Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Coordination complex wikipedia , lookup
Spin crossover wikipedia , lookup
Evolution of metal ions in biological systems wikipedia , lookup
Metalloprotein wikipedia , lookup
Stability constants of complexes wikipedia , lookup
Ring-closing metathesis wikipedia , lookup
Wolff–Kishner reduction wikipedia , lookup
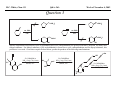
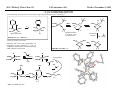
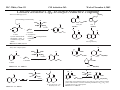
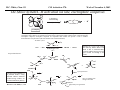
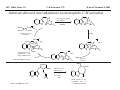
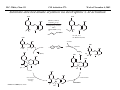
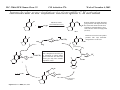
M.C. White, Chem 153 C-H Activation -241- Week of November 4, 2002 The Holy Grail of Catalysis R R CH3 CH2[M] ? R CH2R' C-H activation: Process where a strong C-H bond (90-105 kcal/mol) undergoes substitution to produce a weaker C-M bond (50-80 kcal/mol). Functionalization: Metal-C bond is replaced by any bond except C-H. Methods have been identified to regioselectivity effect C-H activation. Recall that there is both a kinetic and thermodynamic preference to form the less sterically hindered 1o C-M intermediate (see Structure & Bonding; pg. 32). The challenge lies in finding ways to selectively form the C-M intermediate under synthetically useful, mild conditions that enable functionalization and catalyst renewal. ARTHUR: Yes we seek the Holy Grail (clears throat very quietly). Our quest is to find the Holy Grail. KNIGHTS: Yes it is. ARTHUR: And so we’re looking for it. KNIGHTS: Yes we are. BEDEVERE: We have been for some time. KNIGHTS: Yes. ROBIN: Months. ARTHUR: Yes…and any help we get is…is very…helpful. Bergman Acc. Chem. Res. 1995 (28) 154. Exerpt from “Monty Python and the Holy Grail”; 1974. M.C. White, Chem 153 C-H Activation -242- Week of November 4, 2002 Bergman:C-H Activation via Late, Nucleophilic Complexes H H M π-backbonding>> σ-donation Hydrido(alkyl)metal complex M oxidative addition C C σ-complex regioselectivity: sp2 C-H > 1o sp3C-H> 2 o sp 3 C-H >>> 3o sp3 C-H. There is both a kinetic and thermodynamic preference to form the least sterically hindered C-M σ bond. Kinetic preference: activation barrier to σ-complex formation is lower for less sterically hindered C-H bonds and bonds with more s character. Thermodynamic preference: stronger C-M bonds are formed (see Structure and Bonding, pg. 32). MIII H H Me3P M = Ir, 1 Rh, 2 hv or ∆ These hydrido(alkyl)metal complexes are prone to non-productive reductive elimination in the presence of oxidants and non-productive protonolysis in the presence of protic reagents π-donor H2 MI MI ligand dissociation oxidative addition L 16 e- MI CO hv or ∆ coordinatively and electronically unsaturated intermediate H L proposed σ-complex intermediate OC CO low OS metals capable of 18 edonating electrons in σ-bond M = Ir, 3 formation. Highly prone to air Rh, 4 oxidation. Bergman JACS 1982 (104) 352 (Cmp. 1). Bergman OM 1984 (3) 508 (competition exp). Graham JACS 1983 (105) 7190 (Cmp. 3). Bergman JACS 1994 (116) 9585 (Cmp. 4). MIII with acyclic substrates the Rh complex inserts only into 1o C-H bonds arbitrarily set at 1 H L 18 eRelative rate constants for attack at a single C-H bond by 1 and 2 at -60 oC. C-H bond benzene cyclopropane n-hexane (1o) n-hexane (2o) propane (1o) propane (2o) cyclopentane cyclohexane krel (Rh, 2) krel (Ir, 1) 3.9 19.5 2.1 10.4 2.7 5.9 0.2 0 1.5 2.6 0.3 0 1.1 1.8 1.0 1.0 M.C. White, Q. Chen Chem 153 C-H Activation -243- Week of November 4, 2002 Evidence for intermolecular σ-complex formation CD3 D3C CD3 CD3 RhI CO D hv (flash), Kr (165K) OC CO 18 e- RhI RhI [Kr] RhIII D2C OC -1 CD3 D3C CO v (1946 cm ) D3C CD3 CO v (1947 cm-1) Bergman JACS 1994 (116) 9585. CD2 OC OC D CD3 CD3 CO v (2008 cm-1) σ-complex ‡ D Rh ∆G (kcal/mol) OC CD2(C(CD3)3 + 6.9 kcal/mol RhI [Kr] OC + (CD3)4C -3.2 kcal/mol to