Medtronic Bakken Research Center Maastricht. Depolarization wave and mechanics in the paced heart Model and Experiment P ROEFSCHRIFT ter verkrijging van de graad van doctor aan de Technische Universiteit Eindhoven, op gezag van de Rector Magnificus, prof.dr. R.A. van Santen, voor een commissie aangewezen door het College voor Promoties in het openbaar te verdedigen op dinsdag 10 juni 2003 om 16.00 uur door Roy Cornelia Paulus Kerckhoffs geboren te Geleen Dit proefschrift is goedgekeurd door de promotoren: prof.dr.ir. T. Arts en prof.dr.ir. D.H. van Campen Copromotoren: dr.ir. P.H.M. Bovendeerd dr. F.W. Prinzen v This organ deserves to be styled the starting point of life and the sun of our microcosm just as much as the sun deserves to be styled the heart of the world. William Harvey, 1578-1657 vi Contents Summary xi 1 Introduction 1.1 General introduction . . . . . . . . . . . . . . . . . . . . . . . . 1.2 Normal cardiac anatomy and physiology . . . . . . . . . . . . . 1.2.1 Anatomy . . . . . . . . . . . . . . . . . . . . . . . . . . 1.2.2 Depolarization wave propagation . . . . . . . . . . . . . 1.2.3 Contraction . . . . . . . . . . . . . . . . . . . . . . . . . 1.3 Artificial ventricular pacing . . . . . . . . . . . . . . . . . . . . 1.3.1 Global effects . . . . . . . . . . . . . . . . . . . . . . . . 1.3.2 Abnormal patterns of depolarization and contraction . . 1.4 Cardiac excitation-contraction coupling . . . . . . . . . . . . . . 1.4.1 Excitation-contraction from cellular to whole heart level 1.4.2 Cardiac electromechanical mapping . . . . . . . . . . . 1.4.3 Modeling cardiac electromechanics . . . . . . . . . . . . 1.5 Aim of the study . . . . . . . . . . . . . . . . . . . . . . . . . . 1.6 Outline of the thesis . . . . . . . . . . . . . . . . . . . . . . . . 1 2 2 2 4 5 5 5 6 6 6 7 7 8 8 2 Homogeneity of cardiac contraction despite physiological depolarization 2.1 Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . 2.2 Material and Methods . . . . . . . . . . . . . . . . . . . . 2.2.1 Geometry and myofiber orientation . . . . . . . . . 2.2.2 Depolarization wave . . . . . . . . . . . . . . . . . 2.2.3 Modeling mechanical properties of cardiac tissue . 2.2.4 Modeling left ventricular mechanics . . . . . . . . 2.2.5 Numerical implementation . . . . . . . . . . . . . 2.2.6 Simulations . . . . . . . . . . . . . . . . . . . . . . 2.3 Results . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2.3.1 Depolarization wave . . . . . . . . . . . . . . . . . 2.3.2 Cardiac mechanics . . . . . . . . . . . . . . . . . . 2.4 Discussion . . . . . . . . . . . . . . . . . . . . . . . . . . . 2.4.1 Model . . . . . . . . . . . . . . . . . . . . . . . . . vii . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . asynchrony of . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11 12 13 13 13 15 17 17 18 20 20 20 24 25 viii Contents 2.4.2 Physiology . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 28 2.5 Conclusions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 29 2.6 Appendix A, model of active stress development . . . . . . . . . . . . . . . 29 3 Timing of depolarization and contraction in the paced canine left ventricle 3.1 Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3.2 Material and methods . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3.2.1 Experiments . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3.2.2 Simulations . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3.3 Results . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3.3.1 Experiments . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3.3.2 Simulations . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3.4 Discussion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3.4.1 Model setup . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3.4.2 Results . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3.5 Conclusions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 31 32 33 33 33 36 36 36 40 40 41 43 4 Regional contraction in the paced canine left ventricle 4.1 Introduction . . . . . . . . . . . . . . . . . . . . . . . 4.2 Material and methods . . . . . . . . . . . . . . . . . 4.2.1 Experiments . . . . . . . . . . . . . . . . . . . 4.2.2 Simulations . . . . . . . . . . . . . . . . . . . 4.3 Results . . . . . . . . . . . . . . . . . . . . . . . . . . 4.4 Discussion . . . . . . . . . . . . . . . . . . . . . . . . 4.4.1 Results . . . . . . . . . . . . . . . . . . . . . . 4.4.2 Limitations . . . . . . . . . . . . . . . . . . . 4.5 Conclusions . . . . . . . . . . . . . . . . . . . . . . . 45 46 46 46 47 50 54 54 55 55 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5 Intra- and interventricular asynchrony of electromechanics in the ventricularly paced heart 5.1 Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5.2 Material and methods . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5.2.1 Geometry and myofiber orientation . . . . . . . . . . . . . . . . . . 5.2.2 Depolarization and mechanics . . . . . . . . . . . . . . . . . . . . . 5.2.3 Numerical implementation . . . . . . . . . . . . . . . . . . . . . . 5.2.4 Simulations and data analysis . . . . . . . . . . . . . . . . . . . . . 5.3 Results . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5.3.1 Geometry . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5.3.2 Global behavior . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5.3.3 Regional behavior . . . . . . . . . . . . . . . . . . . . . . . . . . . 5.4 Discussion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5.4.1 Physiology . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5.4.2 Limitations . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 57 58 59 59 61 62 62 63 63 63 63 65 65 67 Contents ix 5.5 Conclusions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 68 5.6 Acknowledgements . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 68 6 General discussion 6.1 Introductory remarks . . . . . . . . . . . . . . . . 6.2 Modeling depolarization and contraction in a normally beating left ventricle . . . . . . . . . . . 6.2.1 Mathematical models . . . . . . . . . . . 6.2.2 Model design . . . . . . . . . . . . . . . . 6.2.3 Electromechanics in a normal heartbeat . 6.3 Simulating timing of depolarization during ventricular pacing . . . . . . . . . . . . . . . . . 6.4 Simulating cardiac mechanics during ventricular pacing . . . . . . . . . . . . . . . . . 6.5 A model of cardiac electromechanics in the paced left ventricle . . . . . . . . . . . . . . . . . . . . . 6.6 Heterogeneous electromechanical delay? . . . . . 6.7 Future perspectives . . . . . . . . . . . . . . . . . 6.8 General conclusions . . . . . . . . . . . . . . . . 69 . . . . . . . . . . . . . . 70 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 70 70 71 71 . . . . . . . . . . . . . . 72 . . . . . . . . . . . . . . composite of right and . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 73 73 74 74 75 Bibliography 76 Samenvatting 87 Dankwoord 89 Curriculum Vitæ 91 x Contents Summary The heart is a hollow muscular organ that pumps blood through the vascular system. Myofiber contraction is initiated by depolarization of the cell membrane. A coherent contraction of all myofibers in the cardiac walls is a requisite for an efficient pump function of the heart as a whole. In the heart, a specialized conduction system enables a fast spread of myocyte depolarization. Sometimes, normal depolarization wave propagation is disturbed, by for instance a block in the conduction system and by ventricular pacing. Abnormal ventricular depolarization patterns result in deterioration of pump function and can contribute to the development of heart failure. In the clinic, pacemakers are used to maintain a normal cardiac rhythm, but during the last decade the evidence is increasing that the abnormal depolarization and contraction patterns are a negative side effect of pacing therapy. This side effect might be reduced by choosing better pacing sites than usually employed. Mathematical models may be helpful to find the best pacing site. It is usually assumed that the time interval between depolarization and contraction is constant. However, so-called excitation-contraction coupling is complex and involves several steps. Cardiac excitation-contraction coupling is the process from electrical excitation to mechanical contraction of myofibers. Knowledge of this process is important in understanding physiology and pathophysiology of heart function. Timing of excitationcontraction (electromechanical delay), has been measured in cells, focussing on Calcium movement. Calcium is involved in electrical activity and stress development. In the whole heart, depolarization and cardiac deformation has been measured. MRI tagging has been used to map the temporal evolution of local three-dimensional strain in the entire myocardium in combination with mapping of epicardial depolarization. Knowing strain, timing of mechanical activation can however not be determined unequivocally. Mathematical models are of great help in gaining insight in electromechanical delay. Several mathematical models on cardiac electrophysiology and mechanics are described, but none have been used yet in a combination to study cardiac ventricular pacing. This thesis describes the development and the first results of a three-dimensional finite element model of left and left and right ventricular electromechanics. The model is used to investigate the electromechanical delay in the normal heart, and the relation between depolarization and contraction in the paced heart. The model is tested by comparison of simulated maps of electromechanics with experimentally obtained ones. The model is expected to become a useful tool in optimizing pacing therapy. xi xii Summary The geometry of the left and right ventricle is obtained from MR images of the canine heart. Myofiber orientation and the fast conducting Purkinje system are also incorporated in the model. The arrival time of the depolarization wave is modeled by solving the eikonal-diffusion equation, while considering effects of local anisotropy of the myofiber structure. The passive extracellular matrix of the cardiac material is assumed to be non-linearly elastic and anisotropic. Active myofiber stress depends on time, sarcomere length, and sarcomere shortening velocity. In performing a simulation of a cardiac cycle, local values are obtained of depolarization time, displacement, strain and stress. Global hemodynamics such as right and left cavity volumes, pressures, and flows are also calculated. The pattern of depolarization has been simulated realistically for the normal heart as well as for ventricular pacing. One of the most striking findings was that the time elapsed between the moment of local depolarization and the onset of contraction appeared to vary spatially so that contraction is more synchronous than depolarization. Apparently, there is a mechanism, which adapts electromechanical delay to synchronize contraction in the normal heart. When pacing the heart, asynchrony appeared much more prominent than during sinus rhythm, as found in simulation and experiments. The distribution of electromechanical delay as needed to synchronize the normal heart, was applied to the heart, paced from a site in the ventricle. As a consequence, the simulated distribution of myofiber shortening changed only minimally. Apparently, the narrow physiological range (40 ms) of electromechanical delay is too small to significantly affect asynchrony as it occurs during pacing (120 ms). Finally, intra- and interventricular asynchrony of depolarization, contraction, and pump function has been simulated realistically in a combination of left and right ventricle. The geometry used was obtained from an MR image series of a canine heart. Thus, geometry could be introduced patient-specifically. We conclude that the model of ventricular electromechanics is very succesful in assessing effects of electromechanical delay in the intact beating heart. The model is also promising in realistically simulating various types of pathology such as conduction disturbances, and the effect of proposed treatments such as pacing. Chapter 1 Introduction 1 2 Chapter 1 1.1 General introduction The heart is a hollow muscular organ that pumps blood through the vascular system for transport of oxygen and nutrients to the tissue and metabolic waste from the tissue. Cardiac pump function is generated by contraction of the myofibers in the cardiac walls. Myofiber contraction is initiated by depolarization of the outer cell membrane. A coherent contraction of all myofibers in the cardiac walls is a prerequisite for an efficient pump function of the heart as a whole. In the human heart, a specialized conduction system enables a fast spread of myocyte depolarization across the walls, such that the complete heart is depolarized within about 40-50 ms. The importance of a coherent myofiber contraction becomes apparent in pathological cases (e.g. conduction disorders) in which the time needed to depolarize the complete myocardium is increased. As a result, myofiber contraction becomes more asynchronous, which adversely affects cardiac pump function (71). In the clinic, pacemakers are used to maintain a normal cardiac rhythm, but since the pacing electrode is positioned outside the conduction system, ventricular pacing results in asynchronous depolarization and contraction of the ventricles. While the gross features of the relation between depolarization, contraction and pump function are known, several aspects remain unclear. For example, in the normal heart, contraction seems to be more synchronous than depolarization (22; 106). This finding suggests that the time delay between the moment of depolarization and the onset of contraction, from now on referred to as electromechanical delay (EM delay) varies spatially across the cardiac walls. Also, the choice of the optimal location of a pacing site and the optimal timing of pacing remains a matter of trial and error. In this thesis, a three-dimensional mathematical model of cardiac electromechanics is presented. The model is used to investigate the EM delay in the normal heart, and the relation between depolarization and contraction in the ventricularly paced heart. The model is expected to become a useful tool in optimizing pacemaker therapy. 1.2 Normal cardiac anatomy and physiology 1.2.1 Anatomy The heart consists of two pumps, located side-by-side (figure 1.1). Both sides contain an atrium and a ventricle. The atria collect blood that returns to the heart and facilitate filling of the ventricles. The right ventricle (RV) maintains the pulmonary circulation, the left ventricle (LV) the systemic circulation. The geometry of the left ventricle can be characterized by an ellipsoidal-shaped cavity encapsulated by a thick wall (82). The mitral valve prevents backflow of blood into the left atrium when the left ventricle contracts, and blood is ejected through the aortic valve. The aortic valve prevents backflow from the aorta into the left ventricle. The wall of the right ventricle is thinner and less powerful than that of the left ventricle. The right ventricle is crescent-shaped in Introduction 3 cross-section, wrapped partially around the left ventricle. The tricuspid valve prevents backflow of blood into the right atrium when the right ventricle contracts, and blood is ejected through the pulmonary valve. The pulmonary valve prevents backflow from the pulmonary artery to the right atrium. The interventricular septum separates the left and right ventricles. The outer surface of the heart, the epicardium, is smooth. The surface of the cavities, the endocardium, is irregular, showing many invaginations protruding into the wall up to 30% of its thickness. Papillary muscles originate from the endocardium that support functioning of the mitral and tricuspid valve leaflets. Figure 1.1: Cross-section of the heart, showing atria and ventricles, and the specialized conduction system. A part of the pulmonary artery is cut away. SA node: sino-atrial node, AV node: atrio-ventricular node. Adapted from Ref. (49) The heart wall is largely composed of rod-shaped myofibers. Locally these myofibers are aligned, thus defining a local myofiber orientation. Subepicardial myofibers follow a left-handed helix parallel to the wall. Near the apex, these myofibers cross the wall, and continue in a right-handed helical pathway at the subendocardium. Near the base, the myofibers cross over to the subepicardium again. At midwall the myofiber orientation is predominantly circumferential. Cardiac myocytes contain numerous myofibrils. Neighboring myofibrils have their sarcomeres aligned, which gives them a striated appearance. The sarcomere is considered to be the basic contractile unit of cardiac muscle, consisting of a threedimensional array of the contractile myofilaments actin and myosin. Myofibers have the 4 Chapter 1 ability to generate contractile force. Contraction is initiated by a steep rise in intracellular calcium concentration, caused by depolarization of the myocyte, thus encompassing EM delay. Myocytes are situated within the so-called extra-cellular matrix, which consists, among others, of networks of collagen fibers. Passive material properties of cardiac muscle are largely determined by the composition and structure of this collagen. 1.2.2 Depolarization wave propagation In a normal heart, depolarization starts in the sinoatrial (SA) node (figure 1.1). This node is a cluster of modified myofibers located beneath the endocardium of the right atrium (49). The cell membranes self-depolarize about 70 times a minute. The resulting cellular transmembrane action potential propagates as a wave over neighboring cells, thus depolarizing the atria. The atrio-ventricular (AV) node, located near the septum between the atria and the ventricles, slows down the depolarization wave. Distal to the AV node, the depolarization wave is conducted fast through the His bundle, splitting in a left and right branch. These branches bifurcate into many small branches, thus forming the fast conducting Purkinje system, located in the subendocardium of the left and right ventricle. These Purkinje fibers are insulated with connective tissue. Purkinje fibers enable a fast propagation of depolarization, typically 3-4 m/s (57). Figure 1.2: Depolarization times in the normal heart [ms]. Adapted from Durrer et al (23). Introduction 5 The Purkinje system ends in the Purkinje-muscular junctions (PMJs). Here, the depolarization wave enters the ventricular myocardium. In the myocardium, propagation is much slower (0.6 - 1.0 m/s) (27; 75; 87; 104). Within the walls, the wave propagates mainly from endocardium to epicardium (figure 1.2). From the moment of leaving the Purkinje fibers, the whole ventricular myocardium is depolarized within 40-50 ms (23). 1.2.3 Contraction During the cardiac action potential, calcium (Ca2+ ) enters the cell through depolarizationactivated calcium channels, which contribute to the action potential plateau (10). Calcium entry triggers calcium release from the sarcoplasmatic reticulum. The combination of calcium influx and release raises the free intracellular calcium concentration, allowing it to bind to the myofilament troponin C, located on the actin filament. The resulting conformational change of the actin filament exposes myosin binding sites. A bond between actin and myosin, a crossbridge, is formed when myosin heads attach to the actin binding sites. Depending on the loading of the myocyte, actin filaments slide along the myosin filaments, causing shortening of the sarcomere. The time span between moment of depolarization and moment of crossbridge formation is referred to as electromechanical (EM) delay. In the normal heart, contraction seems to be more synchronous than depolarization (22; 106). This finding suggests that EM delay varies spatially across the cardiac walls. Generated stress depends on the time after depolarization, sarcomere length, and sarcomere shortening velocity. The stress generated in the myofibers is responsible for the pressure rise in the ventricles. Once ventricular pressure exceeds end-diastolic aortic and pulmonary pressure, blood is ejected into the aorta and pulmonary artery, respectively. The amount of blood ejected is directly related to sarcomere shortening. Generally, global left ventricular pump function is characterized by a systolic pressure of about 16 kPa and ejection fraction of the cavity by 60%. 1.3 Artificial ventricular pacing 1.3.1 Global effects Sometimes, depolarization wave propagation is disturbed, for example after AV-block, resulting in a decrease of pump function. In 1957, it was discovered that by combining a pulse generator with a wire electrode attached directly to the heart of a dog, heart rate could be controlled (101). Soon after that, a pacemaker system was applied successfully to patients with a conduction block. During the next three decades of clinical pacing much attention has been paid to proper thresholds (sensing and stimulation) and proper synchronization between atria and ventricles (71). In 2000, more than 150,000 pacemakers were implanted in the U.S. alone (4). Recently, interest in the sequence of ventricular depolarization is growing, because of increasing evidence that chronic pacing 6 Chapter 1 at the conventional pacing site, the right ventricular apex, is associated with decreasing pump function (68), a change in myocardial tissue structure (64), and contributes to heart failure (5; 105). A significant number of studies indicate that the pacing site influences LV pump function. For example, pacing the LV leads in general to better pump function than pacing the RV (for review see Ref. (71)). However, the mechanism why pacing from one site results in better pump function than pacing from another is not clear. 