products RhI OC D D2C CD3 D3C CD3 The reaction of Cp*Rh(CO)2 with neopentane-d12 was monitored using low-temperature IR flash kinetic spectroscopy. The CO stretch at 1946 cm-1 was assigned to the initial intermediate Cp*Rh(CO)(Kr) complex, which after photolysis-mediated formation shows rapid decay. During this time, a second CO stretch at 1947 cm-1 grows in and then decays; this absorption is assigned to a transient intermediate Rh---CD σ-complex. The absorption at 2008 cm-1 is known to correspond to the product Cp*Rh(CO)(D)(C5D11), which increases steadily throughout the course of the reaction. Note that this entire process occurs in less than 1.5 ms. M.C. White, Chem 153 C-H Activation -244- Week of November 4, 2002 Evidence for concerted C-H oxidative addition crossover experiment: evidence in support of a concerted mechanism. H IrI H 3C OC IrIII H IrIII D OC D12 IrI hv CO OC IrI OC CO 18 e- IrI D OC OC D11 D11 σ-complexes Less than 7% of the crossover products were observed by 1 HNMR. This may be indicative of a minor radical pathway. H2C D11 II Ir H IrII + OC Bergman JACS 1983 (105) 3929. OC IrIII D OC IrIII H OC D11 D M.C. White, Chem 153 C-H Activation -245- Week of November 4, 2002 Dehydrogenation of alkanes to alkenes -H2 R R H2 Catalyst requirements: H2, regeneration via olefin dissociation and elimination of H2. H2 must be rapidly and irreversibly removed to avoid olefin hydrogenation and isomerization R H 3L H MLnx MLnx-3 18e- "14e-" H R R oxidative addition metal capable of shuttling between Mn and Mn-2 oxidation states β -hydride R MLn-2x-1 elimination MLn-2x H 16e- H 18e- complex capable of accomodating 3 ligands from the substrate in its coordination sphere mid-cycle The first report: O PPh3 (BF4 Ir(III) H Ph3P (coe) + H O recall: intermediate in cationic hydrogenation catalysts Crabtree JACS 1979 (101) 7738. -) 10 eq o + H CD2Cl2, -60 C Ir(III) Ph3P observed to form quantitatively by NMR PPh3 + (BF4-) o o -10 C->40 C Ir(I) H PPh3 (BF4-) PPh3 75% recall: hydrogenation catalyst M.C. White, Chem 153 C-H Activation -246- Week of November 4, 2002 Crabtree:thermal dehydrogenation of alkanes to alkenes P(p-FC6H4) 3 O H CF3 Ir(III) solvent + 150oC 14 days 355 mM sacrificial H2 acceptor with unusually high heat of hydrogenation 56% (54%) 4% (3%) P(p-FC6H4) 3 7.1 nM t-Bu + + O H 18% (17.5%) yields based on catalyst. + t-B u trans-3-hexene 14% (18.5 %) cis-3-hexene 8% (7.5 %) 2d, 1.4 tn 2d, 3 tn tn = turnover # Proposed Mechanism: H hydrogenation H Ir(III) CF3 F Ir(III) β-hydride elimination "tail-biting" H Ir(III) (p-FC6H4)3P (C6H4p-F)3P OC(O)CF3 CF3 O Crabtree JACS 1987 (109) 8025. (p-FC6H4)3P CF3 H O Ir 14 e- Ir(III) OC(O)CF3 Ir(III) CF3 O t-Bu (p-FC6H4)3P O P(p-FC6H 4) 3 (I) hydrogenation pathway P(p-FC6H 4) 3 t-Bu P(p-FC6H 4) 3 O Ir(I) R isomerization pathway H t-Bu (C6H4p-F)3P OC(O)CF3 P(p-FC6H4) 3 (C6H4p-F)3P (C6H4p-F)3P R Ir(III) R (p-FC6H4)3P H oxidative addition R OC(O)CF3 H O R P(p-FC6H4) 3 H H P(p-FC6H4) 2 H isomerization P(p-FC6H 4) 3 OC(O)CF3 (p-FC6H4)3P Ir(III) H t-Bu P(p-FC6H4) 3 H H O H P(p-FC6H 4) 3 R O Ir(III) 2d, 9 tn P(p-FC6H 4) 3 P(p-FC6H4) 3 R Product distributions of linear alkenes are thought to result from isomerization of the initial kinetic 1-ene product via intermediate Ir hydride species. Subjecting 1-hexene to the reaction conditions gives similar olefin distributions (in parentheses). OC(O)CF3 R only trifluoroacetate complexes were active in alkene dehydrogenations. Their greater lability with respect to acetate may allow more facile interconversion from η3 to η1 necessary to provide an open coordination site for H2 acceptor binding. M.C. White, Chem 153 C-H Activation -247- Week of November 4, 2002 Crabtree:photochemical dehydrogenation of alkanes to alkenes P(Cy)3 (III) CF3 Ir O H solvent + 7.1 nM P(Cy)3 hv (254 nm) 7 days t-Bu tbe 355 mM + t-B u 2.77tn (1.6) + H2 2.19 tn (3.84) 0.85 tn (0.32) 1.26 tn (0.82) tn w/out tbe present (in parentheses). Irradiation with light of the appropriate wavelength promotes reductive elimination of the dihydride catalyst leading directly to the catalytically active 14e- complex. It's interesting to note that no reaction takes place with tbe in the absence of 254 nm light. This implies that tbe acts as a H 2 acceptor from a photochemically excited intermediate. P(Cy)3 R H isomerization pathway P(Cy)3 OC(O)CF3 O Ir(III) CF3 O H P(Cy)3 Ir(III) + + Proposed Mechanism: H Under conditions of hv and tbe, methylcyclohexane is the preferred product. This is thought to result from a kinetic preference to form the sterically less hindered M-C bond. Methylenecyclohexane subjected to the reaction conditions results in only 25% conversion to the thermodynamically more stable 1-methylcyclohexene. Although the reaction proceeds w/out tbe, the product ratios reflect more isomerization activity. O H H Ir(III) hv, 254nm P(Cy)3 OC(O)CF3 H H R (Cy)3P H β -hydride elimination H Ir(III) OC(O)CF3 O O P(Cy)3 O Ir(I) CF3 O (Cy)3P H2 H (Cy)3P oxidative addition R t-B u (Cy)3P R Crabtree JACS 1987 (109) 8025. CF3 Ir(III) H (Cy)3P P(Cy)3 * P(Cy)3 H2 R (Cy)3P (Cy)3P Ir(I) 14 e- OC(O)CF3 t-B u Some free H2 is formed even in the presence of tbe. M.C. White, Chem 153 C-H Activation -248- Week of November 4, 2002 Tanaka: photochemical dehydrogenation OC Rh(I) PMe3 Cl 0.7mM Me3P hv, rt, N 2 27 h, 155 tn (solvent) PMe3/Rh 2 5 5 10 10 Proposed Mechanism: time (h) 1- 1 3 22 3 22 1 12 6 28 10 + + + 1:79:20 138 tn, 17 h A theoretical amount of H2 was detected in the gas phase. When a N2 stream was used, tn increased to 195 tn. H2 hexenes 2311 4 4 4 3.4 2 1 1 1 1 Added phosphine ligand decreases the efficiency of the reaction but increases the regioselectivity towards formation if 1-hexene. Within the same PMe3/Rh ratio, an erosion in regioselectivity is observed upon prolonged reaction times. This is indicative of catalyst mediated alkene isomerization. Could this ratio also be reflective of the rates of olefin hydrogenation? Exposure of 1-hexene to the reaction conditions results in 2-hexene (35%) and hexane (63%) after 22 h. TN 5.4 4.0 18.7 0.6 7.2 OC Added phosphine ligand may take up a vacant coordination site cis to the M-alkyl, preventing formation of the agostic interaction necessary to effect β-hydride elimination. A decrease in both alkane dehydrogenation and olefin isomerization results. Me3P CO Me3P Rh(I) PMe3 R Cl 14 eH R β -hydride elimination R H H Cl hv H2 R PMe3 16 e- light-promoted reductive elimination of H2 ?? reductive elimination Rh(I) PMe3 Rh(III) Cl PMe3 R H PMe3 Rh(III) H Cl PMe3 H PMe3 Rh(III) Cl PMe3 intermediate in Wilkinson hydrogenation 930 tn, 69h N2 stream M.C. White, Chem 153 C-H Activation -249- Week of November 4, 2002 Goldman: Wilkinson’s Catalyst Varient sacrificial alkenes OC Rh(I) Me3P (solvent) + sacrificial alkene PMe3 , 53 tn , 59 tn Cl 0.7mM H2 (1000 psi), 60oC 1.5 h, x tn + t-Bu , 106 tn alkane , 4 tn A variety of sacrificial alkenes work in the dehydrogenation of cyclooctane, an especially reactive substrate. Cyclooctene has a very low heat of hydrogenation probably resulting from transannular steric repulsions in cyclooctane which are less severe in cyclooctene. n-hexane gave hexenes in modest tn (9.6) with norbornene as the H2 acceptor. No mention was made to the isomer distributions. Goldman JACS 1992 (114) 9492. H Proposed Mechanism: OC Rh (I) Me3P PMe3 H2 Cl H Rh(III) Me3P 16 e- PMe3 Cl CO 18eCO Formation of octahedral dihydride complex is thought to initiate ligand dissociation. Wilkinson's hydrogenation catalyst (see hydrogenation, pg. 142), known to dissociate PPh3 upon H2 oxidative addition, is cited as precedent for this. There is no evidence that CO dissociates preferentially over PMe3. The authors invoke this to