1.3.2 Abnormal patterns of depolarization and contraction During ventricular pacing, the depolarization wave propagates considerably slower and less uniform (68; 94; 99) than normal. Depolarization starts by slow propagation through the myocardial tissue near the pacing site. When reaching the subendocardium, depolarization spreads faster (27). Finally, the epicardium of the wall remote from the pacing site is reached generally about 120 to 160 ms after the moment of pacing (94). As a result, the distribution of local amplitude and time course of contraction is considerably affected (69; 99). Generally, during the isovolumic contraction phase, myofibers shorten rapidly in the early-depolarized regions, and are pre-stretched in the late depolarized regions. During ejection, early-depolarized myofibers shorten minimally or may even be stretched. Late depolarized myofibers shorten often twice as much than in the normal heartbeat. The contraction pattern is found to vary systematically with increasing distance from the pacing site (22; 55; 69). 1.4 Cardiac excitation-contraction coupling 1.4.1 Excitation-contraction from cellular to whole heart level Cardiac excitation-contraction coupling is the process from depolarization to contraction of the myocyte (10). Knowledge of this process is important in understanding physiology and pathophysiology of heart function. Calcium is considered an important ion in excitation-contraction coupling. Calcium is involved in both electrical activity and active stress development. Timing of excitation-contraction coupling on the cellular level influences cardiac pump function on the whole heart level. On the cellular level, many measurements have been performed on calcium movement (for an overview, see (10)). At the subepicardium of the intact heart, calcium transients and action potentials were simultaneously measured ex vivo in myocytes by mapping with fluorescent indicators (45). In the entire heart however, these measurements have not been performed yet. Instead, depolarization and cardiac deformation have been measured. Introduction 7 1.4.2 Cardiac electromechanical mapping Near simultaneous measurements of epicardial depolarization and epicardial deformation have been performed during ventricular pacing in dogs using electrode-brushes and video-recordings of optical markers (22), applied to the anterior LV. More recently, MRI tagging (8) has been used to map the temporal evolution of local three-dimensional strain in the entire myocardium in combination with epicardial mapping of depolarization with a multi-electrode sock (26; 106). The moment of electrical activation was defined as the moment halfway of the steep upstroke of the depolarization wave (depolarization occurs generally within 2-3 ms). Time of mechanical activation was defined as the (measurable) moment of onset of circumferential shortening (106) instead of (non-measurable) onset of contraction. However, the relation between onset of contraction and onset of shortening is not unique. This can be illustrated by considering two myofibers, in which crossbridge formation starts simultaneously. Then, the moment of onset of shortening for those myofibers can be very different, since it depends strongly on the force, experienced by those myofibers from the neighboring tissue. Currently, electromechanical delay, from the moments of excitation to contraction, cannot be measured in the entire myocardium. Mathematical models are likely to be of great help in the role of local electromechanical delay and its effect on the cardiac contraction pattern. 1.4.3 Modeling cardiac electromechanics Several mathematical models on cardiac electrophysiology and mechanics have been designed. Mathematical models of cardiac electrophysiology have been used in a variety of applications. They have been developed to investigate cardiac electrophysiological properties (33), the effect of geometry and myofiber orientation on propagation (39; 19; 18), the effect of the Purkinje system on myocardial propagation (18; 88), and to gain insight in several conduction disturbances (9; 47; 107). Mathematical models of cardiac mechanics (6; 11; 20; 37; 86; 90) have been developed to understand the distribution of myofiber stress and strain. This stress cannot be measured accurately, because a force transducer damages the tissue (36). However, knowledge of distribution of myofiber stress and strain is important, since the local stress-strain loop is linearly related to local oxygen consumption (83). Regions with a higher workload than average are likely to be more susceptible to reduction in blood supply, which would be a risk factor for the development of ischemic heart disease. Therefore, models of cardiac mechanics have been used to investigate the effect of myofiber orientation (13; 74; 92) and regional ischemia (12; 54; 85) on myofiber stress and strain. Furthermore, such models have been used to investigate the relation between timing of depolarization and contraction (84; 90) for a normal heart beat, but not yet for ventricular pacing. Also, in the latter models, a homogeneous distribution of EM delay was assumed. 8 Chapter 1 1.5 Aim of the study As mentioned above, there is an increasing need to understand the influence of the site of ventricular pacing for ventricular pump function. This understanding requires the knowledge of propagation of depolarization, the coupling between excitation and contraction, and the importance of coherence of contraction for total pump function. The aim of the present study was to investigate excitation-contraction coupling in the intact heart under normal and ventricular pacing conditions. This was achieved by developing and using a mathematical model of cardiac electromechanics. The mathematical model is used to provide information which cannot be measured (e.g. onset of contraction) and is a flexible tool for optimizing the selection of sites for pacing. The model was tested by comparison of simulations and experimental data of depolarization times and myofiber strain time courses. 1.6 Outline of the thesis In chapter 2 the three-dimensional model of normal LV electromechanics is presented. A finite element method is used to obtain approximate solutions of the equations used. In the unloaded reference state, endocardial and epicardial surfaces of the LV are represented by truncated confocal ellipsoids. Depolarization wave propagation is modeled using the eikonal diffusion equation (19). This equation solves the arrival time of the depolarization wave as a function of space, allowing for effects of anisotropy of wave propagation. Wall mechanics are determined by solving the equations of force equilibrium. Myocardial material is considered anisotropic and non-linearly elastic. Active mechanical properties depend on time, sarcomere length, and sarcomere shortening velocity. The model is used to investigate EM delay in the normal heart. In chapter 3 it is investigated whether depolarization during ventricular pacing can also be modeled with the eikonal-diffusion equation. Simulated patterns of moment of depolarization in the canine LV are matched to the measurement for pacing at the left ventricular free wall. By inverse analysis, propagation properties of the depolarization wave in the LV wall are estimated. With the thus obtained parameters, simulations of electromechanics of pacing at the right ventricular apex are performed. Simulated maps of timing of epicardial depolarization and onset of shortening are compared to measurements, obtained at the epicardial surface of the canine LV. In chapter 4 it is investigated whether, and to what extent, heterogeneous EM-delay, as it occurs during a normal heartbeat, affects regional contraction and total pump function during RVA pacing. The slope from a linear regression analysis of the relation between local depolarization time and local normalized circumferential strain is used as a measure of mechanical synchrony. The thus calculated slope is compared with experimental results, obtained in a MRI-tagging experiment. In chapter 5 a comprehensive finite element model of left and right ventricular electromechanics is presented. The realistic geometry of the heart is fitted on a Introduction 9 set of canine cine-MR short axis images. Focusing on interventricular asynchrony of hemodynamics and motion, simulations of complete cardiac cycles are performed for a normal heartbeat with synchronous mechanical activation and ventricular pacing. Pacing is simulated, while starting depolarization from the right ventricular apex or left ventricular free wall. The model is considered a first step towards patient-specific modeling. 10 Chapter 1 Chapter 2 Homogeneity of cardiac contraction despite physiological asynchrony of depolarization The contents of this chapter are published in the Annals of Biomedical Engineering, 31(5), 536-547, 2003: Homogeneity of cardiac contraction despite physiological asynchrony of depolarization: a model study R.C.P. Kerckhoffs, P.H.M. Bovendeerd, J.C.S. Kotte, F.W. Prinzen, K. Smits, T. Arts 11 12 2.1 Chapter 2 Introduction The normal heart beat is initiated by spontaneous electrical depolarization of the sinus node in the right atrium. The resulting depolarization wave propagates across both atria, passes the atrioventricular node and ventricular conduction system, and finally depolarizes both ventricles. Depolarization is followed by contraction of the myofibers. The normal contraction pattern of the left ventricle (LV) may be affected by underperfusion (2; 28; 52; 98) and abnormal conduction of the depolarization wave (69; 99). With the rapid evolution of methods to determine myocardial deformation noninvasively, deduction of pathology from such measurements is a challenge. Size and location of ischemic regions were assessed by analysis of cardiac images with the help of mathematical models of cardiac mechanics (1; 89). A next interesting challenge is to predict the pattern of contraction from a known pattern of electrical depolarization, or, vice versa, to deduce the sequence of electrical depolarization from non-invasively measured mechanical deformation (106). Understanding of electromechanical interaction becomes especially relevant because of the rapidly increasing interest in cardiac resynchronization. We postulate that mathematical models, which combine wave propagation and wall mechanics, may provide new insights in interpreting cardiac deformation towards cardiac pathology. There are several models on myocardial electrophysiology (for an overview, see (56)) and mechanics (11; 58; 86; 91) separately, but the behaviour of the combination of the two principles in an entire ventricle is not well known. It is known that timing of mechanical contraction determines the distribution of strain. It is also known that this timing is mainly determined by the pattern of depolarization. However, it has not been thoroughly investigated yet whether the combination of both apparently known principles into a comprehensive electromechanical model will result in a realistic pattern of contraction. In the present study, therefore, such combination of models has been designed. Simulations were assessed, focusing on the pattern of deformation. From possible discrepancies with experimental results, characteristics of electromechanical coupling were further unraveled. In designing the model of left ventricular electromechanics, depolarization wave propagation was modeled using the eikonal diffusion equation (19). This equation solves the arrival time of the depolarization wave as a function of space, allowing for effects of anisotropic wave propagation and wave front curvature. The method was preferred over the well-known bidomain model (33) because of computational efficiency. The solutions of the eikonal diffusion equation and the bidomain model were reported to match closely (17). Left ventricular electromechanics were modeled in a 3D finite element mesh. Wall mechanics were solved by equations of force equilibrium. Myocardial material was considered anisotropic, non-linearly elastic and time dependent (7; 37; 86). The aortic input impedance was simulated by a three-element windkessel model (103). First a cardiac contraction cycle was simulated for a regular pattern of depolarization. Homogeneity of cardiac contraction despite physiological asynchrony of depolarization 13 In particular, we assumed that the delay between depolarization and the onset of crossbridge formation was the same for all myofibers. Simulated results on midwall myofiber strain, epicardial myofiber strain, and the distribution of external work were compared with reported experimental findings (21; 32; 69; 70; 106). The sensitivity of wall mechanics to timing of depolarization was investigated with the help of a simulation where effects of time differences were excluded by synchronous depolarization of all myofibers. 2.2 Material and Methods The mathematical model of LV electromechanics has been designed in three modules, describing 1) LV geometry and myofiber orientation, 2) depolarization wave propagation, and 3) LV wall mechanics. 2.2.1 Geometry and myofiber orientation In the reference state, defined by zero transmural pressure, the canine LV wall was represented by a thickwalled truncated ellipsoid (11). Volumes of LV wall, cavity, and papillary muscles were 140 ml, 40 ml, and 4 ml, respectively. Local myofiber orientation was quantified by helix angle h , and the transverse angle t . The helix angle spans the local circumferential direction and the projection of the myofiber orientation on the plane parallel to the wall. The transverse angle spans the local circumferential direction and the projection of the myofiber orientation on the plane perpendicular to the local longitudinal direction. Myofiber orientations were obtained by optimizing for homogeneous myofiber shortening during ejection (74). Thus, the transmural courses of h and t varied throughout the wall according to figure 2.1. 2.2.2 Depolarization wave For the moment of depolarization tdep within the wall the eikonal-diffusion equation ~ tdep ): (rewritten from (19)) was solved for the gradient of tdep (r q ~ tdep M r ~ tdep cf r ~ (M r ~ tdep ) = 1 k0 r (2.1) Parameter cf (0.67 m=s) represents the velocity of the depolarization wave along the myofiber direction. Constant k0 (2:1 10 4 m2 s 1 ) determines the influence of wave front curvature on wave velocity. Dimensionless tensor M describes anisotropy of wave propagation in the global coordinate system. Tensor M is referred to a local coordinate system, aligned with the myofiber, and is related to M by: M = RM RT (2.2) 14 Chapter 2 80 Near apex 60 Base Angle [o] 40 Equator 20 Base 0 Equator −20 Near apex −40 −60 −1 0 <− Endocardium 1 Epicardium −> Figure 2.1: Myofiber orientation as a function of transmural position at several longitudinal positions; the helix (- -) angle and transverse angle (–) are components of myofiber orientation, as defined in the text. Rotation tensor R is associated with the myofiber orientation; index T indicates the transpose. The largest principal component along the myofiber direction is m11 = 1. The other principal components are m22 = m33 = 0:38, indicating that the wave front velocity p perpendicular to the myofiber direction (ct = cf m22 = 0:42 m=s) is slower. Parameters cf , ct , and k0 were derived from (88). To simulate the effect of the fast conducting Purkinje fibers, parameter cf had a 6-fold value in the subendocardial layer between apex and equator (figure 2.2). With the assumption that the LV is electrically insulated, the wave fronts are perpendicular to the boundary ext , being the basal, endocardial, and epicardial surface of the cardiac wall: ~ tdep = 0 at ext ~n M r (2.3) Vector ~n represents the orthogonal on the boundary ext . Normally, depolarization was assumed to start (tdep =0) simultaneously at four locations stim (figure 2.2) in the LV. Three locations are situated at the LV subendocardium (23) and one location at the RV subendocardium (24): tdep = 0 at stim (2.4) Homogeneity of cardiac contraction despite physiological asynchrony of depolarization 15 Figure 2.2: Distribution of depolarization wave velocity cf [m=s] parallel to the myofiber direction. In most of the myocardium cf = 0:67 m=s. At the subendocardium between apex and equator cf = 4 m=s with a narrow, smooth transition zone around.The small circular regions represent the regions of initiation of ventricular depolarization, as indicated by stim in equation 2.4. 2.2.3 Modeling mechanical properties of cardiac tissue The mechanical behavior of the cardiac tissue depended on time as expressed by the constitutive equations. Total Cauchy stress was composed of a passive ( p ) and an active component (a ) along the myofiber direction ~ef : = p + a~ef ~ef Passive tissue stress p was related to the deformation gradient tensor Green Lagrange strain tensor E by: (2.5) F and the 1 F @Wp F T with E = 1 (F T F I ) (2.6) det(F ) @E 2 Symbol I represents the identity tensor. Wp represents the deformation energy density as a function of strain E , being composed of an isotropic component Wi , related p = 16 Chapter 2 to tissue shape change, a component Wf , related to the extra stiffness of the material in the myofiber direction, and a component Wv , related to volume change: Wp = Wi + Wf + Wv (2.7) with Wi = a0 (ea1 I1 +a2 I2 2 1) (2.8) Wf = a3 (ea4 Ef 1) (2.9) Wv = a5 [det(F T F ) 1]2 (2.10) I1 and I2 represent the first and second invariants of E , respectively (51). Material parameter values a0 through a5 are listed in table 2.1. Ef represents the Green Lagrange strain component along the myofiber direction: 2 1 l Ef = [( s )2 2 ls0 1] (2.11) with actual sarcomere length ls , and sarcomere length in the reference state ls0 = 1:9 m. This strain energy formulation is a slight modification of the one proposed earlier (73), i.e. the term a4 Ef2 was removed from the exponent in Wi , and put in a separate term Wf for a better description of the increasing passive stiffness for large sarcomere lengths (37). a0 a1 a2 a3 kPa kPa 0.5 3.0 6.0 0.01 a4 a5 kPa 60 55 Table 2.1: Passive material tissue properties, based on uni- and bi-axial measurements of myocardial tissue slabs (60) and on the pressure-volume relationship of passive inflation of the LV (59). The characteristics of active stress a were modeled using a contractile element, with length lc , in series with an elastic element, with length ls lc (7). The magnitude of a was described by a = f1 (ta ; ls ; lc ) (2.12) where ta represents time elapsed since depolarization. The time course of the lc is described by a first order differential equation: Homogeneity of cardiac contraction despite physiological asynchrony of depolarization @lc = f2 (ls @t lc ) 17 (2.13) The used functions f1 and f2 have been described in the Appendix. 2.2.4 Modeling left ventricular mechanics Within the cardiac wall the equation of conservation of momentum was used while neglecting volumetric and inertial forces: r~ = ~0 (2.14) Pressure loads on the endocardium and epicardium were homogeneous, and equal to LV cavity pressure plv and zero, respectively: ~n ~n = 0 at the epicardium ~n ~n = plv at the endocardium (2.15) (2.16) To prevent rigid body motion, motion in the base to apex direction was set to zero at the base. In 3 points (anterior, posterior, and lateral) at the basal endocardium circumferential motion was also set to zero. LV mitral inflow was simulated by quasi-static increase of pressure in the nonactivated LV from 0 to 1 kPa in pressure steps of 0.05 kPa. LV pressure in the isovolumic contraction and relaxation phases was estimated (11) such that LV cavity volume remained constant within 0.5%. Aortic flow and pressure were related by a realistic three-element Windkessel model (103), being composed of a flow resistance (1.5 107 P a s m 3 ) in series with a compliance (1.0 10 9 m3 P a 1 ) that was parallel to the peripheral flow resistance (1.4 108 P a s m 3 ). The aortic valve opened when LV pressure exceeded aortic pressure, being set at 10 kPa. Reversal of aortic flow closed the valve. 2.2.5 Numerical implementation The eikonal-diffusion equation with boundary conditions (Eq. 2.1-2.4) was solved using a Galerkin type finite element method with 8-noded hexahedral elements with trilinear interpolation. The LV wall was subdivided into 9984 elements, with 11037 degrees of freedom. This resulted in a mean spatial resolution of about 2 mm. The solution was facilitated by gradual increase of the non-linear term in each succesive iteration (3; 88). A classical upwind scheme (Streamline Upwind Petrov Galerkin, (14)) was used to stabilize the finite element calculations (18). 18 Chapter 2 The equations related to mechanics (Eq. 2.14-2.16) were solved using a Galerkin type finite element method with 27-noded hexahedral elements with triquadratic interpolation. The LV wall was subdivided into 108 elements, with 3213 degrees of freedom. A Newton-Raphson iterative procedure was used, and a Newton-Cotes integration scheme (43) was used with the integration points at the node positions. All equations were solved on a 64-bit Origin 200 computer (SGI, Mountain View, CA, USA), using a single processor at 225 MHz. The finite element calculations were performed with the FORTRAN77 compiler based package SEPRAN (SEPRA, Leidschendam, the Netherlands) on a UNIX platform. 2.2.6 Simulations Two simulations were performed, one in which contraction was initiated according to the regular electrical depolarization pattern (NORM simulation) with a constant electromechanical delay of 0 ms for all myofibers and another in which contraction was unphysiologically synchronously initiated (SYNC simulation). Global hemodynamics, three-dimensional distributions of electrical depolarization time, myofiber stress and strain as a function of time, and stroke work density were calculated. Sarcomere length ls at the anterior and posterior midwall and epicardium was calculated as a function of left ventricular volume Vlv , normalized to wall volume Vw . The ratio ls =(Vlv =Vw ) during ejection was estimated by linear regression. Natural myofiber strain f was defined as: l f = ln( s ) ls0 (2.17) Myofiber strain during isovolumic contraction and ejection f;ic and f;ej , respectively, were defined as: f;ic = f;eic f;bic f;ej = f;eej f;eic (2.18) (2.19) where f;bic , f;eic , and f;eej are shown in figure 2.3. Stroke work density Wf [J m 3 ] was defined as Wf = I cardiac cycle f df (2.20) where f is the total Cauchy myofiber stress in the tissue. The effect of restricting wall motion at the base has been investigated by performing a simulation BOUN with an additional restriction of radial motion at the endocardial Homogeneity of cardiac contraction despite physiological asynchrony of depolarization 19 0.25 ε f,eic ε f,bic ε f,eej 0.2 Myofiber strain εf 0.15 0.1 0.05 0 −0.05 fill −0.1 0 ic ej 200 400 Time [ms] ir 600 Figure 2.3: Myofiber strain f during the cardiac cycle. fill filling; ic isovolumic contraction; ej ejection; ir isovolumic relaxation; f;bic myofiber strain at beginning of isovolumic contraction; f;eic myofiber strain at end of isovolumic contraction; f;eej myofiber strain at end of ejection. basal contour. The effect of heterogeneity in the distribution of passive stiffness has been investigated in simulation HETER, having a gradient in parameter a0 (table 2.1) according to figure 2.4, as based on measurements reported earlier (60). The effect of having a transmural gradient in end-diastolic sarcomere length has been investigated in simulation LS0, where initial sarcomere length ls0 (eq. 2.11) varied linearly from 1.84 m at the endocardium to 1.96 m at the epicardium (76). In the simulations BOUN, HETER, and LS0, myofiber contraction was initiated by the depolarization pattern, as calculated in the NORM simulation. The differences of the test simulations with the NORM simulation were expressed in the square root of the volume-weighted mean of the squared differences, as normalized to the volume-weighted mean value (RMS%). To assess whether meshing was sufficiently fine, a simulation of depolarization wave propagation was performed with four times as many elements, resulting in a mean resolution of 1.4 mm. In the transmural direction, where the largest gradients were found, resolution was 0.8 mm. Similarly, for the mechanics, the number of elements was doubled. 20 Chapter 2 0.65 0.55 0 Parameter a [kPa] 0.6 septum 0.5 anterior/posterior 0.45 0.4 −1 lateral −0.5 0 0.5 <−− Endocardium Epicardium −−> 1 Figure 2.4: Distribution of parameter a0 (kPa, table 2.1) in the HETER simulation with heterogeneous passive material properties. 2.3 Results 2.3.1 Depolarization wave For solving the eikonal-diffusion equation calculation time was approximately 4 hours. In figure 2.5 the calculated regular depolarization pattern has been mapped. This map was used to initiate contraction in the NORM simulation, with constant electromechanical delay. The wave started in the 4 regions, shown in figure 2.2, and propagated to apex and base, and from endocardium to epicardium. After 20 ms the epicardium at the left ventricular free wall was reached. The epicardial free wall near the base was depolarized last at 43 ms. 2.3.2 Cardiac mechanics Calculation time for solving the equations related to mechanics was approximately 10 hours. Homogeneity of cardiac contraction despite physiological asynchrony of depolarization 21 Figure 2.5: Map of depolarization times [ms] as simulated for a regular heart beat. Depolarization starts at tdep =0. The left panel represents a cross-section of the septum. The right panel represents a view of the left ventricle with a 90o section removed, exposing a cross-section of the LV free wall and the lateral endocardium. Global hemodynamics In both the NORM and SYNC simulations hemodynamics were similar (figure 2.6, table 2.2). In the NORM simulation depolarization pressure developed about 22 ms later than in the SYNC simulation. This difference can be explained by the fact that in the SYNC simulation contraction started at tdep = 0, whereas in the NORM simulation starting time was distributed over the time interval between 0 to 43 ms. Local mechanics Myofiber strains - In the NORM simulation early depolarized myofibers shortened during the isovolumic contraction phase, while late activated myofibers were prestretched (figure 2.7; figure 2.8, left panel). Both during isovolumic contraction and during ejection phase myofiber shortening was more homogeneously distributed in the SYNC simulation than in the NORM simulation (table 2.3). Myofiber stresses - The distribution of myofiber stress is quite insensitive to the pattern of depolarization (figure 2.8). After a change in depolarization sequence, during the ejection phase relative changes in myofiber stress were considerably smaller than these changes in myofiber strain. Stroke work density - The area of a workloop (figure 2.8, right panel) represents work per unit of tissue volume, or stroke work density (figure 2.9). In the NORM simulation stroke work density in the early-depolarized regions was lowest and gradually increased towards the late-depolarized regions. Comparing the NORM with the SYNC simulation, mean stroke work density appeared similar (figure 2.8). In the NORM 22 Chapter 2 15 plv [kPa] 18 10 16 5 14 0 80 Vlv [ml] plv [kPa] 12 10 60 8 40 6 0.4 qao [l/s] 4 0.2 0 0 2 0 500 Time [ms] 40 60 80 Vlv [ml] Figure 2.6: Global hemodynamics in simulation NORM (- -) and SYNC (–). The left panel represents from top to bottom the left ventricular pressure plv , cavity volume Vlv , and aortic flow qao as a function of time. Dots indicate moments of opening and closure of the valves. The right panel represents the pressure-volume loops. simulation however, stoke work density was much more inhomogeneous than in the SYNC simulation. The mean values of myofiber strain, stress, and work in table 2.3 were calculated in the free wall, transmurally from 1/4 to 3/4 from base to apex. In table 2.4, for the NORM and SYNC simulation the change in sarcomere length per change in cavity volume is shown for various locations in the left ventricle. For comparison, similar data MEAS were obtained from measurements reported on canine hearts (32). For both SYNC and MEAS, myofiber shortening at the epicardium was slightly less than at midwall. Differences between anterior and posterior locations were not significant. For the NORM simulation, myofibers in the epicardium shorten more than at midwall. Effects of transmural redistribution of initial sarcomere length in the LS0 simulation, of different boundary conditions in the BOUN simulation, and of passive stiffness in the HETER simulation were similar (table 2.5). Myofiber shortening during isovolumic Homogeneity of cardiac contraction despite physiological asynchrony of depolarization tic tej plv;max dp=dtmax qao;max Vlv;be =Vw Vlv;ee =Vw Vs EF ms ms kPa kPa/s ml/s ml % NORM 60 196 17.7 370 398 0.60 0.23 48.6 60.9 23 SYNC 44 194 17.8 332 409 0.60 0.23 48.7 61.4 Table 2.2: Hemodynamic variables in the NORM (with regular depolarization) and SYNC (with synchronous depolarization) simulations; tic duration of isovolumic contraction phase; tej duration of ejection phase; plv;max maximum LV pressure; dp=dtmax maximum first time derivative of LV pressure; qao;max maximum aorta flow; Vlv;be and Vlv;ee LV cavity volume at beginning and end of the ejection phase, respectively; Vw LV wall volume; Vs stroke volume; EF ejection fraction Equator Wf f;ej f;ic f;ej Epi f;ic f;ej kJ m kPa - 3 NORM 4.45 1.6 22.9 3 -0.021 0.048 -0.138 0.043 0.052 0.026 -0.190 0.027 SYNC 4.84 0.5 23.8 2 -0.015 0.009 -0.146 0.010 0.000 0.004 -0.135 0.004 MEAS -0.034 0.017 -0.098 0.012 Table 2.3: Myofiber work, stress, and strain (mean sd.) in the transmural equatorial region (Equator) and at the epicardium only (Epi) in the free wall for both the NORM and SYNC simulations and measurements (21): NORM simulation with regular depolarization pattern; SYNC simulation with synchronous depolarization; MEAS epicardial measurements; Wf myofiber regional external work; f;ej myofiber stress during ejection; f;ic myofiber strain during isovolumic contraction; f;ej myofiber strain during ejection. contraction was affected, but myofiber stress and strain during ejection did not change by more than 8.5%. For the depolarization wave, mesh refinement resulted in a root mean square value (RMS) of the change in calculated depolarization time of 1.4 ms. Mesh refinement in solving mechanics changed myofiber strain and stress by less than 0.4% and 0.1%, respectively, during the ejection phase. 24 Chapter 2 Figure 2.7: Distribution of myofiber strain during the isovolumic contraction phase (strain at the end minus strain at the beginning of the isovolumic contraction phase) for the NORM simulation (left) and the SYNC simulation (right) in an anterior view of the LV with septum on the left, and free wall on the right side. Notice that in this figure early shortening and prestretch occur as negative and positive strains, respectively. The circular dots indicate positions in the LV free wall, for which myofiber stress and strain are plotted in figure 2.8. Epicardium, anterior Epicardium, posterior Midwall, anterior Midwall, posterior NORM 1.0935 0.006 1.1725 0.009 0.6562 0.0017 0.7229 0.0031 SYNC 0.7800 0.006 0.7815 0.006 0.8423 0.002 0.8476 0.003 MEAS 0.653 0.1 0.743 0.05 0.797 0.13 0.868 0.09 Table 2.4: Sarcomere shortening ls per ejected volume Vlv , normalized to wall volume Vw , after linear regression ls =(Vlv =Vw ) [m] for the NORM and SYNC simulation (with a 95% confidence interval); MEAS measurements of Ref. (32). 2.4 Discussion Simulations with our model predict that a physiological sequence of depolarization results in an unphysiologically non-uniform contraction pattern. Making the sequence of depolarization unphysiologically synchronous, the simulated contraction pattern appears physiologic. First we will discuss to what extent this discrepancy might be Homogeneity of cardiac contraction despite physiological asynchrony of depolarization a) Myofiber strain c) Workloops 8 16 33 4 12 29 Myofiber stress 4 10 25 40 40 30 30 20 20 10 10 Myofiber strain 0.15 b) Myofiber stress [kPa] Epicardium Myofiber stress Endocardium 0.1 0.05 0 −0.05 0 500 Time [ms] 25 0 0 500 Time [ms] 0 0 0.15 Myofiber strain Figure 2.8: a) Myofiber strain as a function of time for 3 positions transmurally and 3 positions from base (top) to apex (bottom). Plots were obtained for the NORM simulation (-) and the SYNC simulation (–). The circles in the curves indicate moments of opening and closure of cardiac valves. The printed numbers are the electrical depolarization times [ms] for the NORM simulation; b) Myofiber stress as a function of time for the same positions and simulations as in a); c) Myofiber stress-strain loops for the same positions and simulations as in a). attributed to the simplifying assumptions in the mathematical model. Subsequently, it will be discussed if the findings can be explained by physiological processes, not yet implemented in the model. 2.4.1 Model The LV geometry was simplified to a prolate ellipsoid. Near the base, the ellipsoid is truncated, thus requiring appropriate boundary conditions for motion of the wall. In the NORM and SYNC simulations rotational motion was prohibited at the endocardial basal contour, while radial motion of the base was allowed. Due to these boundary conditions, the wall tended to orient perpendicularly to the basal plane. This resulted in 26 Chapter 2 Figure 2.9: Stroke work density [kJ m 3 ] for the NORM and SYNC simulations in an anterior view of the LV with septum on the left, and free wall on the right side. f;ej BOUN HETER LS0 kPa 22.2 2.6 22.9 2.4 23.0 2.4 RMS% % 4.4 5.8 5.1 f;ic -0.016 0.047 -0.023 0.048 -0.023 0.044 RMS% % 28.4 8.5 21.7 f;ej -0.149 0.042 -0.138 0.044 -0.142 0.053 RMS% % 8.1 1.31 8.5 Table 2.5: Myofiber stress (mean sd.) during ejection (f;ej ) and myofiber strain (mean sd.) during isovolumic contraction (f;ic) and ejection (f;ej ) for the test simulations BOUN, HETER, and LS0. All simulations were performed with a regular depolarization pattern (NORM). The differences with the NORM simulation were expressed in the root mean square (RMS%), relative to the volume-weighted means of the NORM simulation. some prestretching at the base in the SYNC simulation (figure 2.7). To estimate the effect of constraining motion of the base, the results of the NORM simulation were compared to that of a simulation BOUN by which radial motion at the base was also prohibited. The distribution of stress and strain was severely affected near the base but much less in other regions of the LV. During ejection, below the equator a change of the boundary condition at the base affects both myofiber stress and myofiber strain by 8.1%. In simulations, LV wall mechanics appear sensitive to the choice of myofiber Homogeneity of cardiac contraction despite physiological asynchrony of depolarization 27 orientation (13). In view of a limited accuracy of available experimental data and the presence of biological variation, myofiber orientation is obtained by optimization on a homogeneous distribution of myofiber stress in the wall (74). The used distribution of myofiber orientation is within the range of reported anatomical measurements (29; 82). The effect of fast propagation in the Purkinje system has been modeled in a continuum approach, characterized by a high depolarization wave velocity near the endocardium. More accurate descriptions cannot be made easily, because biological variance in the Purkinje system is considerable (53; 57; 93). Our description has been based on an average of experimental data (23; 24; 80; 79). Normally, for a detailed description of depolarization wave propagation, a resolution is needed of about 0.2 mm, especially near strong wave front curvatures. For the conditions of our simulations, a 2 mm resolution appeared sufficient. Mesh refinement did not change the solution significantly. Passive myocardial tissue is orthotropic due to the sheet structure of the myocardium (46). Sheets have less influence on deformation in systole than in diastole (91). Focusing on systole, the sheet structure has been neglected by considering passive myocardial tissue to be transversely isotropic. Active material was modeled uniaxially. An active component of transverse stiffness is shown to exist (48), but appears not to affect the pattern of early myofiber shortening and prestretching as shown in a model study (90). We assumed homogeneneity of material properties throughout the LV wall. However, some studies indicate heterogeneity of passive material properties (60). In the HETER simulation, myofiber strain and stress during the isovolumic and ejection phase seems practically not affected by the introduced heterogeneity of material properties (table 2.5). Residual stress and strain (63) might affect systolic stresses through a transmural variation of resting sarcomere length (76). Such a variation affected myofiber strain and stress in systole in the LS0 simulation (table 2.5), but did not affect the pattern of early shortening and prestretch, as observed in the NORM simulation Muscular tissue is about as incompressible as water, as shown by the high wave velocity for ultrasound (1550-1650 m s 1 , (102)). During a cardiac cycle, total tissue volume may change by no more than 3% (97; 108) due to shifts of coronary blood volume. We set compressibility parameter a5 in equation 2.10 to 55 kPa, thus limiting tissue volume changes during the cardiac cycle to about 3%. The real cardiac geometry with a left and right ventricle is much more complex than the truncated ellipsoid we used. The load the RV exerts on the LV wall through RV pressure and force transmission in the LV-RV attachment were neglected. As RV pressure develops earlier than LV pressure, timing of stretch in the LV wall might be affected. However, the difference in timing of pressure (0-15 ms, (96)) is small with respect to the range of depolarization times during sinus rhythm. Also, due to the confocal description of the LV geometry, the apex is too thick. To minimize the influence of the right ventricle, apical region, and boundary conditions, we focused on deformation in the equatorial region of the LV free wall. It is unlikely that the used LV geometric imperfections have a significant impact on timing of contraction in this part of the left ventricle. In summary, inaccuracies introduced by the assumptions made in the model may 28 Chapter 2 influence the pattern of myocardial contraction. None of these imperfections can however explain the differences in time course of systolic myofiber strain as found in comparing the NORM and SYNC simulation. 2.4.2 Physiology In the NORM simulation propagation of depolarization was physiological (figure 2.5) in the following aspects. The LV depolarizes within 40-55 ms (24; 23; 71). Depolarization starts at the lower third of the LV endocardium, propagating to apex and base (57). The LV free wall and septum depolarized practically simultaneously (79). Complete depolarization of the left, and right septal endocardium occurs within 21 and 40 ms, respectively (24). The LV free wall is activated transmurally within 15-18 ms (80). Despite the similarity in depolarization, strain patterns are significantly different between the NORM simulation and experiments. In experiments with sinus rhythm or atrial pacing, where depolarization is similar to that of the NORM simulation, systolic strains (figure 2.8, table 2.3, and table 2.4) and stroke work (figure 2.9) have been shown to be distributed evenly throughout the ventricular wall (21; 32; 70; 106). Experimental results are not different from the results of the SYNC simulation on sarcomere shortening (table 2.4) during ejection. Myofiber strains during isovolumic contraction and ejection in the SYNC simulation are closer to measured strains (table 2.3), than myofiber strains in the NORM simulation. In the NORM simulation, however, the considerable differences in systolic strain pattern (figure 2.8) closely resembles the pattern as found in experiments during ventricular pacing (106), a situation of asynchronous depolarization. Under these circumstances, in early systole myofibers shorten in the early-activated regions (figure 2.8, endocardium) and stretch in the late activated regions (figure 2.8, epicardium). Furthermore, both myofiber strain and stroke work are low in the early-activated regions, and high in the late activated regions. Exactly, these characteristics are also found in the mechanics of the NORM simulation. Thus our model predicts an unphysiological non-uniform contraction pattern during a physiological pattern of depolarization, and a physiological contraction pattern during an unphysiological synchronous depolarization. This finding suggests that myocardial tissue is able to synchronize contraction despite asynchrony of depolarization. It is not known yet what mechanism may be responsible for this behavior. Probably, there is a local controlling mechanism that makes contraction more synchronous than depolarization by proper variation of local electro-mechanical delay time. Actually, the SYNC simulation can be interpreted as a simulation with a normal depolarization pattern and a heterogeneous distribution of electro-mechanical delay times, which completely compensates for asynchrony of local depolarization. Until now there is no direct experimental evidence for regional differences in electromechanical delay. However, the finding that expression of the Ito -channel is higher in the subepicardial layers than in the subendocardial layers (77) may reflect the Homogeneity of cardiac contraction despite physiological asynchrony of depolarization 29 physiology of such mechanism. The Ito channel has a prominent role in modulating the systolic calcium transient by enhancing the rise of the intra- cellular calcium concentration. As a result, the time elapsed between depolarization and contraction may be shorter in the subepicardial layers than in the subendocardial ones. Instead of phenomenologically adding electro-mechanical delay times, incorporating a ion-based model of heterogeneously distributed action potential (30; 66) into our model may yield better results also. However, computation time would increase dramatically, losing the benefit of shorter computation time by using the eikonal-diffusion equation. 2.5 Conclusions A three dimensional finite element model of LV depolarization wave propagation and mechanics has been developed. The simulated depolarization wave appeared physiologic. Then, the systolic myofiber strain distribution was unphysiologically inhomogeneous. When simulating LV mechanics with unphysiological synchronous depolarization myofiber strain was more homogeneous and more physiologic. Apparently, the delay between depolarization and onset of crossbridge formation is distributed such, that contraction is more synchronous than depolarization. Furthermore, it was found that variations in timing of depolarization caused larger relative changes in the distribution of myofiber strain than in that of myofiber stress. 