arrive at the same 14 eintermediate proposed in Tanaka's photochemical system. H H Rh (III) PMe3 Cl PMe3 H H 16 eH PMe3 Rh (III) H Cl PMe3 Ph3P Me3P Rh (I) PMe3 Cl Tanaka's 14 e- intermediate Rh (III) PPh3 Cl M.C. White, Chem 153 C-H Activation -250- Week of November 4, 2002 Substrate directed dehydrogenation via C-H activation Possible intermediates: H3CO O N + (OTf-) + + H3CO (OTf-) O N H3CO (OTf-) O N H N PtII CF3CH2OH CH3 N 70oC, 60 h N N Sames constructs a ligand for the metal from the requisite functionality of the target that directs C-H activation towards only one of the 2 ethyl susbstituents. This results in selective dehydrogenation to give the platinum hydride in >90% yield. The reaction is stiochiometric in platinum and the metal must be removed via treatment with aqueous potassium cyanide. N PtIV CH3 CH4 PtII + H 3CO H3CO O N N N H O Rhazinilam Sames JACS 2000 (122) 6321. H N Pt N H O N (OTf-) M.C. White, M.S. Taylor Chem 153 C-H Activation -251- Week of November 4, 2002 Dehydrogenation of n-alkanes to terminal olefins R = t-Bu, i-Pr A R R P H Ir H P (0.5 mol%) R R Longer reaction times 150°C sacrificial hydrogen acceptor (norbornene, t-butylethylene, or 1-decene) At low conversions, 1-octene is the major product of the dehydrogenation reaction (90 to >95% selectivity at 5% conversion, depending upon the acceptor used). Ethylene was not a suitable acceptor, resulting in inhibition of catalysis due to formation of a stable Ir-ethylene complex. As the reaction proceeds, olefin isomerization via sequential hydrometallation and β-hydride elimination erodes the kinetic selectivity, resulting in a mixture of olefin isomers. R R P H Ir H Although the nature and the concentration of the sacrificial hydrogen acceptor had little effect on the reaction rate, these factors had a large effect on the observed distribution of double bond isomers in the product. The authors propose that the observed isomer distribution is largely determined by the competition between the sacrificial acceptor and the product olefin for insertion into the Ir-H bond of the dihydride intermediate. A P R R R R P n-Oct Ir H R R P P R R P R R Ir H R R P Ir A Goldman, A. JACS 1999, 121, 4086. P R R A M.C. White, Chem 153 C-H Activation -252- Week of November 4, 2002 Direct carbonylation of benzene The first report: OC + (solvent) Soon afterwards: O CO 1 atm P h3P Rh(I) O PPh3 OC H Cl 7.2 mM + hv (295-420), rt, 40h Eisenberg JACS 1986 (108) 535. Postulated mechanism: 1 atm (solvent) 3 tn RhCl(CO)(PPh3)2 is a photochemical decarbonylation catalyst at rt. CO Rh(I) PMe 3 PBu3 PEt3 P(i-Pr)3 P(p-tolyl)3 PPh3 P(OMe) 3 hv (295-420), rt, 33h 73 tn 1970 1955 1957 1947 1979 1982 2011 H Ph Me3P Cl OC Rh(III) H PMe3 18 e- Rh(I) Me3P PMe3 CO hv Cl 16 eCl O 14 e- Me3P Cl PMe3 PMe3 CO OC Rh(I) Me3P Rh(III) H PMe3 18 e- Cl CO Rh (III) H PMe3 16 e- TN 73 19 17 2 3 2 2 O OC H Cl 0.21 mM Me3P CO (cm -1) Phosphine PMe3 PMe3 is thought to increase the effectiveness of the Rh catalyst both by increasing electron density at the metal thereby promote oxidative addition and by decreasing tail-biting of the complex. Tanaka Chem. Lett. 1987 249. Tanaka JACS 1990 (112) 7221. M.C. White, Chem 153 OC + CO PMe3 Rh(I) Cl 0.21 mM Me3P 1 atm hv (295-420), rt, 33h 295-420 >325 aldehyde tn (1-decanal, 2-, 3-, 4-) nonene tn 610 (85:5:4:2:3) 126 (8:45:17:15:16) 319 0 R Revised proposed catalytic cycle: H 27 tn Effects of irradiation wavelength: Flash photolysis revealed loss of CO (thought to lead to the catalytically active 14e- species for C-H oxidative addition) is the dominant photoreaction of RhCl(CO)(PPh3) 2 at >330 nm. Metal-to-ligand charge transfer band of Rh-CO @ 365 nm. Ford JACS 1989 (111) 1932. Absorption of non-conjugated aldehydes appear at ~285 nm. It was hypothesized by Tanaka that cutting of the short-wavelength region capable of aldehyde excitation