2.6 Appendix A, model of active stress development Active stress was dependent on sarcomere length ls , length lc of the contractile element, and time ta , elapsed since the moment of electrical depolarization: a = f1 (ta ; ls ; lc ) = ls f (l )f (t ; l )(l ls0 iso c twitch a s s lc )Ea (2.21) where ls0 represents the sarcomere length in the reference state, and Ea is the stiffness of the serial elastic element. The dependency of isometrically developed active stress on lc was represented by fiso (lc ) = 0 T0 tanh2 (a6 (lc lc a7 a7 )) lc > a7 (2.22) which is similar to measurements of (42) on rat cardiac trabecula at an intracellular calcium concentration of approximately 7 M . The dependency of myofiber stress on ta and ls was represented by 30 Chapter 2 8 ta < 0 <0 ftwitch (ta ; ls) = tanh2 ( ttar ) tanh2 ( tmaxtd ta ) 0 ta tmax :0 ta > tmax (2.23) with tmax = b(ls ld ) (2.24) where tr is the activation rise time constant, td the activation decay time constant, and tmax the activation duration. Parameter b relates activation duration to the length of a sarcomere ls . ld is the sarcomere length at which this duration is 0. The time course of the contractile element length lc was simulated by a first order differential equation (function f2 in eq. 2.13): @lc = (Ea (ls lc ) 1)v0 @t where v0 represents the unloaded shortening velocity. (2.25) The values of the parameters are listed in table 2.6. a6 a7 T0 Ea v0 ls0 tr 1 1 1 m m kPa m m s m s 2.0 1.5 180 20 7.5 1.9 0.075 td s 0.075 b ld 1 s m m 0.15 -0.4 Table 2.6: Active material properties used. Equation 2.25 was solved using an Adams-Bashfort-Moulton multi-step integration scheme (43). Chapter 3 Timing of depolarization and contraction in the paced canine left ventricle The contents of this chapter are submitted to Journal of cardiovascular electrophysiology Timing of depolarization and contraction in the paced canine left ventricle: model and experiment R.C.P. Kerckhoffs, O.P. Faris, P.H.M. Bovendeerd, F.W. Prinzen, K. Smits, E.R. McVeigh, T. Arts 31 32 3.1 Chapter 3 Introduction In the normal heart, the depolarization wave propagates through the atrioventricular node to the His bundle, through the right and left bundle branches into a network of fast-conducting Purkinje fibers (3 to 4 m/s) near the endocardium (57). At the Purkinjemuscular junctions (PMJs), the depolarization wave enters the ventricular myocardium, where propagation is much slower (0.6 - 1.0 m/s) (27; 75; 87; 104). From this moment on, the wave propagates mainly from endocardium to epicardium. After 4050 ms the whole myocardium has been depolarized (23). Upon depolarization, crossbridge formation in the myofibers is initiated. The combined stress development in all myofibers leads to an increase in ventricular pressure, and finally blood is expelled from the ventricular cavity. Ventricular pacing induces abnormal depolarization patterns in the ventricles (68; 94; 99). Under these conditions, a major part of ventricular depolarization occurs through slower myocardial conduction rather than through the Purkinje system. The resulting abnormal and more asynchronous depolarization is followed by asynchronous contraction of the myofibers. Thus, both pump function (68), and on the long run, structure (64) are deteriorated. Moreover, some clinical studies indicate that patients, being paced at the right ventricle (RV), have an increased risk of heart failure (5; 105). Therefore, investigators are seeking for better sites of pacing. Positioning of the pacing electrode by trial and error is cumbersome. In assessing various pacing sites for minimal mechanical asynchrony, realistic mathematical models of cardiac electromechanics in the ventricles are likely to be useful tools. The goal of this study was to design a mathematical model of electromechanics in the left ventricle (40) during pacing. As a compromise between accuracy and computation time, we have chosen to describe the propagation of the depolarization wave through the myocardium by the eikonal-diffusion equation (19). In a simple setup, the model needs only a few parameters values to be known. For instance, myocardial tissue is anisotropic, causing the wave to travel faster along the myofiber direction than perpendicular to it. Furthermore, in the subendocardium the wave is propagating faster than in the rest of the myocardium. The related parameters, being velocities of wave propagation perpendicular to the myofibers and along the myofibers in the subendocardial layers, respectively, were estimated by fitting the model to experiments (26) in which the heart was paced at the lateral free wall of the left ventricle (LVFW). Next, the predictive quality of the model was assessed, in simulating pacing of the left ventricle from the right ventricular apex (RVA). Simulated mechanics for LVFW and RVA pacing were evaluated in terms of onset of myofiber shortening. Timing of depolarization and contraction in the paced canine left ventricle 3.2 33 Material and methods 3.2.1 Experiments Maps of timing of depolarization at the epicardium were obtained after pacing at the left ventricular free wall (LVFW) and right ventricular apex (RVA) in 2 dogs, as described before (26). In brief, socks with 128 electrodes were placed over the ventricular epicardium of anesthetized dogs. Bipolar epicardial pacing electrodes were placed at the RVA and LVFW. Epicardial recordings were obtained at an acquisition rate of 1000 Hz. After the electrical data were obtained, the animals were euthanized and the hearts excised. The hearts were filled with vinyl polysiloxane in order to maintain an enddiastolic shape and the sock electrode locations were recorded using a 3D digitizer. Unipolar voltage readings from each electrode were averaged over approximately 20 heartbeats. Depolarization time was determined as the steepest downstroke of the electrode voltage reading. 3.2.2 Simulations Design of the model of wave propagation The LV wall at end diastole was represented by a thickwalled truncated prolate ellipsoid (40). Volumes of LV wall and cavity were 140 ml and 80 ml, respectively. The distribution of helix and transverse angles of myofiber orientation was realistic, based on literature (74) (figure 3.1). Figure 3.1: Ellipsoidal geometry of the LV, with superimposed myofiber orientation. A: epicardial view; B: close up of endocardium and several myofibers showing the transmural rotation. The moment of depolarization tdep within the wall was determined by solving the ~ tdep ): eikonal-diffusion equation for the gradient of tdep (r 34 Chapter 3 q ~ tdep M r ~ tdep kr ~ (M r ~ tdep ) = 1 c r (3.1) Parameter c [m=s] represents the velocity of the depolarization wave along the myofiber direction. Parameter k [m2 =s] determines the influence of wave front curvature on wave velocity. The ratio of k=c represents a characteristic radius of wave curvature, below which wave propagation velocity severely depends on wave curvature. Dimensionless, transversely isotropic tensor M describes anisotropy of wave propagation. The largest principal direction coincides with myofiber orientation, having a related principal value equal to 1. Both other principal values, which are related to the transverse principal directions, are equal to a 2 . The value of a represents the ratio of longitudinal to transverse velocity of wave propagation. Wave velocity c and the anisotropy ratio a may vary across the myocardial wall. In the model we distinguished between the velocity cm in the myocardium, and a velocity ce at the subendocardium (figure 3.2, table 3.1). Anisotropy at the subendocardium ae was allowed to be different from anisotropy in the myocardium am . Figure 3.2: Distribution of depolarization wave velocity c and of anisotropy ratio a. The velocity and ratio cm and am , respectively, in the myocardium was allowed to be different from velocity and ratio ce and ae , respectively, at the subendocardium. The LV subepicardium at the septal side represents the subendocardium of the RV. The locations for pacing at the left ventricular free wall, and right ventricular apex are denoted by LVFW, and RVA, respectively. The LV was assumed electrically insulated. Depolarization was started at t = 0 at the pacing regions LV F W or RV A (figure 3.2). Timing of depolarization and contraction in the paced canine left ventricle 35 Modeling mechanical properties of cardiac tissue Wall mechanics were determined from solving equations of force equilibrium. Myocardial material was considered anisotropic, non-linearly elastic and time dependent (40). Local active force developed at the moment of depolarization. LV pressure was determined from the interaction of the LV with an aortic input impedance (103). For LVFW and RVA pacing, a complete cardiac cycle was simulated. Numerical implementation The equations were solved using the finite element package SEPRAN (SEPRA, Leidschendam, the Netherlands) on a 64-bit Origin 200 computer (SGI, Mountain View, CA, USA), using a single processor at 225 MHz on a UNIX platform. The eikonal-diffusion equation was discretized with 8-noded Galerkin-type hexahedral elements having trilinear interpolation. Two meshes were used to describe the LV wall: a coarse mesh (9984 elements, 11037 degrees of freedom) with a mean spatial resolution 2.4 mm and a dense mesh (44064 elements, 46987 degrees of freedom) with a mean spatial resolution 1.4 mm. The equations related to mechanics were discretized with 27-noded hexahedral elements with triquadratic interpolation. The LV wall was subdivided into 108 elements, with 3213 degrees of freedom. Simulations and data analysis The myocardial wave velocity cm and subendocardial anisotropy ratio ae were fixed at values of 0.75 m/s and 1.5, respectively (table 3.1). Parameter k was set to a realistic value of 2.1 10 4 m2 s 1 for the simulations on the dense mesh. For the, less time consuming, simulations on the coarse mesh, it was increased to 7.5 10 4 m2 s 1 to enable numerical convergence. For the subendocardium three values for wave velocity ce were used (table 3.1). Also three values for the anisotropy factor am in the myocardium were applied. Thus, simulations were obtained for all nine combinations of the latter values, while pacing from a position on the free wall of the left ventricle (LVFW, figure 3.2). The calculated maps were compared with the experimentally measured map for LVFW pacing. Using criteria on the moment of breakthrough, and timing of the wave front arrival at a point near the base, as far as possible from the pacing site, the best combination of parameter settings was chosen. Next, keeping the thus found parameter setting, a simulation was performed when pacing from the right ventricular apex (RVA). The results of the simulation were compared to the map as measured with RVA pacing. The maps of depolarization timing for LVFW pacing that best matched the experiments and the map for RVA pacing were used as input in the simulation of mechanics. Timing of mechanics was characterized by the moment of onset of myofiber shortening in the midwall (106). 36 Chapter 3 parameter model values cm [ m s 1 ] 0.75 1 ce [ m s ] 0.75, 1.2, 1.8 am [-] 1.5, 2.0, 2.5 ae [-] 1.5 2 1 k [m s ] 2.1 10 4 , 7.5 10 4 literature values source 0.6 - 1.0 (27; 75; 87; 104) 1.2 (27) 2.1 - 3.3 (27; 75; 87; 104) 1.5 (27) 2.1 10 4 (88) Table 3.1: Parameter values in the model, and reported in literature. All reported measurements were done in the LV, except for the measurement in the RV denoted by the asterisk . The simulations with parameter variations, performed on a coarse mesh (see Numerical implementation), were calculated with k = 7.5 10 4 m2 s 1 , to enable convergence. The simulations, performed on the dense mesh were calculated with k = 2.1 10 4 m2 s 1 . cm wave velocity parallel to the myofibers in the myocardium; ce wave velocity parallel to the myofibers in the subendocardium (see also figure 3.2); am anisotropy ratio of depolarization wave velocities parallel, and perpendicular to the myofibers in the myocardium; ae anisotropy ratio of depolarization wave velocities parallel, and perpendicular to the myofibers in the subendocardium; k diffusion constant that determines the influence of wave front curvature on wave velocity. 3.3 Results 3.3.1 Experiments In figure 3.3a a map is shown of the moment of depolarization on the epicardial surface of the heart during LVFW pacing. The inner circle represents the border of the region of the measurement, which is approximately the equatorial region. Epicardial isochrones in the early depolarized region resembled quasi-ellipses with a long to short axis ratio of about 2.5. Isochrones were initially close together, indicating slow apparent propagation, whereas after approximately 60 ms this distance increased. At the right ventricular free wall the wave converged towards the base, where the maximum depolarization time was 117 ms. In the same heart, for RVA pacing, apparent wave velocity increased in the late depolarized regions also. At the left ventricular free wall the wave converged towards the equatorial region, where the maximum depolarization time was 113 ms (figure 3.3b). In the other heart (figure 3.3c), a similar pattern was found, having a maximum depolarization time of 125 ms. 3.3.2 Simulations For solving the eikonal-diffusion equation calculation times were approximately 2, and 12 hours for the coarse, and dense mesh (figure 3.4), respectively. For LVFW pacing, isochrones in the early depolarized regions resembled ellipses, with Timing of depolarization and contraction in the paced canine left ventricle 37 Figure 3.3: Maps of measured timing of epicardial depolarization [ms] for LVFW (a), and RVA pacing (b and c). The maps are represented as bull’s-eye maps with the apex in the center. The inner circles represent the border of the region of the measurement, which is approximately the equatorial region. Maps obtained from a) and b) are from the same heart. The location and maximum depolarization time tdep;max are denoted in the plot. P posterior; RV right ventricular side. their major axes aligned with the myofiber direction (figure 3.4). In figure 3.5 the epicardial depolarization timing patterns are represented as bull’s eye plots. Increase of the ratio of myofiber to cross-fiber wave velocity in the myocardium am caused the long to short axis ratio of the isochrones near the pacing site to increase proportionally. Furthermore, the time needed for occurrence of breakthrough, increased with am . Maximum depolarization time tdep;max increased also with an increase in anisotropy. The main effect of an increase of endocardial wave velocity ce was a proportional increase of the apparent wave velocity at locations far from the pacing site. Increasing ce resulted in a decrease in maximum depolarization time. Subendocardial wave velocity did not affect ellipticity of isochrones. All waves ended at the septal base. 38 Chapter 3 Figure 3.4: Timing of depolarization [ms] in the LV for a simulation of LVFW pacing. Parameter values were: cm = 0.75 m=s; am = 2.5; ce = 1.2 m=s; ae = 1.5; k = 2.1 10 4 m2 =s. A part of the anterior free wall is removed, showing the septal endocardium, and a cross-section of the midseptum. Notice that the long axes of the ellipsoidal wave fronts in the early depolarized region are aligned with the epicardial myofiber orientation (figure 3.1). In comparing numerical and experimental results, one should realize that the simulations hold for the left ventricular epicardium and the septal part of the RV endocardium. So, the part between RVA and right ventricular side of the base cannot be used for comparison. Comparing figures 3.3a and 3.5, the best match was found for a subendocardial wave velocity ce = 1.2 m=s and a myocardial anisotropy am = 2.5. With these settings, and a smaller k , the simulation was repeated in the dense mesh (figures 3.4 and 3.6a). The results were virtually identical to those in the coarse mesh, indicating that further mesh refinement was not needed, and that the value of k was not critical. With the same parameter settings and dense mesh, RVA pacing was simulated. Near the RVA pacing site, the long axes of the isochrones of depolarization were aligned with the myofiber direction (figure 3.6). The simulation of RVA pacing matched the measured maps in the following aspects: apparent wave velocity in the posterior region was larger than anteriorly, the equatorial circle was reached at the same time (after 113-125 ms in the experiments and after 120 ms in the simulation), and the wave converged in a similar region in the LVFW. Timing of midwall myofiber shortening closely followed the timing of epicardial Timing of depolarization and contraction in the paced canine left ventricle 39 Figure 3.5: Bull’s eye plots of simulated maps of timing of epicardial depolarization [ms] for LVFW pacing. The purple circles indicate a similar region in which the measurements were performed. The arrows in the bottom left map indicate the right ventricular region. The dotted line indicates the LV/RV attachment. From bottom to top, subendocardial wave velocity ce is increased. From left to right, myocardial anisotropy ratio am is increased. Parameter k , subendocardial anisotropy ratio ae , and myocardial wave velocity cm along the myofiber were fixed at 7.5 10 4 m2 =s, 1.5, and 0.75 m/s, respectively. In the bottom row, subendocardial wave velocity equaled myocardial wave velocity ce = cm = 0.75 m=s and ae = am , implying no influence of the Purkinje system. A anterior; P posterior; S septum. 40 Chapter 3 Figure 3.6: Bull’s eye plots of simulated maps of timing of epicardial depolarization (top) and timing of onset of midwall myofiber shortening (bottom) for RVA and LVFW pacing [ms]. The simulations of wave propagation were performed in the dense mesh. The circles encompass the regions that were measured in the experiments. cm = 0.75 m=s, ce = 1.2 m=s, am = 2.5, ae = 1.5. A Anterior; S Septum; P Posterior. depolarization (figure 3.6). 3.4 Discussion With a relatively simple model setup, depolarization timing patterns for pacing at the LV free wall and RV apex were simulated succesfully in the left ventricle with a realistic myofiber orientation. We will discuss to what extent simplifying assumptions in the model might affect the obtained results. 3.4.1 Model setup In the real heart, the influence of the Purkinje system on myocardial propagation velocity in ventricular pacing depends on the distribution and density of the Purkinjemuscular junctions (PMJs), which has been reported to be variable (16; 23; 53; 72). Timing of depolarization and contraction in the paced canine left ventricle 41 Furthermore, it has been reported that retrogade propagation delay is generally shorter than antegrade. Antegrade delays from 1 ms to infinity (in the latter case the wave could not leave the Purkinje system) during pacing have been reported (35; 57; 78). In our model, PMJ density, distribution, and propagation delay are condensed into effective subendocardial wave velocities of 1.2 m/s and 0.8 m/s along and perpendicular to the myofiber, respectively. This approach seems realistic if the distribution of PMJs is dense. Consequently, only one wavefront is formed. However, in case of a coarse distribution of PMJs (9; 18; 107), it is also possible that the depolarization wave reaches a remote area of myocardium through the Purkinje fibers before the wave that propagates through the myocardium. In that case, so-called secondary wavefronts can occur, which cannot be described by our model. The real geometry of the heart, including the right ventricle (RV), is more complex than the truncated ellipsoid we used. Although the faster RV subendocardial wave velocity is accounted for, the model lacks the RV free wall. The RV free wall also contains Purkinje fibers, which might affect the depolarization pattern. For LVFW pacing, neglection of the RV will affect the depolarization pattern only far from the pacing site. For RVA pacing, we expect that, due to the fast wave velocity in the septum, neglection has a minor influence on the LV depolarization pattern. For the LV subendocardium, a rotationally symmetric region of faster propagation was assumed. Near the His bundle in the upper septum however, PMJs are not present (57). For the present study this does not seem critical, but for studying pacing near the His bundle a more detailed approach may be required. Myocardium is organized in laminar sheets (100) four to eight cells thick. Due to this sheet structure, macroscopic myocardial electrophysiological properties may be orthotropic (46), in contrary to the assumed transverse isotropy. Although there are some indications for orthotropy from numerical models (34), accurate measurements of orthotropic electrophysiological properties have not been performed yet. The eikonal-diffusion equation solves for depolarization times. Simulations of pathologies like for example bundle branch block, WPW syndrome, and pacing are possible. Pathologies related to repolarization however (e.g. fibrillation, as in the model of Berenfeld et al (9)) cannot be simulated with the eikonal-diffusion equation. 3.4.2 Results The model of wave propagation contains 5 parameters, 3 of which were fixed. Myocardial wave velocity cm parallel to the myofiber was fixed at 0.75 m/s, because of the small range of reported wave velocities (table 3.1) in canine myocardium. Despite limited reports on the subendocardial anisotropy ratio (27), ae was fixed at 1.5, because we expect that the final solution is relatively insensitive to this parameter. Namely, the subendocardial layer appears important for what happens after breakthrough. Late in the period of depolarization, the depolarization wave in the endocardium is directed 42 Chapter 3 mainly from apex to base, thus about coinciding with the subendocardial fiber direction. Since the transverse component of propagation is small under these circumstances, the final solution will not be affected severely by inaccuracy of ae . The setting of the varied parameter am was determined (am = 2.5) such, that breakthrough occured around 60 ms as in the experiment. The other varied parameter, the wave velocity near the endocardium, was set to 1.2 m/s, predominantly on the basis of apparent epicardial wave velocity after breakthrough. Finally, in a simulation with a relatively coarse grid, where parameter k was elevated, the solution appeared very similar to the solution with a finer grid and a more physiologic value of k . Thus, k appears not to be critical for ventricular wave propagation during pacing. The current model has been succesfully tuned to LVFW pacing in one experiment. Application of the resulting settings in the model enabled an accurate description of the depolarization wave with a different pacing site, located at the RV apex. For a detailed knowledge of parameter values more experiments are likely to be needed. In a previous study where a normal depolarization pattern during sinus rhythm was simulated (40), parameter values were different. Subendocardial wave velocity ce was 4 m/s, whereas it was 1.2 m/s in the present study. In the previous study the wave was started in 4 regions of earliest depolarization as measured (23). The Purkinje wave velocity of 4 m/s accounted for a fast spread of depolarization. With the current obtained value of ce , simulation of a normal depolarization pattern is possible by a increasing the area of the early depolarized regions. Thus, the model is capable of simulating normal and abnormal patterns, with an equal set of parameters. The pattern of onset of myofiber shortening was very similar to that of depolarization. This finding agrees with experimental data (106) where moments of onset of shortening and depolarization were found to be linearly related, with a slope of about unity. To model the timing of cardiac depolarization, also the bidomain model (33) could be used. This yields transmembrane and extracellular potentials as a function of time and space. However, because of the need of a very dense mesh and small time steps to represent the steep depolarization upstroke, on the whole heart level, the bidomain model is computationally very demanding. The computationally less demanding eikonaldiffusion equation has been used before (18; 88). For LVFW pacing, Colli-Franzone et al (18) found patterns of depolarization which are similar to our patterns. However, in their model, two simulations are needed. The moment when the depolarization wave reaches a PMJ (central PMJ) is determined in the first simulation, neglecting the Purkinje system. In the second simulation other PMJs are activated, according to the distance to the central PMJ and Purkinje velocity. With our current model set up, only one simulation is needed. In another study, Tomlinson (88) computed depolarization times for sinus rhythm with the eikonal-diffusion equation. The Purkinje system was modeled by prescribing depolarization times at the entire endocardium, as measured. In this approach, for pacing, the role of the Purkinje system would be completely neglected. Timing of depolarization and contraction in the paced canine left ventricle 3.5 43 Conclusions With a relatively simple model setup using the eikonal diffusion equation, depolarization timing patterns for pacing at the LV free wall and RV apex were simulated succesfully in a left ventricle with a realistic myofiber orientation. Timing of breakthrough and total time needed to depolarize the LV in the simulations and experiments were similar. Within the myocardium, cross-fiber velocity of wave propagation is estimated to be 0.4 times the velocity along the myofiber direction (0.75 m/s). Near the endocardium, wave propagation is about 1.6 times faster than in the rest of the myocardium, but about 3 times slower than found in Purkinje fibers. Maps showing timing of onset of shortening were similar to previously published measured maps in which contraction wave velocity was similar to depolarization wave velocity. 44 Chapter 3 Chapter 4 Regional contraction in the paced canine left ventricle R.C.P. Kerckhoffs, O.P. Faris, P.H.M. Bovendeerd, F.W. Prinzen, K. Smits, E.R. McVeigh, T. Arts 45 46 4.1 Chapter 4 Introduction In the normal heart, the depolarization wave propagates from the atrioventricular node to the His bundle, through the right and left bundle branches into a network of fastconducting Purkinje fibers near the endocardium. At the end of Purkinje fibers, the depolarization wave enters the ventricular myocardium. From this moment on, the wave propagates mainly from endocardium to epicardium. After 40-50 ms the whole myocardium has been depolarized. Upon depolarization, cross-bridge formation in the myofibers is initiated. The combined stress development in all myofibers leads to an increase in ventricular pressure, and finally blood is expelled from the ventricular cavity. When pacing from a site in the ventricles, depolarization spreads more slowly and less uniformly (68; 94; 99). The resulting more asynchronous contraction of the myofibers affects pump function (68), myocardial tissue structure (64), and may even contribute to the development of heart failure (5; 105). Realistic mathematical models of cardiac electromechanics in the ventricles are likely to be useful tools to better understand these phenomena. In sinus rhythm, a map of electromechanical (EM) delays, opposite to the depolarization timing, makes contraction practically synchronous, and results in a physiologic map of circumferential strain (40). The hypothesis was introduced that this map of EM-delay may be a property of the cardiac tissue as a result of a long term adaptation. In the present study it was investigated whether, and to what extent, heterogeneous EM-delay, as it occurs during a normal heart beat, would affect regional contraction and total pump function during ventricular pacing. To that purpose, a finite element model of left ventricular electromechanics was used (40). Ventricular pacing was simulated by using the eikonal-diffusion equation (19; 41), while starting depolarization from the right ventricular apex (RVA). First, the electromechanical (EM) delay was kept constant, implying the sequence of depolarization to be equal to the sequence of contraction. The calculated characteristics of contraction were compared with experimental results, obtained in MRI- tagging experiments. Secondly, pacing was simulated, but now the spatial distribution of EM-delay was chosen such, that during sinus rhythm contraction is synchronous. By comparing these results with the experiments, it was tested, whether and to what extent EM-delay could explain the found distribution of myofiber contraction. 4.2 Material and methods 4.2.1 Experiments Maps of subepicardial depolarization timing and a map of circumferential midwall strain for RVA pacing were measured in one dog, as described before (25). In brief, a sock with 128 electrodes was placed over the ventricular epicardium of an anesthetized dog. A bipolar epicardial pacing electrode was placed at the RVA. The animal was positioned in Regional contraction in the paced canine left ventricle 47 a MR scanner to obtain tagged cine images of the short and long axes of the heart during pacing (106). Between image acquisitions, electrical recordings were obtained at an acquisition rate of 1000 Hz. The animal was then euthanized and the heart excised. End-diastolic heart shape was preserved by filling the heart with vinyl polysiloxane. The sock electrode locations were then recorded using a 3D digitizer. From the tagged images, midwall circumferential strain was calculated throughout the cardiac cycle (65). Unipolar voltage readings from each electrode were averaged over approximately 20 heartbeats. Depolarization time was determined as the steepest downstroke of the electrode voltage reading. Electrode locations in digitizer coordinates were transformed to scanner coordinates using a rigid-body rotation and translation. 4.2.2 Simulations Setup of the model Details of the model of LV electromechanics have been previously published (40). In short, the reference state was defined by zero transmural pressure. The LV wall was represented by a thickwalled truncated prolate ellipsoid with a realistic myofiber orientation (figure 4.1A). The moment of depolarization tdep for RVA pacing (figure 4.1B) was determined realistically (41) from solving the eikonal-diffusion equation (19). Wall mechanics were determined by solving the equations of force equilibrium. Passive myocardial material was considered anisotropic and non-linearly elastic (figure 4.1C). Stress development in the myofibers depended on time, sarcomere length, and sarcomere shortening velocity (figure 4.1D and E). Local active force development was initiated either at the moment of depolarization, or after an additional time delay. During ejection, LV pressure was determined from the interaction of the LV with an aortic input impedance (103). Numerical implementation The eikonal-diffusion equation was solved using a Galerkin finite element method with 8-noded hexahedral elements with trilinear interpolation of the field of depolarization times. The LV wall was subdivided into 44064 elements with 46987 degrees of freedom, resulting in a spatial resolution of about 1.4 mm. The equations related to mechanics were solved using a Galerkin finite element method with 27-noded hexahedral elements with triquadratic interpolation of the displacement field. The LV wall was subdivided into 108 elements, with 3213 degrees of freedom. All equations were solved on a 64-bit Origin 200 computer (SGI, Mountain View, CA, USA), using a single processor at 225 MHz. The finite element calculations were performed with the FORTRAN77 compiler based package SEPRAN (SEPRA, Leidschendam, the Netherlands) on a UNIX platform. 48 Chapter 4 Figure 4.1: Overview of the model (40). A: Myofiber orientation in the left ventricle, plotted on epicardium, endocardium, and partly through midwall. B: Depolarization times [ms] for RVA pacing. Time step between isochrones is 10 ms. C: Stress [kPa] of passive material parallel (–) and perpendicular (- -) to the myofiber for biaxial stretching. D: Active myofiber stress [kP a] as a function of time [ms] for isometric twitches at sarcomere lengths of 1.6, 1.9, and 2.2 m. E: Linear (default in model) and Hill relation between sarcomere shortening velocity [m=ms] and myofiber force, normalized to maximum force. For the Hill relation: see Discussion. Simulations and data analysis Three separate simulations were performed. In the first simulation, normal sinus rhythm (SR) was simulated as in Ref (40). The EM-delays that make contraction synchronous in the SR simulation, are shown in the right panel of figure 4.2. Next, mechanics after Regional contraction in the paced canine left ventricle 49 pacing at the RVA (RV0 simulation) was simulated, in which timing of initiation of contraction was identical to that of depolarization (figure 4.1B). In a third simulation (RVEM simulation), we introduced the EM delay from the SR simulation. The resulting map of initiation of contraction of the RVEM simulation is shown in figure 4.2A. Figure 4.2: A: Moments of onset of contraction in the RVEM simulation. B: Electromechanical delay times from the SR simulation, and used in the RVEM simulation. The EM-delay is distributed such, that for a normal heart beat (SR simulation), contraction is synchronous (40). Notice that the depolarization times, depicted in figure 4.1B, plus the here depicted EM-delays in B, result in the mechanical activation times in A. For all simulations, global hemodynamics and LV midwall circumferential natural strains cc were computed as a function of time. Circumferential strain cc was defined as cc = ln( lmc ) lmc0 (4.1) with lmc the actual length of an infinitesimal line element, oriented in the circumferential direction in the reference situation, with a reference length lmc0 . Circumferential strain during ejection cc;ej was defined as the change in cc from the beginning to the end of ejection. To facilitate comparison between model and experiment, ejection strain was divided by mean ejection strain, to yield the normalized strain ^cc;ej . Thus, differences in ventricular preload, afterload, and myofiber contractility between experiment and simulation were corrected for. 50 Chapter 4 The moment of depolarization is an important determinant of the regional contribution of the myocardium to total cardiac pump function (22). The slope from a linear regression analysis (plus a 95% confidence interval) of the relation between local depolarization time and local normalized circumferential strain has been used as a measure of mechanical synchrony. Midwall circumferential strain is a practical measure from an experimental point of view. In the midwall, myofibers are oriented almost completely circumferential. Myofiber strain f throughout the complete LV wall is more relevant for physiological function. Stroke work density Wf [J m 3 ] was defined as Wf = I cardiac cycle f df (4.2) where f is total Cauchy myofiber stress. 4.3 Results Calculation time for simulation of a complete cardiac cycle was approximately 7 hours. Maximum pressure and ejection fraction in both the RV0 (14.5 kPa and 36.7%, respectively) and RVEM (14.5 kPa and 36.9%, respectively) simulations were similar. These values were lower than in a simulated normal heart beat (17.8 kPa and 61.4%, respectively). See figure 4.3. In both the simulated and measured paced ventricle, strain patterns depended on the pattern of depolarization time (range 0 to 124 ms in the simulations and 21 to 125 in the experiment). In figure 4.4, circumferential strain cc is shown as a function of time for both pacing simulations and experiment for early, mid, and late depolarized regions. Early depolarized regions - In the RV0 simulation, cc (tdep ranging from 11 to 12 ms) was negative (-0.17) during isovolumic contraction. During ejection, cc;ej was positive (0.06). Stretching continued in the isovolumic relaxation phase. In the experiment (tdep from 21 to 36 ms), strains were negative (-0.05 to -0.07) during the isovolumic contraction phase. During the ejection phase, cc became positive up to 0.05 just before mid-ejection. Next, strain decreased slowly. In early depolarized regions, cc;ej was positive. Mid depolarized regions - In the RV0 simulation (tdep ranging from 70 to 74 ms), maximum cc was slightly positive (ranging from 0.02 to 0.05) in the isovolumic contraction phase, but became negative at the end of this phase. cc;ej was -0.10. In the experiment (td ep ranging from 70 to 74 ms), maximum cc was slightly positive (0 to 0.02) in the isovolumic contraction phase, but cc became negative at the end of this phase (-0.07). During ejection, strain either continued to decrease, or first became positive, and then slowly decreased later in this phase. cc;ej during ejection was slightly negative (-0.02 to -0.05). Regional contraction in the paced canine left ventricle 18 10 16 plv [kPa] 15 5 51 14 0 80 Vlv [ml] plv [kPa] 12 10 60 40 8 6 0.4 qao [l/s] 4 0.2 0 0 2 0 500 Time [ms] 40 60 80 Vlv [ml] Figure 4.3: Global hemodynamics for the RV0 (–), RVEM (- -) and SR simulation ( ). The left panel represents from top to bottom the left ventricular pressure plv , cavity volume Vlv , and aortic flow qao as a function of time. Dots indicate moments of opening and closure of the valves. The right panel represents the pressure-volume loops. Notice the decreased systolic pressure and ejection fraction for the pacing simulations, compared to the simulated normal heart beat from (40). Late depolarized regions - In the RV0 simulation (tdep ranging from 107 to 116 ms), cc was positive up to 0.08 during the isovolumic contraction phase. During ejection, strain decreased by 0.22 to 0.24. Shortening continued in the beginning of isovolumic relaxation. In the experiment (tdep ranging from 112 to 113 ms) maximum cc was 0.06 to 0.1 during isovolumic contraction. During ejection, cc;ej ranged from -0.20 to -0.23. In the beginning of isovolumic relaxation, strain decrease continued. Circumferential strain for the RVEM simulation was similar to that of the RV0 simulation (mid panel of figure 4.4). cc during isovolumic contraction was slightly larger in the early depolarized regions (-0.15). In regions with mid depolarization times, maximum cc was also slightly larger (ranging from 0.02 to 0.06). At the beginning of ejection, the dependence of myofiber strain on depolarization time throughout the complete LV wall is similar to the dependence of midwall circumferential strain on depolarization time (figure 4.5A). Chapter 4 midwall circumferential strain 52 0.1 0 −0.1 −0.2 0 ejection ejection 200 Time [ms] 0 200 Time [ms] ejection 0 200 Time [ms] Figure 4.4: Circumferential strain as a function of time in the RV0 simulation (left), RVEM simulation (mid), and experiment (right) during the isovolumic contraction, ejection, and isovolumic relaxation phase. Strains in early (green –), mid (blue - -), and late (red ) depolarized regions are shown. The vertical lines denote the beginning and end of ejection. Figure 4.5: A: Myofiber strain at the beginning of ejection f and B: stroke work density [kJ m 3 ] for the RV0 simulation. In combination with figure 4.1B, it is observed that myofiber strain at the beginning of ejection is negative in early depolarized regions, and positive in the late ones. In the RV0 simulation, stroke work density Wf [kJ m 3 ] depended also on Regional contraction in the paced canine left ventricle 53 depolarization time (figure 4.5B). In the early depolarized regions, stroke work density was negative (-3.35 kJ m 3 ). In the late depolarized regions, stroke work density was 7.47 kJ m 3 . The relation between circumferential strain and depolarization is further illustrated in figure 4.6, where normalized circumferential strain during ejection ^cc;ej is plotted as a function of depolarization time tdep for the RV0 simulation and the related experiment. normalized myofiber ejection strain 2 1 0 −1 −2 −3 −4 −100 −50 Depolarization time [ms] 0 Figure 4.6: Midwall circumferential strain during ejection ^cc;ej normalized to mean strain during ejection as a function of epicardial depolarization time tdep [ms] for the simulation (Æ) and measurement () for RVA pacing. Depolarization time was related to the moment of dp=dtmax (22). The solid line represents a linear fit for the simulation with ratio -47.7 2.9 s 1. The dashed line represents a linear fit for the experiment with ratio -37.6 3.0 s 1. The slope, that relates ^cc;ej and tdep , was slightly steeper for the RV0 simulation (-47.7 2.9 s 1) than the slope for the experiment (-39.5 3.0 s 1 ). The slope for the RVEM simulation (with heterogeneous EM-delay) was slightly less steep (-36.7 1.8 s 1 ) than the slope for the experiment. So an EM delay, resulting in full synchronization during sinus rhythm, slightly synchronized contraction during RVA pacing. 54 4.4 Chapter 4 Discussion With a mathematical model, electromechanics in the left ventricle for pacing at the RV apex was simulated realistically. The introduction of the EM-delay in the RVA simulation made contraction in the RVEM simulation slightly more synchronous. It is likely that the narrow physiological range of electromechanical delay to synchronize contraction in a normal heart is too small to significantly affect asynchrony during pacing. 4.4.1 Results We wanted to test the effect of the hypothesis, put forward in a previous paper (40), that in a normally stimulated LV, a heterogeneously distributed EM-delay exists, such that contraction is initiated synchronously. We assumed that the EM-delay does not change when the LV is acutely paced after normal heart beats. The slope in the simulation with EM-delay (RVEM simulation) (-36.7 1.8 s 1 ) is close to the experimental finding (39.5 3.0 s 1 ). It may be very well that during longer lasting ventricular pacing, EM delays would adapt to the situation of pacing, but this is not clear. Because measured asynchrony during ventricular pacing is much larger than during sinus rhythm, this adaptation is probably not very effective during ventricular pacing. Electrical and mechanical activation have been measured before (68; 106). For practical reasons, timing of mechanical activation was defined as the (measurable) moment of onset of circumferential shortening instead of onset of contraction. These definitions have different meanings. This can be illustrated by considering two myofibers, in which crossbridge formation starts simultaneously. Then, the moment of onset of shortening for those myofibers can be very different, since it depends strongly on the force, experienced by those myofibers from the neighbouring tissue. Currently, true electromechanical delay, from the moments of excitation to contraction, cannot be measured in the entire myocardium. Our model is a helpful tool in gaining insight in excitation and contraction. In mid and late depolarized regions, time courses of circumferential strain cc in the experiment and both pace simulations were similar (figure 4.4). Differences between experiment and simulations were observed in the early depolarized regions. During isovolumic contraction, early depolarized myofibers shorten less in the experiment than in the simulation. In the experiment, these myofibers are located in the RV free wall. In the simulation, early depolarized myofibers are located in the septum. Possibly, RV diastolic filling in the experiment was relatively less than LV diastolic filling in the simulation, and thereby diastolic stretching in the experiment was less (this was not measured). Then, shortening of early depolarized myofibers was also smaller. By dividing ejection strain by mean ejection strain, differences of filling in the experiment and simulation were ruled out. The pattern of stroke work density in the RV0 simulation is similar to that earlier reported in literature (70), with negative work in early depolarized regions and large Regional contraction in the paced canine left ventricle 55 positive work in late depolarized regions. 4.4.2 Limitations The real cardiac geometry with a LV and RV is more complex than the truncated ellipsoid we used in the simulations. The load the RV exerts on the LV wall through RV pressure and force transmission in the LV-RV attachment were neglected. For RVA pacing, RV pressure develops earlier than LV pressure, as observed in patients with left bundle branch block (31), and stretch in the LV wall might be affected. Since we did not include the RV, its influence remains unclear. In the model, the relation between active stress and myofiber shortening velocity is assumed linear. The slope of the relation between ^cc;ej and tdep for a simulation with RVA pacing and a hyperbolic Hill relation (42) (figure 4.1E) was -46.3 2.5 s 1 , which is not very different from the slope of the RV0 simulation (-47.7 2.9 s 1 ). In comparing model and experimental results, we focused on the ejection phase. The choice of the beginning and end of the ejection phase influences cc;ej . In the simulations, determination of duration of this phase is trivial. In the experiment however, in the absence of a registration of aortic flow, determination of beginning and end of ejection is not trivial. Assuming ventricular volume is closely related to mean circumferential strain, the onset of mean strain decrease and minimum mean strain were chosen as the beginning (t = 73 ms) and end (t = 163 ms) of ejection, respectively. In the simulation, these moments coincided with the moments of opening and closing of the aortic valve, respectively. Moreover, in the experiment, variation of beginning of ejection by +15 and -15 ms changed the slope of the linear fit between normalized myofiber ejection strain and depolarization time slope only by -8 % and -4 %, respectively. 4.5 Conclusions A three-dimensional finite element model of LV electromechanics for ventricular pacing has been developed. Experiments were performed to test the model. Model and experiment agreed in the following aspects: 1) Both systolic pressure and ejection fraction decreased relative to natural sinus rhythm. 