would improve yields of the desired aldehyde. wavelength (nm) Week of November 4, 2002 Direct carbonylation of alkanes Aliphatic hydrocarbons: (solvent) C-H Activation -253- H O H hv 285 nm Photo-induced Norrish Type II Chemistry H O Tanaka Chem. Comm. 1987 758. + CH3CHO 92 tn H While Norrish Type II reactions leading to dehomologated terminal alkenes were suppressed by going to a longer wavelength, carbonylation selectivity towards the 1o position of the alkane was lost and catalytic activity was diminished. These results imply that photo-induced CO dissociation may not be the major pathway in this system for generating the complex capable of C-H activation of linear aliphatic alkanes. Tanaka JACS 1990 7221. OC Rh (I) PMe3 Cl 16 e- R The rate of benzene carbonylation catalyzed by RhCl(CO)(PMe3)2 irradiated at >290 nm (ca. 314 nm, a wavelength where Rh-CO does not absorb) is proportional to CO pressure. Goldman proposes a photoelectronically excited intermediate as the species effecting C-H activation. Goldman JACS 1994 (116) 9498. 0.6 tn O Me3P O The carbonylation reaction is highly regioselective towards primary C-H bonds to give linear aldehydes with high selectivities. Unfortunately, the aldehydes formed readily undergo a secondary photochemical reaction (Norrish Type II) to give a dehomologated terminal alkene and acetaldehyde in large quantities. + * Irradation Me3P Cl OC Rh(III) OC O Rh(I) Me3P H PMe3 Cl 16 e- PMe3 18 ePMe3 R OC Rh CO (III) Cl H PMe3 18 e- R of a solution of RhCl(CO)(PMe3)2 /C 6H6 in the absence of CO at -40oC afforded two isomers of the 18 ealkylhydrido complexes which were fully characterized by NMR (1H, 31P, 13C NMR). Fields JACS 1994 (116) 9492. M.C. White, Chem 153 C-H Activation -254- Week of November 4, 2002 Direct formation of aldimines R The first report: N OC + (solvent) RNC 55 mM Rh (I) PMe3 Rh(I) + CyNC H Cl 0.7 mM Me3P OC (solvent) hv, rt, 36h 3 tn R = cyclohexyl, 5 tn Me, 38%/Rh t-Bu, 3%/Rh Cy PMe3 N Cl 0.7 mM Me3P hv, rt, 17h 6.0 mM low conversions may be due in part to the low solubility of the isocyanide under the rxn conditions. Selectivities not reflective of C-H activation via an organometallic intermedaite. H 6%/Rh + N Cy + 12%/Rh 12%/Rh N Tanaka Chem. Lett. 1987 2373. H Cy R RNC C N PPh3 Rh(I) Cl 0.2 mM Ph3P + N H hv, rt, 36h 4 tn R= neopentyl 1.0 mM Proposed mechanism: RNC Rh(I) PPh3 -PMe3 Cl + PMe3 Ph3P 16 e- RNC N Rh(I) Jones notes that this system (unlike the one reported by Tanaka) is completely ineffective at aldimine formation from aliphatic hydrocarbons. PPh3 Cl R 14 eH R H N H CNR Rh(III) RNC Rh(III) PPh3 PPh3 Cl Cl 16 e- 16 eCNR Jones OM 1990 (9) 718. M.C. White, Chem 153 C-H Activation -255- Week of November 4, 2002 Direct Borylation of Alkanes: Stoichiometric (solvent) BCat' 83%/W 100% regioselectivity Bcat' OC OC WII B Selectivity between activation/ functionalization of 1o vs. 2o C-H bonds is high. Reactions of the tungsten complex with cyclohexane resulted in 22% yield based on W. This system appears to be highly sensitive to sterics as demonstrated in it's ability to discriminate between the linear and branched 1o C-H bonds of isopentane. O hv (solvent) BCat' COO + Cat'B 55%/W 18 estoichiometric BCat' (solvent) lesser reactivity was also observed with the Ru and Fe analogs 74%/W The exact mechanism of C-H activation/functionalization is unclear. Two possibilities are likely: 1. oxidative addition followed by reductive elimination, 2. σ-bond metathesis. The first possibility would require loss of a second CO or Cp* slippage to create a site of electronic unsaturation at the W to accomodaite both the alkyl and hydride substituents. Alternatively, σ-bond methathesis could occur directly with the shown 16e- intermediate. Alkane dehydrogenation followed by anti-Markovnikov hydroboration is excluded since aliphatic alkenes result in vinylborates