2) In early depolarized regions, both shortening and stroke work declined; in the late depolarized regions changes were opposite. The relation between moment of epicardial depolarization and normalized midwall circumferential strain was linear, with a slope of -47.7 2.9 s 1 in the model and -39.5 3.0 s 1 in the experiment for RV apex pacing. The introduction of the EM-delay in the RVA simulation hardly affected asynchrony, with a slope of -36.7 1.8 s 1 . We conclude that the narrow physiological range of electromechanical delay is too small to significantly affect asynchrony during ventricular pacing. 56 Chapter 4 Chapter 5 Intra- and interventricular asynchrony of electromechanics in the ventricularly paced heart R.C.P. Kerckhoffs, P.H.M. Bovendeerd, F.W. Prinzen, K. Smits, T. Arts 57 58 5.1 Chapter 5 Introduction The heart consists of two pumps, located side-by-side. The right ventricle (RV) pumps the blood to the lungs and the left ventricle (LV) to the rest of the body. Pressure is about 4 times higher in the LV than in the RV. Depolarization is normally initiated by selfdepolarization of the sinoatrial (SA) node in the right atrium. Next, the depolarization wave propagates over the atria through the atrioventricular node to the His bundle. The His bundle splits in a right and left branch, depolarizing the right and left ventricle, respectively, via networks of fast-conducting Purkinje fibers. The whole myocardium is depolarized within 40-50 ms (23). The LV is activated somewhat earlier than the RV (50; 96). Stress development in all myofibers leads to an increase of ventricular pressures. Equal amounts of blood are ejected by the LV and RV cavities. The normal contraction pattern of the heart may be affected by abnormal conduction of the depolarization wave (69; 99), for instance when the left bundle branch is blocked (31). The normal contraction pattern is affected by abnormal propagation of the depolarization wave, as is the case during ventricular pacing. In the early years of clinical pacing, most attention has been paid to proper thresholds and synchronization between atria and ventricles (71). Recently, interest for a proper sequence and synchrony of ventricular depolarization is growing. In a ventricularly paced heart, depolarization is not as synchronous as in a normal heart and stroke work density is distributed non-uniformly (70). The resulting more pronounced asynchrony of contraction of the myofibers affects pump function (68), myocardial tissue structure (64), and may even contribute to the development of heart failure (5; 105). An interesting challenge is to develop mathematical models incorporating patient specific characteristics with the goal to optimize timing and location of ventricular pacing. Models of cardiac mechanics have been used to investigate the effect of myofiber orientation (13; 74; 92) and regional ischemia (12; 54; 85) on the distribution of myofiber stress and strain. Such models have been used to investigate the relation between timing of depolarization and contraction (40; 90) for a normal heart beat, but not yet for ventricular pacing. The aim of this study was the extension of an existing three-dimensional finite element model of LV electromechanics (40) with the right ventricle, and demonstration of the potential for patient-specific modeling of cardiac electromechanics during ventricular pacing. We focused on timing of LV and RV hemodynamics, asynchrony of depolarization, myofiber shortening, stroke work, and systolic septal wall motion. The geometry of the LV and RV was obtained from non-invasively acquired MR short axis images of the canine heart. The myofiber helix angle (82) was assumed to vary linearly across the wall. Values of parameters describing depolarization wave propagation and mechanical properties were as presented before (40; 41). Complete cardiac cycles were simulated for a normal heart with synchronous activation and ventricular pacing. Pacing was initiated from the right ventricular apex (RVA) or from the left ventricular free wall (LVFW). Intra- and interventricular asynchrony of electromechanics in the ventricularly paced heart 5.2 59 Material and methods 5.2.1 Geometry and myofiber orientation LV and RV geometry were derived from five cine-MR short axis images (1.17 1.17 8.0 mm3 voxel volume) in end-diastolice state of an anesthetized dog (m=26 kg). Interpolation was performed between the images in the long axis (z-)direction with custom software (Analyze, Mayo Clinic), thus obtaining the contours of the heart in 20 short axis sections. The contours of the RV endocardium were subdivided in a RV free wall partition and a RV septum partition. Thus, 4 sets of datapoints were obtained for the epicardial (20 contours), LV endocardial (18 contours) and RV free wall and septal (each 13 contours) surfaces (figure 5.1). Figure 5.1: Fitted contours (lines) for 4 cross-sections from base (left) to near the apex (right). The dots represent the original data, as obtained manually from the MRI short axis images. The position ~r of each data point was written in Cartesian coordinates fx; y; z g. To enable an efficient mathematical description of the cardiac surfaces, Cartesian coordinates were transformed to prolate ellipsoidal coordinates f; ; g: x = C sinh( ) sin() cos() y = C sinh( ) sin() sin() z = C cosh( ) cos() (5.1) where C is the focal length of the system, the radial coordinate, the circumferential angle, and the longitudinal angle. The origin of both the Cartesian and the ellipsoidal coordinate system was defined by the intersection of the LV long axis and the most basal slice. All 4 surfaces were described by a prolate ellipsoidal harmonic series (61; 62): 60 Chapter 5 (; ) = L X m=l X l=0 m= l alm Plm (cos)eim (5.2) Here Plm are associated Legendre polynomials, and alm are coefficients, resulting from a least squares fit (with highest order L) to the experimental datasets. Using the fit, for each arbitrary point on each arbitrary surface, an orthonormal wall bound ellipsoidal coordinate system f~er ; ~ec ; ~el g was defined (figure 5.2): @~r @~r @~r @ @ ~er = @~r ; ~ec = @~r ; ~el = @ @~r k k @ k k @ k k @ (5.3) Figure 5.2: The mesh of the left and right ventricle, showing epicardium and RV and LV endocardium. The local wall bound coordinate system for an epicardial node is shown. ~er radial direction; ~ec circumferential direction and ~el longitudinal direction. Here the base vectors ~er , ~ec and ~el point in the local radial (transmural), circumferential and longitudinal direction, respectively. For points within the cardiac wall, the local coordinate system was obtained by interpolation of the coordinate systems at the nearest points on the inner and outer surfaces. The coordinate system was used to define the orientation of the myofibers in the wall. Myofibers were assumed to be in Intra- and interventricular asynchrony of electromechanics in the ventricularly paced heart 61 planes, spanned by the local circumferential and longitudinal direction. The in plane myofiber orientation was characterised by the helix angle, defined as the angle between the myofiber direction and the local circumferential direction (82). The helix angle was assumed to vary linearly from 70Æ at the epicardium and 70Æ at both the RV and LV endocardium. Similarly, in the septum the helix angle ranged from 70Æ at the RV side to 70Æ at the LV side. 5.2.2 Depolarization and mechanics The applied myocardial material properties have been published previously (40; 41). In short, the moment of depolarization tdep for RVA and LVFW pacing (figure 5.3) was determined from solving the eikonal-diffusion equation (19). Parameter values were taken from Ref. (41). Subendocardial wave velocities parallel and perpendicular to the myofiber (1.2 and 0.8 m/s, respectively) were higher than those in the rest of the myocardium (0.75 and 0.3 m/s, respectively) (figure 5.3). Figure 5.3: Distribution of depolarization wave velocity c and of anisotropy ratio a. The locations for pacing at the left ventricular free wall, and right ventricular apex are denoted by LVFW, and RVA, respectively. The geometry, as obtained from the MR images, was defined as the reference state of zero transmural pressure in the simulations of cardiac mechanics. Myocardial material was considered anisotropic and non-linearly elastic. Mechanical active stress in the myofiber direction depended on time elapsed after depolarization, sarcomere length, and sarcomere shortening velocity (40). Local active force development started at the moment of depolarization. 62 Chapter 5 Within the cardiac wall the equation of conservation of momentum was used while neglecting volumetric and inertial forces. Pressure loads on the LV and RV endocardium were assumed to be homogeneous, and equal to LV cavity pressure plv and RV cavity pressure prv , respectively, while load on the epicardium was assumed zero. To prevent rigid body motion, motion in the base to apex direction was set to zero at the base. In 3 points (anterior, posterior, and lateral) at the LV basal endocardium circumferential motion was also set to zero. LV mitral and RV tricuspid inflows were simulated by prescribing realistic increase of pressures in the non-activated ventricles from 0 to 1 kPa in the LV and from 0 to 0.25 kPa in the RV. LV and RV pressures in the isovolumic contraction and relaxation phases were estimated (11) such that LV and RV cavity volumes remained constant within 0.5%. During ejection, LV and RV pressures were related to aortic and pulmonary flow, respectively, with three-element Windkessel models (103). For the LV and RV, a characteristic input impedance (1.2 107 and 0.6 107 P a s m 3 , respectively) was put in series with a compliance (1.4 10 9 and 5.8 10 9 m3 P a 1 ) that was parallel to the peripheral flow resistance (1.2 108 and 1.65 107 P a s m 3 ). The aortic and pulmonary valves opened when cavity pressures exceeded end-diastolic aortic and pulmonaric pressures, being set at 10 and 2 kPa, respectively. Reversal of flows closed the valves. 5.2.3 Numerical implementation The equations were solved using the finite element package SEPRAN (SEPRA, Leidschendam, the Netherlands) on a 64-bit Origin 200 computer (SGI, Mountain View, CA, USA), using a single processor at 225 MHz on a UNIX platform. The eikonaldiffusion equation was discretized with 8-noded Galerkin-type hexahedral elements having trilinear interpolation of the field of depolarization times. The ventricles were subdivided into 33536 elements with 37405 degrees of freedom with a mean spatial resolution 1.1 mm. The equations related to mechanics were discretized with 27noded hexahedral elements with triquadratic interpolation of the displacement field. The ventricles were subdivided into 224 elements, with 6723 degrees of freedom. Calculation time for simulation of a complete cardiac cycle was approximately 30 hours. 5.2.4 Simulations and data analysis In total 3 simulations were performed. A normal cardiac cycle was simulated in which myofibers were activated synchronously (SYNC simulation) as described earlier (40). Furthermore, simulations of pacing at the RV apex (PACERV simulation) and LV free wall (PACELV simulation) were performed. Global hemodynamics and myofiber natural strains f were computed as a function of time (40). Stroke work density Wf [J m 3 ] was computed from f and Cauchy myofiber Intra- and interventricular asynchrony of electromechanics in the ventricularly paced heart 63 stress f by Wf = 5.3 I cardiac cycle f df (5.4) Results 5.3.1 Geometry Fit orders L (equation 5.2) for the epicardial, LV endocardial, and RV free wall and septal surface (figure 5.1) were set to 6, 6, 5, and 5, respectively. For these surfaces, the root mean squared distance between the original and fitted position of the data points was 0.80 mm, 0.86 mm, 0.36 mm and 0.48 mm, respectively. LV cavity, RV cavity, LV wall (LV free wall and septum), and RV free wall volumes were 26.2, 17.2, 57.6, and 7.90 ml, respectively (figure 5.2). 5.3.2 Global behavior Maximum ventricular pressures and ejection fractions in the SYNC simulation were slightly larger than in both simulations of pacing (figure 5.4, table 5.1). dp=dtmax for both the LV and RV was largest in the SYNC simulation as compared to the simulations of pacing, except for the LV in the PACELV simulation. When the LV was paced, left ventricular dp=dtmax was larger than left ventricular dp=dtmax when the RV was paced. Similarly, when the RV was paced, right ventricular dp=dtmax was larger then right ventricular dp=dtmax when the LV was paced. In both the SYNC and PACERV simulations, dp=dtmax was reached earlier in the RV than for the LV. In the PACELV simulation, dp=dtmax was reached earlier in the LV. In all simulations, stroke volume was larger for the LV than the RV. In the SYNC and PACERV simulations, the RV entered the ejection phase 24 and 26 ms before the LV, respectively (table 5.1). In the PACELV simulation, the LV entered this phase 12 ms before the RV. The ejection phase lasted longer for the LV than for the RV in all simulations. 5.3.3 Regional behavior Depolarization times for the SYNC, PACERV, and PACELV simulations are shown in the top row of figure 5.5. Depolarization for the SYNC simulation was completely synchronous, with depolarization times tdep = 0 for all myofibers. Complete depolarization of the heart was faster in the PACERV than in the PACELV simulation: maximum depolarization time in the PACERV and PACELV simulations was 92 and 129 ms, respectively. In the PACERV and PACELV simulations, the waves ended at the base of the left and right ventricular free wall, respectively. 64 Chapter 5 Figure 5.4: Global hemodynamics in the LV (–) and RV (- ) in the SYNC, PACERV, and PACELV simulations. The left panels for each simulation represent from top to bottom: cavity pressure [kPa]; cavity volume [ml] and flow [l/s] as a function of time. Mitral and tricuspid inflow were defined as negative and aortic and pulmonary outflow as positive. Dots indicate moments of opening and closure of the valves. The right panels for each simulation represent the pressure-volume loops. The sudden decrease in pressure and (as a result) increase in flow at the end of ejection in the PACERV simulation was due to a bug in the software. Sarcomere length [m] at the beginning of LV ejection and stroke work density [kJ m 3 ] are shown in the mid and bottom row of figure 5.5, respectively. For the SYNC simulation, sarcomere length was larger in the LV subendocardium than in the RV subendocardium. Sarcomere length and stroke work were distributed relatively homogeneously, except for a region in the LV base. Furthermore, in both simulations of pacing, sarcomere length and stroke work were dependent on the sequence of depolarization: low values were observed in the early depolarized regions, and high values in late ones. In the PACERV simulation, the septum and RV free wall thickened early, whereas the LV free wall was still relatively thin. The LV apex moved towards the RV side. In the PACELV simulation, the LV free wall thickened early, whereas the septum and RV free wall were still relatively thin. The LV apex moved away from the RV. For a better view on septal motion, a short-axis view of the basal area has been depicted in figure 5.6 for the beginning of LV ejection. Relative to the septum in the SYNC simulation, the septum in the PACERV moved towards the LV side. In the PACELV simulation, the septum moved towards the RV side. Intra- and interventricular asynchrony of electromechanics in the ventricularly paced heart pmax dp=dtmax qmax Vbe =Vw Vee=Vw Vs EF tdp=dtmax tic tbej tej kPa kPa/s ml/s ml % ms ms ms ms SYNC LV 13.2 206 221 0.95 0.54 23.5 42.9 234 58 258 176 RV 3.2 84.9 207 3.25 1.31 15.3 59.7 230 34 234 164 PACERV LV 12.9 137 199 0.95 0.57 22.3 40.5 282 106 306 174 RV 3.15 73.5 190 3.26 1.33 15.2 59.1 280 80 280 170 PACELV LV 13.1 240 213 0.95 0.55 23.4 42.5 300 114 314 174 65 RV 2.98 55.1 159 3.29 1.34 15.3 59.1 314 212 326 164 Table 5.1: Hemodynamic variables in the SYNC, PACERV, and PACELV simulations; pmax maximum cavity pressure; dp=dtmax maximum first time derivative of cavity pressure; qmax maximum outflow; Vbe and Vee cavity volume at beginning and end of the ejection phase, respectively; Vw wall volume; Vs stroke volume; EF ejection fraction; tdp=dtmax moment of dp=dtmax ; tic duration of isovolumic contraction phase; tbej beginning of ejection phase; tej duration of ejection phase 5.4 Discussion We have presented a three-dimensional finite element model of RV and LV electromechanics. With the model, complete cardiac cycles of a normal heart beat and pacing at the RVA and LVFW were simulated. Myofiber strain and stroke work density were determined by the sequence of depolarization. The earliest activated ventricle had the earliest start of ejection, while at the beginning of ejection the septum moved towards the last activated ventricle. The potential of the model in simulating conduction disturbances in a realistic cardiac geometry has been demonstrated: the geometry of the mesh was based on non-invasively obtained short axis MR images of a dog. 5.4.1 Physiology The depolarization sequence for LVFW pacing was in agreement with previously reported measurements (81). In the simulations with ventricular pacing, during the isovolumic contraction phase, myofibers shortened rapidly in the early depolarized regions, whereas in the late depolarized regions, myofibers were pre-stretched, as has been reported previously (26; 106). The pattern of stroke work density was also similar to experimentally determined patterns (70). In both simulations of pacing, maximum pressure and dp=dtmax decreased as compared to normal sinus rhythm, as has been reported earlier also (67). 66 Chapter 5 Figure 5.5: Anterior views of the heart, showing cross-sections of the walls, and LV and RV endocardium. Top panel: depolarization times [ms] for the SYNC, PACERV, and PACELV simulations, represented on the deformed mesh at end-diastole. Mid panel: sarcomere length [m] for the latter mentioned simulations. Sarcomere length is represented on the deformed mesh at the beginning of LV ejection. Bottom panel: stroke work density [kJ m 3 ] for the simulations. The stroke work is represented on the undeformed mesh in the reference state. Considering interventricular asynchrony, the following agreements with experiments were also observed. In the SYNC simulation the RV entered the ejection phase before the LV, as also has been reported previously in the canine heart (96). In simulating Intra- and interventricular asynchrony of electromechanics in the ventricularly paced heart 67 30 20 y [mm] 10 0 −10 −20 −30 −40 −50 −40 −20 0 x [mm] 20 Figure 5.6: Basal contours of the epicardium and LV and RV endocardium at the beginning of LV ejection for the SYNC (–), PACERV ( ), and PACELV (- ) simulations. Notice the movement of the septum to the LV and RV side relative to the SYNC simulation for RV apex pacing and pacing at the LV free wall, respectively. pacing of the LV, this ventricle entered the ejection phase first (96), while the septum moved towards the RV. In simulating pacing of the RV, the opposite was observed: the RV entered the ejection phase before the LV. The behavior as found in the simulations has also been reported in measurements on patients with a left bundle branch block (LBBB), where the depolarization sequence is similar to RV apex pacing (31). The septum flattened and moved towards the LV (31; 50). Therefore, excellent qualitative and good quantitative agreements were found between model and experiment, not only for regional mechanical behavior, but also for the timing and degree of pressure development and timing of volume ejection as well as septal displacement. 5.4.2 Limitations In the SYNC simulation, stroke work was more inhomogeneous than in earlier performed simulations (40; 74; 95) due to a simplified myofiber orientation. Filling and stroke volume were not similar for both ventricles. For one heart beat it is possible that one ventricle ejects slightly more blood than the other. However, on the average, filling and ejection have to be equal for the LV and RV. The difference in filling might be due to the geometry, which was referred to end-diastole as the zero-pressure 68 Chapter 5 state in stead of a state somewhere in mid-diastole. The difference in LV and RV stroke volume may also be related to inaccuracies in parameter values of aortic and pulmonary impedance parameters. The reference geometry corresponds to the end-diastolic state of a canine heart. Since the depolarization wave propagates mainly in this phase, the latter geometry is correct for solving timing of depolarization. However, for solving mechanics, the assumption that this state corresponds to the unloaded reference state is wrong. In the model, the cavity to wall volume ratios in the reference state were 0.51 and 2.31 for the LV and RV, respectively. In the real LV, cavity to wall volume ratio in the state of zero cavity pressure is about 0.30 (21). Finally, we assumed mechanical and electrical properties to be the same in the ventricles. The RV and LV are however somewhat different in electromechanical properties (15). 5.5 Conclusions A three-dimensional finite element model of electromechanics in the left and right ventricles has been developed. In the model, the ventricular geometry was obtained from available MRI measurements of a canine heart, with a rms error of about 0.8 mm. During sinus rhythm ventricular ejection started earlier for the right side than for the left side. In simulations with ventricular pacing, results agreed with experimental findings in the following aspects: 1) depolarization sequence; 2) the spatial distributions of sarcomere length and stroke work density depended mainly on timing of depolarization; 3) maximum pressure and maximum increase of pressure were lower than during sinus rhythm; 4) the earliest activated ventricle had the earliest start of ejection, and 5) the septum moved towards the last activated ventricle at the onset of systole. 