rather than the observed alkylborate esters. Proposed mechanism: II O OC W B OC COO 18 e- 2%/W hv CO OC W II O R B O OC H 16 e- ? II OC W H OC + Cat'B R 16 e- stoichiometric Photolysis in the presence of PMe3 results in the formation of Cp*W(CO) 2(PMe 3)Bcat'. This was taken as evidence for the photo-induced loss of CO to generate coordinatively unsaturated 16 eintermediate that may interact with the alkane. Hartwig Science 1997 (277) 211. M.C. White, Chem 153 C-H Activation -256- Week of November 4, 2002 Direct Borylation of Alkanes: Catalytic The first catalytic report: note similarity w/ Bergman stiochiometric C-H activation complexes. IrIII Me3 P O + H BPin 17 mol% HB + H2 150 oC, 5 d O 1 eq (solvent) BPin 53% (based on borane) BPin = pinacolborane Smith JACS 1999 (121) 7696. Hartwig runs with it... facile thermal alkene dissociation forms coordinatively unsaturated complexes IrI 10 mol% 200 oC, 10 d Bpin 2 C6H13 + 2 H2 58%/B O 2 C6H13 (solvent) + O B O The rate acceleration observed in going from a 3rd row metal complex to an analogous 2nd row complex may be accounted for by a weakening of M-C bonds which may promote turnover steps in the catalytic cycle. Rh I B O (pinBBpin) 5 mol% 150 oC, 5 h Bpin 2 C6H13 + 2 H2 85%/B Rh I Hartwig Science 2000 (287) 1996. 1 mol% o 150 C, 80 h Bpin 2 C6H13 72%/B + 2 H2 100% regioselectivity for the terminal borane was consistently observed. The linear borane is thought to be the kinetic product. Exposure of secondary alkyl boranes to reaction conditions does not result in isomerization. 2-Methylheptane resulted exclusively in products formed from primary C-H bond activation with the less sterically hindered terminal methyl group becoming functionalized selectively. M.C. White, Chem 153 C-H Activation -257- Week of November 4, 2002 Mechanism of direct borylation of alkanes HBpin, generated under the rxn conditions, is equally effective as source of borane Rh I O + C6H13 O B B O (solvent) 5 mol% O C6H13 Bpin Rh I O + H 5 mol% B Bpin C6H13 O C6H13 (pinBBpin) 85%/B 150 oC, 5 h Hartwig Science 2000 (287) 1996. Hartwig's mechanistic proposal Rh I 18 e- ∆ Rh I 14eRh I To validate his mechanistic proposal that invokes high energy Rh(V) intermediates, Hartwig synthesizes what he claims is an Ir(V) dihydrido bisboryl species (the high reactivity of the Rh complex has precluded its isolation/characterization). Although Hartwig argues against a σ-complexed borane Ir(III) species, his evidence does not conclusively eliminate it as a possibility. The independently synthesized intermediate was an effective alkane borylation reagent, resulting in similar yields and the same selectivities observed in the catalytic system. via RhIII intermediates 18 e- R-Bpin H X = H, Bpin H X Bpin Bpin X Rh III R-H Rh V H R 18eRh(V) is a very high energy oxidation state: controversial intermediates. X III Ir H Bpin Bpin H 200oC Rh III ? or IrV C6H13 (solvent) Bpin Bpin C6H13 pinBX Rh V X Bpin H X 45%/B HX Hartwig JACS 2001 (123) 8422. H Bpin M.C. White, Chem 153 C-H Activation -258- Week of November 4, 2002 Direct Arene borylation: Suzuki precursors Towards synthetic utility... In several examples the authors were able to achieve intermolecular C-H activation/functionalization without using the substrate as the solvent. Some substrates were borylated under neat conditions while others employed cyclohexane as solvent. η5-indenyl complex capable of rearranging to η3 and η1 IrI Cl Cl Cl Cl O B H 16h Cl 1.5 eq Ph2P Cl aryl-H: HBpin (1:2) 69% yield, 4h Cl PPh2 dppe Recall that, in general, Ir complexes are less reactive than the corresponding Rh complexes towards alkane borylation. Aryl C-H bonds are more reactive towards C-H activation than alkyl C-H. The factors favoring activation of aryl C-H bonds are the high degree of s character in the Csp2-H bond which favors σ-complexation to the metal and the strength of the resulting aryl Csp2-M bond after oxidative addition. Bpin excellent regioselectivities for functionalization of sterically less hindered sites 1).HBpin, 2 mol% (Ind)Ir(COD), 2 mol% dppe, 100oC, 16 h 2). 3-bromotoluene, 2 mol% Pd(PPh3)4, K3PO4, DME, 80oC, 17 h. Cl MeO2C aryl-H: HBpin (1:2) 95% yield, 25h 89% based on arene MeO 1 eq Smith Science 2002 (295) 305. A related study that uses the bpy ligand in conjunction with IrI: Hartwig JACS 2002 (124) 390. BPin MeO aryl-H: HBpin (1:3) 62% yield, 95h, dmpe Consecutive aryl borylation/Suzuki: Cl BPin BPin dppe 2mol%, 100oC O 1 eq N 2mol% Cl Cl 80% yield based on dichlorobenzene M.C. White, Chem 153 C-H Activation -259- Week of November 4, 2002 Question 1: Catalytic Indole Production Propose a catalytic cycle for the following Ru system that affords indoles in good yields from 2,6-xylyl isocyanide. PMe3 Me3 P H Ru II Me3 P PMe3 CN 20 mol% benzene, 120oC, 94 h HN M.C. White, Chem 153 Q&A -260- Week of November 4, 2002 Question 2 Provide a detailed mechanism for the following transformation. O 3-5 eq. O Co 2(CO) 8 2 mol% HSiEt 2Me (1.2 eq), CO (50 atm) MeEt2SiO H 53% M.C. White, Chem 153 Q&A -261- Week of November 4, 2002 Question 3 PdL nI PdL nI cat. PdLn I cat. PdLn I CO CO PdL nI PdL nI O O When a competition exists between cyclic acylpalladation and cyclic carbopalladation, the preferred outcome is different for alkenyl and alkynyl substrates. For alkenyl substrates, cyclic acylpalladation is favored over cyclic carbopalladation, and for alkynyl substrates, this preference is reversed. Given these empirical observations, predict the products of the following transformations: n-Bu OH E E I OH 5% Cl2Pd(PPh3)2 NEt3 ( 2eq), CO (1 atm) MeOH, 70oC I Me 5% Cl2Pd(PPh3)2 NEt3 ( 2eq), CO (1 atm) MeOH, 70oC I Me E E 5% Cl2Pd(PPh3)2 NEt3 ( 2eq), CO (1 atm) MeOH, 70oC M.C. White,Q. Chen Chem 153 C-H Activation -262- Week of November 4, 2002 Cyclometallation Cyclometallation: intramolecular C-H activation of supporting metal ligands (a.k.a. "tail-biting")... H Ph3P H ∆ C 6D 6 Ph2P PPh3 IrI P h3P Cl 16 eagostic interaction PtII Ph3P Ph2P IrIII P h3P PPh3 H -PPh3 P h3P PPh3 H Ph3P PtII OA ligand dissociation to create an open coordination site Cl PtIV rate-limiting step: RE Bennnett JACS (91) 1969 6983. Chelate-assisted C-H activation: Ph3P Substrates with Lewis basic functionality can temporarily become appended to a site of coordinative unsaturation on a metal and undergo chelate assisted C-H activation. EtO H P h3P RuII Ph3P H O PPh3 (excess) PPh3 PPh3 H Ph3P hydrogenation OEt O EtO O O OA RuII PPh3 P h3P PPh3 H OEt O RuII Ph3P OEt Ph3P Whitesides OM 1982 (1) 13 H Ibers JACS 1976 (98) 3874. Ph3P Ph3P Ph3P Ru0 H PPh3 16 e- Ph3P Ru0 14 e- PPh3 PtII PPh3 Ph3P -PPh3 PPh3 PtII M.C. White, Chem 153 C-H Activation -263- Week of November 4, 2002 Chelate assisted Csp2-H-olefin reductive coupling Murai's breakthrough system... metal chelating LB functionality H Ph3P O RuII OC R1 R2 H + Y SiMe3 PPh3 O PPh3 2 mol% >99% >99% yield Si(OEt)3 R2 toluene, reflux O O Si(OEt)3 R1 O O 2-10 mmol 2 mmol C sp2-H 4 atoms from LB functionality results in 5-membered ring metal chelate Si(OEt)3 O Y Y = H (ethylene) "privileged olefin" Si(OEt)3 CH2SiMe3 t-Bu >99% >99% 100% regioselectivity Murai Nature 1993 (366) 529. Many other examples follow: O H O O Ph3P RuII OC R + H Y O PPh3 6 mol% R O t-Bu O toluene, reflux 2-10 mmol 2 mmol O PPh3 O C6H13 98% t-Bu 73% Internal alkynes also add... Ph3P RuII OC + R Murai Chem. Lett. 1995 681. H R PPh3 CF3 R Si(OEt)3 Aryl esters: R H Si(OEt)3 >99% O Y Murai Chem. Lett. 1995 679. O O Et O PPh3 2 mol% H O Ph3 P OMe + toluene, reflux R = Pr (72%), E/Z = 16/1 Ph (85%), E/Z = 9/1 Ru II OC H Si(OEt)3 PPh3 PPh3 2 mol% CF3 O OMe toluene, reflux Si(OEt)3 Only aromatic esters substituted with CF 3 or F groups (m,p,and o) resulted in coupled product. Other