5.6 Acknowledgements We thank Ryan Lahm at Medtronic, Minneapolis, MN for kindly providing the MR data set. Chapter 6 General discussion 69 70 6.1 Chapter 6 Introductory remarks In this thesis, a three-dimensional mathematical model of cardiac electromechanics has been designed. The model was used to investigate the EM delay in the normal heart, and the relation between depolarization and contraction in the ventricularly paced heart. The model is expected to become a useful tool in optimizing resynchronization therapy. For a normal heartbeat, a discrepancy was found. The model predicted that a physiological sequence of depolarization resulted in an unphysiologically non-uniform contraction pattern. When the sequence of depolarization was unphysiologically synchronous, the contraction pattern appeared physiologic. This finding suggests that myocardial tissue is able to synchronize contraction despite the presence of asynchrony of depolarization with a heterogeneous distribution of EM-delay. To obtain a realistic pattern of timing of depolarization, parameters of wave depolarization were estimated by fitting simulated to measured maps of depolarization time for LVFW pacing. The thus obtained parameters were tested in a simulation of RVA pacing. Then the simulated map of depolarization time appeared similar to experimentally obtained maps. Next, we wanted to investigate whether, and to what extent, heterogeneous EM-delay, as it occurs during a normal heartbeat, would effect regional contraction and total pump function during ventricular pacing. With the model, electromechanics in the left ventricle for pacing at the RV apex was simulated realistically. Pump function decreased as compared to normal sinus rhythm. The introduction of the EM-delay in the simulation of RV pacing made contraction slightly more synchronous. It is likely that the narrow physiological range of electromechanical delay to synchronize contraction in a normal heart is too small to significantly affect asynchrony during pacing. In a first step towards patientspecific modeling, a cardiac geometry, as measured in a dog with MRI, was applied in the model. With the model, simulations of depolarization wave propagation and cardiac mechanics were performed for a normal heartbeat, and for RVA and LVFW pacing. Myofiber strain and stroke work density were found to be determined by the sequence of depolarization. The earliest activated ventricle had the earliest start of ejection, while the septum moved towards the last activated ventricle at the beginning of ejection. 6.2 Modeling depolarization and contraction in a normally beating left ventricle 6.2.1 Mathematical models Understanding of electromechanical interaction is becoming more relevant because of the rapidly increasing interest in cardiac resynchronization (38; 96). There are several models on myocardial electrophysiology (for an overview, see Ref. (56)) and mechanics (11; 58; 86; 91) separately, but the behavior of the combination of the two principles in an entire ventricle is not well-known. In a model of cardiac mechanics (11), in which General discussion 71 timing of depolarization was prescribed for a normal heartbeat, unphysiological prestretching of late depolarized myofibers occurred. The same phenomena occurred in a model of cardiac electromechanics presented previously (90), but none of these studies focused on the subject of timing of contraction. Therefore, we developed a mathematical model of cardiac electromechanics in the LV to investigate the relation of the propagation of the depolarization wave and cardiac mechanics for a normal heartbeat and during pacing. 6.2.2 Model design Depolarization wave propagation was modeled using the eikonal diffusion equation (19). This equation solves the arrival time of the depolarization wave as a function of space, allowing for effects of anisotropic wave propagation. The method was preferred over the well-known bi-domain model (33) because of computational efficiency. Solutions of the eikonal diffusion equation and the bidomain model were reported to match closely (17). Normally, for a detailed description of depolarization wave propagation, a spatial resolution is needed as high as 0.2 mm, especially near strong curvatures of the wave front. For the conditions used in our simulations, a 2.4 mm resolution appeared sufficient: mesh refinement did not change the solution significantly. The propagation of depolarization appeared physiological as compared to several earlier reported measurements (23; 24; 57; 79; 80). Wall mechanics were determined by solving the equations of force equilibrium. Myocardial passive material was considered anisotropic and non-linearly elastic. Mechanical active stress in the myofiber direction was described as a function of time elapsed after depolarization, sarcomere length, and sarcomere shortening velocity. 6.2.3 Electromechanics in a normal heartbeat Two simulations of complete cardiac cycles were performed. First a cardiac cycle was simulated, in which local active force development was initiated at the moment of depolarization, as calculated for regular wave propagation during sinus rhythm. In a second simulation, active force was initiated synchronously. For both simulations, the time course of myofiber strains and distribution of stroke work density [kJ m 3 ] were compared to earlier reported measurements (21; 32; 69; 70; 106). The model predicted that a physiological sequence of depolarization resulted in an unphysiological, non-uniform contraction pattern. Making the sequence of depolarization unphysiologically synchronous, the simulated contraction pattern appeared physiologic. This finding suggests that myocardial tissue is able to synchronize contraction despite asynchrony of depolarization. It is not known yet what mechanism may be responsible for this behavior. Probably, there is a local controlling mechanism that makes contraction more synchronous than depolarization by proper variation of local electro-mechanical delay time in response to mechanical load. Actually, the simulation 72 Chapter 6 with synchronous depolarization can be interpreted as a simulation with a normal depolarization pattern and a heterogeneous distribution of electro-mechanical delay times, thus synchronizing for local contraction. 6.3 Simulating timing of depolarization during ventricular pacing During pacing, much time is needed for ventricular depolarization by slow conduction through the myocardium, rather than through the Purkinje system (68; 94; 99). In the ventricularly paced heart, the influence of the Purkinje system on myocardial propagation velocity depends on the distribution and density of the Purkinje-muscular junctions (PMJs), which has been reported to be variable (16; 23; 53; 72). Furthermore, it has been reported that the time needed for the depolarization wave to propagate from the myocardium into the Purkinje system is generally shorter than vice versa. All these characteristics of the Purkinje system result in a slower apparent myocardial wave velocity, than the wave velocity in the Purkinje system itself. In our model, details of PMJ density, distribution, and propagation delay were not included. Instead, these parameters were represented by choosing subendocardial wave velocity considerably larger than wave velocity in the rest of the myocardium. In chapter 3 it was investigated whether the timing of depolarization during pacing could be described with the eikonal-diffusion equation. First, parameter settings were determined for a best fit between simulation and experiment for the case of LVFW pacing. The most well-known parameters values were taken from literature (27; 75; 87; 104), being wave velocity parallel to the myofiber [m/s] and subendocardial anisotropy. Parameter k , related to the influence of wave front curvature on wave front velocity, was set to a realistic value of 2.1 10 4 m2 s 1 (88). The remaining two parameters, myocardial anisotropy am and effective wave velocity along the myofibers in the subendocardial layers ce , respectively, were estimated by adjusting the parameter values for getting a best fit between simulation and experimental findings (26) on the map of timing of epicardial for pacing at the left ventricular free wall. Optimal agreement between model and experiment was found by setting am = 2.5 and ce = 1.2 m/s. The setting of k appeared not to be critical for ventricular wave propagation during pacing. An increase of k did not change the timing of epicardial depolarization significantly. Substitution of the estimated parameter values in the model resulted in an accurate description of the depolarization wave with a different pacing site, located at the RV apex. Thus we conclude that the eikonal-diffusion is suitable to simulate depolarization wave propagation during ventricular pacing realistically, taking into account that wave propagation is faster in the subendocardium. General discussion 6.4 73 Simulating cardiac mechanics during ventricular pacing With the model, cardiac deformation was simulated during pacing at the RV apex. The simulated LV deformation pattern appeared realistic, when comparing the pattern with deformation patterns as measured in MRI tagging experiments on dogs. Both systolic pressure and ejection fraction decreased as compared to natural sinus rhythm. In early-depolarized regions, both shortening and stroke work declined. In the late depolarized regions, changes were opposite. The relation between moment of depolarization and the magnitude of normalized midwall circumferential strain was linear, with a slope of -47.7 2.9 s 1 in the model and -39.5 3.0 s 1 in the experiment for RV apex pacing. The sequence of onset of myofiber shortening was very similar to that of depolarization. This finding agrees with earlier reported experimental data (106) on ventricular pacing where moments of onset of shortening and depolarization were found to be linearly related, with a slope of about unity. The map of stroke work density in the simulation of RV apex pacing was similar to that reported earlier (70). In early-depolarized regions work appeared negative and work was about doubled in late depolarized regions. 6.5 A model of cardiac electromechanics in the paced composite of right and left ventricle During ventricular pacing, interventricular asynchrony affects pump function of both ventricles. A three-dimensional finite element model of LV and RV electromechanics was developed. The geometry was derived from the LV and RV of an anesthetized dog (m=26 kg). Cine-MR short axis images were obtained, ranging from base to LV apex. Simulations of complete cardiac cycles were performed for a normal heartbeat with synchronous depolarization and ventricular pacing. Pacing was simulated for pacing sites at the right ventricular apex or left ventricular free wall. In simulations with ventricular pacing, results agreed with experimental findings in the following aspects: 1) depolarization sequence was similar (81); 2) the spatial distributions of sarcomere length and stroke work density depended mainly on timing of depolarization (26; 70; 106); 3) maximum pressure and maximum increase of pressure were lower than during sinus rhythm (67); 4) the earliest activated ventricle had the earliest start of ejection (31; 50; 96), and 5) the septum moved towards the last activated ventricle at the onset of systole (31; 50). However, not all results were physiological. Due to simplifications, in the simulation with synchronous depolarization, distribution of stroke work density was less uniform than normal (74; 95), and stroke volume was not similar for both ventricles. We attribute these discrepancies to an inadequate 74 Chapter 6 setting of various model parameters. For example we assumed that in the RV and LV mechanical and electrical material properties were the same. It has been reported however, that electromechanical properties (15) in the RV wall are somewhat different than in the LV wall. Also, the distribution of myofiber orientation was simplified. Myofiber orientation is a major determinant of distribution of wall mechanics (13). It is not possible yet to accurately measure myofiber orientation in vivo, but can be optimized for homogeneous myofiber shortening (74). 6.6 Heterogeneous electromechanical delay? With a normal depolarization sequence, the onset of contraction is likely to be more synchronous than depolarization as shown in chapter 2. Apparently, there is a mechanism by which EM-delay adapts to enhance synchrony of contraction. Until now there is no direct experimental evidence for regional differences in electromechanical delay. However, the finding that expression of the Ito -channel is higher in the subepicardial layers than in the subendocardial layers (77) may reflect the physiology of such mechanism. The Ito channel has a prominent role in modulating the systolic calcium transient by enhancing the rise of the intra- cellular calcium concentration, which is reflected by the early repolarization in the action potential of subepicardial myocytes. Also, it has been measured that calcium transient is heterogeneously distributed from endocardium to epicardium (44). As a result, the time elapsed between depolarization and contraction may be shorter in the subepicardial layers than in the subendocardial ones. It was investigated whether, and to what extent, heterogeneous EM-delay, as it occurs during a normal heartbeat, would affect regional contraction during ventricular pacing. The introduction of the EM-delay in the simulation of RVA pacing, made contraction slightly more synchronous. It is likely that the narrow physiological range of electromechanical delay to synchronize contraction in a normal heart is too small to significantly affect asynchrony during ventricular pacing. 6.7 Future perspectives The model is likely to be a useful tool in the investigation of the influence of interventricular interaction on heart function during ventricular pacing. Furthermore, effects of left bundle branch block on contraction and on heart function in combination with resynchronization therapy can be investigated. We demonstrated the potential for modeling patient-specific electromechanics during ventricular pacing by inserting a measured cardiac geometry in the model. In general, every mammalian cardiac geometry can be implemented. Currently, it is possible to accurately measure the geometry of a heart non-invasively with MRI techniques with a slice thickness of only a few mm. This is also the resolution that is obtained in the General discussion 75 model, preserving acceptable computation times of a few hours. However, distribution of myofiber orientation, a major determinant of distribution of wall stress and strain (13), can only be measured accurately post-mortem (29). Adaptation models could be used to estimate a patient’s myofiber orientation. With the obtained patient’s cardiac geometry, the presented model of cardiac electromechanics may be useful in optimizing ventricular pacing therapy, or to understand cardiac deformation for various forms of pathology. Conduction disturbances like Wolf-Parkinson-White syndrome and bundle branch blocks can be simulated. Although not part of this study, it is also possible to simulate for example regional (acute or chronic) ischemia, aortic or pulmonary stenosis, myofiber disarray, or dilated hearts. 6.8 General conclusions A three-dimensional finite element model of LV and RV depolarization wave propagation and mechanics has been developed, suitable to simulate ventricular pacing. From the model, it appeared that the delay between depolarization and contraction (electromechanical delay) is distributed such, that during normal sinus rhythm, contraction is more synchronous than depolarization. Within the myocardium, cross-fiber velocity of wave propagation was estimated to be 0.4 times the velocity along the myofiber direction. Near the endocardium, wave propagation is faster than in the rest of the myocardium, but slower than as found in Purkinje fibers. The narrow physiological range of electromechanical delay (43 ms) to synchronize contraction in a normal heart is too small to significantly affect asynchrony during ventricular pacing (120 ms). The LV was succesfully extended with the RV. We demonstrated the potential for modeling patient-specific electromechanics during ventricular pacing by inserting a measured cardiac geometry in the model. 76 Chapter 6 Bibliography [1] Aelen, F. W. L., T. Arts, D. G. M. Sanders, G. R. P. Thelissen, A. M. M. Muijtjens, F. W. Prinzen, and R. S. Reneman. Relation between torsion and cross-sectional area change in the human left ventricle. J. Biomech. 30(3):207–212, 1997. [2] Aelen, F. W. L., T. Arts, D. G. M. Sanders, G. R. P. Thelissen, F. W. Prinzen, and R. S. Reneman. Kinematic analysis of left ventricular deformation in myocardial infarction using magnetic resonance cardiac tagging. Int. J. Card. Imaging 15(3):241–251, 1999. [3] Allgower, E. L. and K. Georg. Numerical continuation methods: an introduction. Springer-Verlag, Berlin, 1990. [4] American Heart Association. Heart Disease and Stroke Statistics - 2003 Update. American Heart Association, Dallas, Tex., 2002. [5] Andersen, H. R., J. C. Nielsen, and P. E. B. Thomsen. Long-term follow-up of patients from a randomised trial of atrial versus ventricular pacing for sick-sinus syndrome. Lancet 350:1210–1216, 1997. [6] Arts, T., P. C. Veenstra, and R. S. Reneman. A model of the mechanics of the left ventricle. Ann. Biomed. Eng. 7:299–318, 1979. [7] Arts, T., P. C. Veenstra, and R. S. Reneman. Epicardial deformation and left ventricular wall mechanics during ejection in the dog. Am. J. Physiology 243:H379–H390, 1982. [8] Axel, L. and L. Dougherty. MR imaging of motion with spatial modulation of magnetization. Radiology 171(3):841–845, 1989. [9] Berenfeld, O. and J. Jalife. Purkinje-muscle reentry as a mechanism of polymorphic ventricular arrhythmias in a 3-dimensional model of the ventricles. Circ. Res. 82:1063–1077, 1998. [10] Bers, D. M. Cardiac excitation-contraction coupling. Nature 415:198–205, 2002. 77 78 BIBLIOGRAPHY [11] Bovendeerd, P. H. M., T. Arts, D. H. van Campen, and R. S. Reneman. Dependence of local left ventricular wall mechanics on myocardial fiber orientation: a model study. J. Biomechanics 25(10):1129–1140, 1992. [12] Bovendeerd, P. H. M., T. Arts, T. Delhaas, J. M. Huyghe, D. H. van Campen, and R. S. Reneman. Regional wall mechanics in the ischemic left ventricle: numerical modeling and dog experiments. Am. J. Physiol. pp. H398–H409, 1996. [13] Bovendeerd, P. H. M., J. M. Huyghe, T. Arts, D. H. van Campen, and R. S. Reneman. Influence of endocardial-epicardial crossover of muscle fibers on left ventricular wall mechanics. J. Biomechanics 27(7):941–951, 1994. [14] Brooks, A. N. and T. J. R. Hughes. Stream-line upwind/Petrov-Galerkin formulation for convection dominated flows with particular emphasis on the incompressible Navier-Stokes equations. Comput. Methods Appl. Mech. Engrg 32:199–259, 1982. [15] Capasso, J. M., E. Puntillo, G. Olivetti, and P. Anversa. Differences in load dependence of relaxation between the left and right ventricular myocardium as a function of age in rats. Circ. Res. 68(3):1499–1507, 1989. [16] Cates, A. W., W. M. Smith, R. E. Ideker, and A. E. Pollard. Purkinje and ventricular contributions to endocardial activation sequence in perfused rabbit right ventricle. Am. J. Physiol. 281:H490–H505, 2001. [17] Colli-Franzone, P. and L. Guerri. Spreading of excitation in 3-D models of the anisotropic cardiac tissue. I. Validation of the eikonal model. Math. biosc. 113:145–209, 1993. [18] Colli-Franzone, P., L. Guerri, M. Pennacchio, and B. Taccardi. Spreading of excitation in 3-D models of the anisotropic cardiac tissue. II. Effects of fiber architecture and ventricular geometry. Math. biosc. 113:145–209, 1998. [19] Colli-Franzone, P., L. Guerri, and S. Tentoni. Mathematical modeling of the excitation process in myocardial tissue: influence of fiber rotation on wavefront propagation and potential field. Math. Biosc. 101:155–235, 1990. [20] Costa, K. D., P. J. Hunter, J. M. Rogers, J. M. Guccione, L. K. Waldman, and A. D. McCulloch. A three-dimensional finite element method for large elastic deformations of ventricular myocardium: II - prolate spheroidal coordinates. J. biomech. eng. 118:464–472, 1996. [21] Delhaas, T., T. Arts, P. H. M. Bovendeerd, F. W. Prinzen, and R. S. Reneman. Subepicardial fiber strain and stress as related to left ventricular pressure and volume. Am. J. Physiol. 264:H1548–H1559, 1993. BIBLIOGRAPHY 79 [22] Delhaas, T., T. Arts, F. W. Prinzen, and R. S. Reneman. Relation between regional electrical activation time and subepicardial fiber strain in the canine left ventricle. Pflügers Arch. 423:78–87, 1993. [23] Durrer, D., R. T. van Dam, G. E. Freud, M. J. Janse, F. L. Meijler, and R. C. Arzbaecher. Total excitation of the isolated human heart. Circ. 41:899–912, 1970. [24] Durrer, D., J. P. Roos, and J. Büller. The spread of excitation in the canine and human heart. In: International symposium on electrophysiology of the heart, edited by G. Marchetti and B. Taccardi, Pergamon Press, Oxford. 1964. [25] Faris, O., F. Evans, C. Ozturk, D. Ennis, J. Taylor, and E. McVeigh. Correlation between electrical and mechanical activation in the paced canine heart. In: Proc. Intl. Soc. Mag. Reson. Med. 9. ISMRM 2001, 2001. [26] Faris, O. P., F. J. Evans, D. B. Ennis, P. A. Helm, J. L. Taylor, A. S. Chesnick, M. A. Guttman, C. Ozturk, and E. R. McVeigh. Novel technique for cardiac electromechanical mapping with MRI tagging and an epicardial electrode sock. Ann. Biomed. Eng. 31(4):430–440, 2003. [27] Frazier, D. W., W. Krassowska, P. Chen, P. D. Wolf, N. D. Danieley, W. M. Smith, and R. E. Ideker. Transmural activations and stimulus potentials in three-dimensional anisotropic canine myocardium. Circ. Res. 63(1):135–146, 1988. [28] Gallagher, K. P., G. Osakada, O. M. Hess, A. Koziol, W. S. Kemper, and J. Ross, jr. Subepicardial segmental function during coronary stenosis and the role of myocardial fiber orientation. Circ. Res. 50(3):352–359, 1982. [29] Geerts, L., P. Bovendeerd, K. Nicolay, and T. Arts. Characterization of the normal cardiac myofiber field in goat measured with MR-diffusion tensor imaging. Am. J. Physiol. 283:H139–H145, 2002. [30] Greenstein, J. L., R. Wu, S. Po, G. F. Tomaselli, and R. L. Winslow. Role of the calcium-independent transient outward current Ito1 in shaping action potential morphology and duration. Circ. Res. 87:1026–1033, 2000. [31] Grines, C. L., T. M. Bashore, H. Boudoulas, S. Olson, P. Shafer, and C. F. Wooley. Functional abnormalities in isolated left bundle branch block. Circ. 79:845–853, 1989. [32] Guccione, J. M., W. G. O’Dell, A. D. McCulloch, and W. C. Hunter. Anterior and posterior left ventricular sarcomere lengths behave similarly during ejection. Am. J. Physiology 272(1 Pt 2):H469–H477, 1997. [33] Henriquez, C. S. Simulating the electrical behavior of cardiac tissue using the bidomain model. Crit. Rev. Biomed. Eng. 21(1):1–77, 1993. 80 BIBLIOGRAPHY [34] Hooks, D. A., K. A. Tomlinson, S. G. Marsden, I. J. LeGrice, B. H. Smaill, A. J. Pullan, and P. J. Hunter. Cardiac Microstructure - Implications for electrical propagation and defibrillation in the heart. Circ. Res. 91:331–338, 2002. [35] Huelsing, D. J., K. W. Spitzer, J. M. Cordeiro, and A. E. Pollard. Conduction between isolated rabbit Purkinje and ventricular myocytes coupled by a variable resistance. Am. J. Physiol. 274:H1163–H1173, 1998. [36] Huisman, R. M., G. Elzinga, and N. Westerhof. Measurement of left ventricular wall stress. Cardiovasc. Res. 14:142–153, 1980. [37] Hunter, P. J., A. D. McCulloch, and H. E. D. J. ter Keurs. Modelling the mechanical properties of cardiac muscle. Progr. Biophys. Mol. Biol. 69:289–331, 1998. [38] Kass, D. A., C. H. Chen, C. Curry, M. Talbot, R. Berger, B. Fetics, and E. Nevo. Improved left ventricular mechanics from acute VDD pacing in patients with dilated cardiomyopathy and ventricular conduction delay. Circ. 99(12):1567– 1573, 1999. [39] Keener, J. P. and A. V. Panfilov. The effects of geometry and fibre orientation on propagation and extracellular potentials in myocardium. In: Computational biology of the heart, edited by A. V. Panfilov and A. V. Holden, John Wiley & Sons Ltd., Baffins Lane, Chichester, chapter 8, pp. 235–258. 1997. [40] Kerckhoffs, R. C. P., P. H. M. Bovendeerd, J. C. S. Kotte, F. W. Prinzen, K. Smits, and T. Arts. Homogeneity of cardiac contraction despite physiological asynchrony of depolarization: A model study. Ann. Biomed. Eng. 31(5):1–12, 2003. [41] Kerckhoffs, R. C. P., O. P. Faris, P. H. M. Bovendeerd, F. W. Prinzen, K. Smits, E. R. McVeigh, and T. Arts. Timing of depolarization and contraction in the paced canine left ventricle: model and experiment. Submitted to J. Cardiovasc. Electrophysiol. 2003. [42] ter Keurs, H. E. D. J., J. J. J. Bucx, P. P. de Tombe, P. Backx, and T. Iwazumi. The effects of sarcomere length and Ca++ on force and velocity of shortening in cardiac muscle. In: Molecular mechanisms of muscle contraction, edited by H. Suga and G. H. Pollack, Plenum Publishing Corporation, pp. 581–593. 1988. [43] Kreyszig, E. Advanced engineering mathematics. John Wiley & Sons, Inc., 605 third av. NY, 8 edition, 1999. [44] Laurita, K. R., R. Katra, B. Wible, X. Wan, and M. H. Koo. Transmural heterogeneity of calcium handling in canine. Circ. Res. 92:DOI:10.1161/01.RES.0000062468.25308.27, 2003. BIBLIOGRAPHY 81 [45] Laurita, K. R. and A. Singal. Mapping action potentials and calcium transients simultaneously from the intact heart. Am. J. Physiol. Heart Circ. Physiol. 280:H2053–H2060, 2001. [46] LeGrice, I. J., B. H. Smaill, L. Z. Chai, S. G. Edgar, J. B. Gavin, and P. J. Hunter. Laminar structure of the heart: ventricular myocyte arrangement and connective tissue architecture in the dog. Am. J. Physiology 269:H571–H582, 1995. [47] Leon, L. J. and B. M. Horáček. Computer model of excitation and recovery in the anisotropic myocardium III. Arrhythmogenic conditions in the simplified left ventricle. J. Electr. 24(1):33–41, 1991. [48] Lin, D. H. S. and F. C. P. Yin. A multiaxial constitutive law for mammalian left ventricular myocardium in steady-state barium contracture or tetanus. J. Biomech. Eng. 120:504–517, 1998. [49] Lindsay, D. T. Functional human anatomy. Mosby-Year Book, Inc, St. Louis, Missouri 63146, 1996. [50] Little, W. C., R. C. Reeves, J. Arciniegas, R. E. Katholi, and E. W. Rogers. Mechanisms of abnormal interventricular septal motion during delayed left ventricular activation. Circ. 65:1486–1491, 1982. [51] Malvern, L. E. Introduction to the mechanics of a continuous medium. Prentice Hall, Inc., Englewood Cliffs, NJ, 1969. [52] Marcus, J. T., M. J. W. Götte, A. C. van Rossum, J. P. A. Kuijer, R. M. Heethaar, L. Axel, and A. Visser. Myocardial function in infarcted and remote regions early after infarction in man: assessment by magnetic resonance tagging and strain analysis. Magn. Res. Med. 38:803–810, 1997. [53] Massing, G. K. and T. N. James. Anatomical configuration of the His bundle and bundle branches in the human heart. Circulation 53(4):609–621, 1976. [54] Mazhari, T., J. H. Omens, J. W. Covell, and A. D. McCulloch. Structural basis of regional dysfunction in acutely ischemic myocardium. Cardiov. Res. 47:284–293, 2000. [55] McVeigh, E. R., F. W. Prinzen, B. T. Wyman, J. E. Tsitlik, H. R. Halperin, and W. C. Hunter. Imaging asynchronous mechanical activation of the paced heart with tagged MRI. Magn. Res. Med. 39:507–513, 1998. [56] Muzikant, A. L. and C. S. Henriquez. Validation of three-dimensional conduction models using experimental mapping: are we getting closer? Progr. Biophys. Mol. Biol. 69:205–223, 1998. 82 BIBLIOGRAPHY [57] Myerburg, R. J., K. Nilsson, and H. Gelband. Physiology of canine intraventricular conduction and endocardial excitation. Circ. Res. 30:217–243, 1972. [58] Nash, M. P. and P. J. Hunter. Computational mechanics of the heart. From tissue structure to ventricular function. J. Elast. 61:113–141, 2000. [59] Nikolić, S., E. L. Yellin, K. T. Tamura, H. Vetter, T. Tamura, J. S. Meisner, and R. W. M. Frater. Passive properties of canine left ventricle: diastolic stiffness and restoring forces. Circ Res 62(6):1210–1222, 1988. [60] Novak, V. P., F. C. P. Yin, and J. D. Humphrey. Regional mechanical properties of passive myocardium. J. Biomech. 27(4):403–412, 1994. [61] O’Dell, W. G. and A. D. McCulloch. Imaging three-dimensional cardiac function. Annu. Rev. Biomed. Eng. 2:431–456, 2000. [62] O’Dell, W. G., C. C. Moore, W. C. Hunter, E. A. Zerhouni, and E. R. McVeigh. Threedimensional myocardial deformations: calculation with displacement field fitting to tagged MR images. Radiology 195:829–835, 1995. [63] Omens, J. H. and Y. C. Fung. Residual strain in rat left ventricle. Circ. Res. 66(1):37–45, 1990. [64] Oosterhout van, M. F. M., F. W. Prinzen, T. Arts, J. J. Schreuder, W. Y. R. Vanagt, J. P. M. Cleutjens, and R. S. Reneman. Asynchronous electrical activation induces inhomogenous hypertrophy of the left ventricular wall. Circ. 98:588–595, 1998. [65] Ozturk, C. and E. R. McVeigh. Four-dimensional B-spline based motion analysis of tagged MR images: introduction and in vivo validation. Phys. Med. Biol. 45:1683– 1702, 2000. [66] Pandit, S. V., R. B. Clark, W. R. Giles, and S. S. Demir. A Mathematical Model of Action Potential Heterogeneity in Adult Rat Left Ventricular Myocytes. Biophys. J. 81:3029–3051, 2001. [67] Peschar, M., H. de Swart, K. J. Michels, R. S. Reneman, and F. W. Prinzen. Left ventricular septal and apex pacing for optimal pump function in canine hearts. JACC 41(7):1218–1226, 2003. [68] Prinzen, F. W., C. H. Augustijn, M. A. Allessie, T. Arts, T. Delhaas, and R. S. Reneman. The time sequence of electrical and mechanical activation during spontaneous beating and ectopic stimulation. Eur. Heart J. 13:535–543, 1992. [69] Prinzen, F. W., C. H. Augustijn, T. Arts, M. A. Allessie, and R. S. Reneman. Redistribution of myocardial fiber strain and blood flow by asynchronous activation. Am. J. Physiol. 259:H300–H308, 1990. BIBLIOGRAPHY 83 [70] Prinzen, F. W., W. C. Hunter, B. T. Wyman, and E. R. McVeigh. Mapping of regional myocardial strain and work during ventricular pacing: experimental study using magnetic resonance imaging tagging. JACC 33(6):1735–1742, 1999. [71] Prinzen, F. W. and M. Peschar. Relation between the pacing induced sequence of activation and left ventricular pump function in animals. PACE 25(4 Pt 1):484– 498, 2002. [72] Rawling, D. A., R. W. Joyner, and E. D. Overholt. Variations in the functional electrical coupling between the subendocardial Purkinje and ventricular layers of the canine left ventricle. Circ. Res. 57:252–261, 1985. [73] Rijcken, J., P. H. M. Bovendeerd, A. J. G. Schoofs, D. H. van Campen, and T. Arts. Optimization of cardiac fiber orientation for homogeneous fiber strain at beginning of ejection. J. Biomech. 30:1041–1049, 1997. [74] Rijcken, J., P. H. M. Bovendeerd, A. J. G. Schoofs, D. H. van Campen, and T. Arts. Optimization of cardiac fiber orientation for homogeneous fiber strain during ejection. Ann. biomed. eng. 27:289–297, 1999. [75] Roberts, D. E. and A. M. Scher. Effect of tissue anisotropy on extracellular potential fields in canine myocardium in situ. Circ. Res. 50:342–351, 1982. [76] Rodriguez, E. K., J. H. Omens, L. K. Waldman, and A. D. McCulloch. Effect of residual stress on transmural sarcomere length distribution in rat left ventricle. Am. J. Physiology 264:H1048–H1056, 1993. [77] Sah, R., R. J. Ramirez, and P. H. Backx. Modulation of Ca2+ release in cardiac myocytes by changes in repolarization rate - role of phase-1 action potential repolarization in excitation-contraction coupling. Circ. Res. 90:165–173, 2002. [78] Salama, G., A. Kanai, and I. R. Efimov. Subthreshold stimulation of Purkinje fibers interrupts ventricular tachycardia in intact hearts. Circ. Res. 74:604–619, 1994. [79] Scher, A. M., A. C. Young, A. L. Malmgren, and R. V. Erickson. Activation of the interventricular septum. Circ. Res. 3:56–64, 1955. [80] Scher, A. M., A. C. Young, A. L. Malmgren, and R. R. Paton. Spread of electrical activity through the wall of the left ventricle. Circ. Res. 1:539–547, 1953. [81] Spach, M. S. and R. C. Barr. Analysis of ventricular activation and repolarization from intramural and epicardial potential distributions for ectopic beats in the intact dog. Circ Res 37:830–843, 1975. [82] Streeter, D. D. Gross morphology and fiber geometry of the heart. In: Handbook of Physiology - The cardiovascular system I, edited by R. M. Berne, Am. Physiol. Soc., Bethesda, chapter 4, pp. 61–112. 1979. 84 BIBLIOGRAPHY [83] Suga, H. Total mechanical energy of a ventricular model and cardiac oxygen consumption. Am. J. Physiol. 236:H498–H505, 1979. [84] Suga, H., Y. Goto, H. Yaku, S. Futaki, Y. Ohgoshi, and O. Kawaguchi. Simulation of mechanoenergetics of asynchronously contracting ventricle. Am. J. Physiol. 259:R1075–R1082, 1990. [85] Taber, L. A. and W. W. Podszus. A laminated shell model for the infarcted left ventricle. Int. J. Solids & Structures 34(2):223–241, 1997. [86] Taber, L. A., M. Yang, and W. W. Podszus. Mechanics of ventricular torsion. J. Biomechanics 29(6):745–752, 1996. [87] Taccardi, B., E. Macchi, R. L. Lux, P. R. Ershler, S. Spaggiari, S. Baruffi, and Y. Vyhmeister. Effect of myocardial fiber direction on epicardial potentials. Circ. 90(6):3076–3090, 1994. [88] Tomlinson, K. A. Finite element solution of an eikonal equation for excitation wavefront propagation in ventricular myocardium. Ph.D. thesis, The University of Auckland, 2000. [89] van der Toorn, A., P. Barenbrug, G. Snoep, F. H. van der Veen, T. Delhaas, F. W. Prinzen, and T. Arts. Transmural gradients of cardiac myofiber shortening in aortic valve stenosis patients using MRI-tagging. Am. J. Physiol. Heart. Circ. Physiol. 283(4):H1609–H1615, 2002. [90] Usyk, T. P., I. J. LeGrice, and A. D. McCulloch. Computational model of threedimensional cardiac electromechanics. Comput. Visual Sci. 4:249–257, 2002. [91] Usyk, T. P., R. Mazhari, and A. D. McCulloch. Effect of laminar orthotropic myofiber architecture on regional stress and strain in the canine left ventricle. J. Elast. 61:143–164, 2000. [92] Usyk, T. P., J. H. Omens, and A. D. McCulloch. Regional septal dysfunction in a three-dimensional computational model of focal myofiber disarray. Am. J. Physiol. 281:H506–H514, 2001. [93] Vassall-Adams, P. R. Ultrastructure of the human atrioventricular conduction tissues. Eur. Heart J. 4:449–460, 1983. [94] Vassallo, J. A., D. M. Cassidy, J. M. Miller, A. E. Buxton, F. E. Marchlinski, and M. E. Josephson. Left ventricular endocardial activation during right ventricular pacing: effect of underlying heart disease. J. Am. Coll. Cardiol. 7:1228–1233, 1986. [95] Vendelin, M., P. H. M. Bovendeerd, J. Engelbrecht, and T. Arts. Optimizing ventricular fibers: uniform strain or stress, but not ATP consumption, leads to high efficiency. Am. J. Physiol. Heart Circ. Physiol. 283(3):H1072–H1081, 2002. BIBLIOGRAPHY 85 [96] Verbeek, X. A. A. M., K. Vernooy, M. Peschar, T. van der Nagel, A. van Hunnik, and F. W. Prinzen. Quantification of interventricular asynchrony during LBBB and ventricular pacing. Am. J. Physiol. 283:H1370–H1378, 2002. [97] Vergroesen, I., M. I. M. Noble, and J. A. E. Spaan. Intramyocardial blood volume change in first moments of cardiac arrest in anesthetized goats. Am. J. Physiol. 253:H307–H316, 1987. [98] Villareal, F. J., W. Y. W. Lew, L. K. Waldman, and J. W. Covell. Transmural myocardial deformation in the ischemic canine left ventricle. Circ. Res. 68(2):368– 381, 1991. [99] Waldman, L. K. and J. W. Covell. Effects of ventricular pacing on finite deformation in canine left ventricles. Am. J. Physiol. 252:H1023–H1030, 1987. [100] Waldman, L. K., D. Nosan, F. Villareal, and J. W. Covell. Relation between transmural deformation and local myofiber direction in canine left ventricle. Circ. Res. 63:550–562, 1988. [101] Weirich, W. L., V. L. Gott, and C. W. Lillehei. The treatment of complete heart block by the combined use of a myocardial electrode and an artificial pacemaker. Surg. Forum 8:360–363, 1957. [102] Wells, P. N. T. Physical principles of ultrasonic diagnosis. Academic Press, New York, 1969. [103] Westerhof, N., G. Elzinga, and G. C. van den Bos. Influence of central and peripherical changes on the hydraulic input impedance of the systemic arterial tree. Med. Biol. Eng. 11:710–723, 1973. [104] Wikswo Jr. , J. P., T. A. Wisialowski, W. A. Altemeier, W. A. Balser, H. A. Kopelman, and D. M. Roden. Virtual effects during stimulation of cardiac muscle. Twodimensional in vivo experiments. Circ. Res. 68(2):513–530, 1991. [105] Wilkoff, B. L., J. R. Cook, and A. E. Epstein. Dual-chamber pacing or ventricular back-up pacing in patients with an implantable defibrillator. JAMA 288:3115– 3123, 2002. [106] Wyman, B. T., W. C. Hunter, F. W. Prinzen, and E. R. McVeigh. Mapping propagation of mechanical activation in the paced heart with MRI tagging. Am. J. Physiol. 276:H881–H891, 1999. [107] Xu, Z., R. M. Gulrajani, F. Molin, M. Lorange, B. Dubé, P. Savard, and R. A. Nadeau. A computer heart model incorporating anisotropic propagation III: Simulation of ectopic beats. J. Electr. 29(2):73–90, 1996. [108] Yin, F. C. P., C. C. H. Chan, and R. M. Judd. Compressibility of perfused passive myocardium. Am. J. Physiol. 271:H1864–H1870, 1996. 86 BIBLIOGRAPHY Samenvatting Het hart is een holle spier dat bloed door het vatenstelsel pompt. Contractie van de hartspiervezels wordt geı̈nitieerd door depolarisatie van het celmembraan. Een coherente contractie van alle spiervezels in de hartwand is vereist voor een efficiënte pompfunctie van het gehele hart. Een gespecialiseerd geleidingssysteem in het hart zorgt voor een snelle verspreiding van de depolarisatie-golf over de wand. Soms is de normale geleiding van de depolarisatie golf verstoord, bijvoorbeeld na een blokkade in het geleidingssysteem. Abnormale ventriculaire depolarisatie patronen leiden tot een afname in pompfunctie en kunnen een bijdrage leveren aan hartfalen. In de kliniek worden pacemakers gebruikt om een normaal hartritme te onderhouden, maar gedurende de laatste tien jaar zijn er steeds meer aanwijzingen dat de abnormale depolarisatie en contractie patronen negatieve bijwerkingen zijn van pacing therapie. Deze bijwerkingen zouden verminderd kunnen worden door betere pacing lokaties te kiezen, dan gebruikelijk is. Wiskundige modellen zouden een bijdrage kunnen leveren om de beste pacing lokatie te vinden. Normaal wordt het tijds-interval tussen depolarisatie en contractie als constant verondersteld. Echter, zgn. excitatie-contractie koppeling is complex en bestaat uit verscheidene stappen. Cardiale excitatie-contractie koppeling is het proces van electrische excitatie tot mechanische contractie van hartspiervezels. Een goede kennis van dit proces is belangrijk om fysiologie en patho-fysiologie van hart functie te begrijpen. Timing van excitatie-contractie (oftewel electromechanische delay) is gemeten in cellen, waarbij vooral op calcium transport geconcentreerd werd. Calcium is zowel betrokken bij electrische processen als ook bij het opbouwen van kracht. In het complete hart zijn depolarisatie en deformatie gemeten. MRI tagging is gebruikt om lokale rek in drie dimensies te meten als functie van de tijd in combinatie met metingen van epicardiale depolarisatie. Met alleen rek is het niet mogelijk om het moment van mechanische activatie eenduidig te bepalen. Wiskundige modellen kunnen daarbij een handig hulpmiddel zijn in het begrijpen van electromechanische delay. Er zijn meerdere wiskundige modellen beschreven die betrekking hebben cardiale electrofysiologie en mechanica, maar er is nog geen gecombineerd model gebruikt om cardiaal ventriculair pacen te bestuderen. In dit proefschrift wordt de ontwikkeling en eerste resultaten van een drie-dimensionaal eindige elementen model beschreven, dat electrofysiologie en mechanica beschrijft in de linker hartkamer alleen en linker en rechter hartkamers. Het model wordt gebruikt om het electromechanisch delay te 87 88 Samenvatting onderzoeken in het normale en gepacede hart. Het model is getest door gesimuleerde patronen van electromechanica met experimenteel verkregen patronen te vergelijken. Verwacht wordt, dat het model een gereedschap zal worden om pacing therapiën te optimaliseren. De geometrie van de linker en rechter hartkamer is verkegen uit MRI metingen van een hondenhart. De vezelrichting en Purkinje syteem zijn ook geı̈mplementeerd. De depolarisatie-tijd is gemodelleerd door de eikonal-diffusie vergelijking op te lossen, rekening houdend met de lokale anisotropie van de vezelstructuur. De passieve extra-cellulaire matrix van het hartweefsel wordt niet-lineair elastisch en anisotroop verondersteld. Actieve vezelspanning is een functie van tijd, sarcomeerlengte en sarcomeer verkortings-snelheid. Lokale waarden als depolarisatie tijd, verplaatsing, rek en spanning voor een hele hartyclus worden uit het model verkregen. Globale hemodynamische waarden als rechter en linker volumes van de hartkamers, drukken en debieten worden ook berekend. Het depolarisatie-patroon voor een gezond hart en voor een gepaced hart is realistisch gesimuleerd. Een van de meest opvallende bevindingen was dat de tijd die verstrijkt tussen het moment van lokale depolarisatie en begin van lokale contractie heterogeen verdeeld bleek te zijn over de hartwand. Er bestaat blijkbaar een mechanisme, dat het electromechanisch delay zo aanpast, dat contractie synchroner is dan depolarisatie in het gezonde hart. Depolarisatie was veel asynchroner in de simulaties van ventriculair pacen dan in een normale hartslag, wat overeenkomt met metingen. De distributie van electromechanisch delay, die nodig is om contractie in een normale hartslag te synchroniseren, werd toegepast in een simulatie van ventriculair pacen. De gesimuleerde distributie van vezelverkorting veranderde hierdoor maar zeer weinig. Blijkbaar is de verdeling van electromechanisch delay (40 ms) te klein om de asynchronie in een gepaced hart (120 ms) significant te beı̈nvloeden. Tenslotte werd de intra- en interventriculaire asynchronie van depolarisatie, contractie en pomp-functie in een combinatie van de linker- en rechterhartkamer realistisch gesimuleerd. Daarbij werd een geometrie gebruikt die verkregen was uit MRI beelden van een hondenhart. Op deze manier kan een geometrie in het model gebruikt worden, die patiënt specifiek is. We concluderen dat het model van ventriculaire electromechanica succesvol gebruikt is in het bepalen van de rol van electromechanisch delay in het gehele kloppende hart. Verwacht wordt dat het model verscheidene pathologiën realistisch zal kunnen simuleren, zoals geleidingsstoornissen en het effect van behandelingen, zoals pacen. Dankwoord Dit proefschrift zou niet tot stand zijn gekomen zonder hulp van een aantal mensen. Peter en Theo, allereerst wil ik jullie bedanken voor de goede samenwerking en de vele interessante discussies (ook op niet-wetenschappelijk gebied). Frits, jouw enthousiasme gaf altijd weer een impuls in mijn motivatie. De discussies met Karel waren steeds een goed moment om alles even op een rij te zetten. Dick, vooral in het begin betrokken, bedankt voor het optreden als tweede promotor. En dan wil ik natuurlijk alle stagiair(e)s Jiska, Judith, Lotte, Mirjam, Petra, Wilco en Wilma bedanken voor jullie bijdrage. Owen and Elliot (I enjoyed the hike and the bear-encounter), thanks for kindly providing the experimental data and giving me a chance to talk at NIH. Also, the hospitality of the people at Medtronic is gratefully acknowledged: Josée, (the Art Crawl was great), Lee (cool boat!), Ryan (I’m glad the Twins won), and Walt (great time in Atlanta!). Alle collega’s maar vooral m’n ex-kamergenoten Hans, Liesbeth en Lyosha en de leden van de family-room Debby, Emiel en Leonie: bedankt voor alle serieuze discussies, maar alle onzin was natuurlijk het leukst. Marjolein, de squash partijen waren altijd een goede uitlaatklep. Patrick en Leo, bedankt voor jullie hulp bij alle computer hard- en software-problemen. Tot slot wil ik Barry bedanken voor zijn opbeurende droge e-mails, Mandy voor de spannende verhalen betreffende haar werk, en pap en mam voor alle steun. 89 90 Dankwoord Curriculum Vitæ 3 maart 1973 1985-1991 1991-1995 1995-1998 1998-2003 2003 Geboren te Geleen VWO, Stella Maris SG, Meerssen Werktuigbouwkunde, Hogeschool Heerlen, Sector Techniek Verkorte opleiding werktuigbouwkunde Technische Universiteit Eindhoven Assistent in Opleiding, Biofysica, Universiteit Maastricht en Biomedische Technologie, Technische Universiteit Eindhoven Onderzoeker, Biofysica, Universiteit Maastricht en Biomedische Technologie, Technische Universiteit Eindhoven 91