benzoates w/electron withdrawing substituents o-NO 2, p-NO 2, o-CN, o-CO2Me failed to give coupled product. Murai Chem. Lett. 1996 109. M.C. White, Chem 153 C-H Activation -264- Week of November 4, 2002 Oxygen chelate assisted Csp2-H-olefin reductive coupling Cyclic and acylic vinyl esters : Si(OEt)3 O O OR H OR R1 Ph3P OC O + R2 RuII H R1 PPh 3 5 mol% H Si(OEt)3 OR PPh 3 A high degree of functional group tolerance O is demonstrated through the substrates tested. OEt Si(OEt)3 A lack of reactivity is observed when the β Csp2-H bond is trans to the ester carbonyl O O NHCH3 80% R = (CH2 )5CH 2 OAc, 85% (CH2 )5CH2 OTBS, 91% (CH2 )5CH2 Br, 54% O R2 toluene, reflux Ph Si(OEt)3 Si(OEt)3 O O Trost JACS 1995 (117) 5371. H RuII OC SiR 3 PPh3 SiR 3 OR OR' PPh3 O CO H CO loss is supported by the observation that the reaction is inhibited in the presence of CO. 70% Hydrogenated product is observed by GC Proposed mechanism: Ph3P O O Ph3P SiR3 OR' H Ru0 PPh3 OR' Ph3P P h3P 14 e- reductive elimination O O Ph3P RuII SiR 3 Ph3P Ru0 H PPh3 PPh3 OR' migratory insertion OR' O Ph3P O R3Si RuII SiR3 Ph3P Ph3P RuII PPh3 P h3P H PPh3 H M.C. White/Q.Chen Chem 153 C-H Activation -265- Week of November 4, 2002 Nitrogen chelate assisted Csp2-H-olefin reductive coupling t-Bu t-Bu N H t-Bu N N Ru 3(CO) 12 (2 mol%) + Si(OEt)3 tol, 135 C, 24h 10% RuH 2(CO)(PPh3) 3 26% (2 mol%) 8% Ru 3(CO) 12 130oC heptane N H Si(OEt)3 81% N (CO) 3 Ru H + Si(OEt)3 Muria Chem. Lett. 1996 111. 2 CO H o Ru(CO)3 Ru(CO)4 1 Ru3(µ-H)(m-C13H8N)(CO)10 Fish OM 1986 (5) 2193. Some Ru-H is formed via the dehydrogenative coupling. M.C. White, Chem 153 C-H Activation -266- Week of November 4, 2002 Nitrogen chelate assisted Csp2-H/CO/olefin reductive coupling Tolerates sensitive functionality: Ph Ph Ph N N + O O Ru3(CO)12 (4 mol%) O N + N N CO (20 atm) tol, 160oC, 24h N O O O 72% (linear:branched; 97:3) Olefin isomerization occurs under the reaction conditions: N N N + + N Ru 3(CO) 12 (4 mol%) or N N CO (20 atm) tol, 160 oC, 24h O O 1-hexene; 68% (linear:branched; 94:6) 2-hexene; 41% (linear:branched; 94:6) Proposed mechanism: N R Ru3(CO)12 + R N O N O Ru(CO)3 N Rux(CO)n N N (CO)4 Ru N N R or N (OC)3Ru CO N Ru(CO)3 H H Ru (CO) 3 R H Ru(CO)n R N N N N Murai JACS 1996 (118) 493. Rux(CO)n O O M.C. White, Chem 153 C-H Activation -267- Week of November 4, 2002 Indole synthesis via isonitrile chelation/ C-H bond activation Propose a catalytic cycle for the following Ru system that affords indoles in good yields from 2,6-xylyl isocyanide. PMe3 Me3P Ru H II Me3 P PMe3 CN 20 mol% HN benzene, 120 oC, 94 h heat promoted RE HN Me3 P Ru N H CN Me3 P Me3 P H Ru 0 P Me3 P Me3 II Me3 P PMe3 PMe3 NC Ru 0 PMe3 PMe3 PMe3 isomerization Me3 P Me3 P PMe3 Ru II N H H PMe3 H Me3 P PMe3 Me3 P tautomerism Ru II N PMe3 H NC PMe3 PMe3 Jones JACS 1986 (108) 5640. NC Me3 P PMe3 migratory insertion Ru II Ru 0 P Me3 Me3 P H OA Me3 P PMe3 PMe3 M.C. White, Chem 153 C-H Activation -268- Week of November 4, 2002 Oxidative functionalization of alkanes The methane to methanol challenge: Synthesizing "liquid gold": Current industrial process consumes significant amounts of energy: Ni/Al2O3 CO (g) + H2 ∆Ho = 49.3 kcal/mol CH4 (g) + H2O (g) 700oC CO (g) + 2 H2 (g) zeolite cat. ∆ CH3OH Direct oxidation is thermodynamically favorable. catalyst ? CH4 (g) + 1/2 O 2 (g) CH3OH ∆Ho = -30.7 kcal/mol overoxidation to CO2 is major problem w/methane oxidation o ∆H = -21.7 kcal/mol Nature does it: Methane Mono-Oxygenase (MMO): Pseudomonos Oleovorans Mono-Oxygenase (POM): MMO oxidizes methane to methanol with 100% chemoselectivity (no overoxidized product results). Oxidizes linear alkanes with 100% regio- and chemoselectivity CH4 + O 2 + NADPH + H+ MMO M. Capsulatus 12 min CH3OH + NADP+ + H2O 84 tof tof = nmol product/min/mg enzyme n-alkanes + O2 + NADPH + H + C6-C12 POM 1-alcohols + NADP+ + H2O 1-octanol, 590 tof Higher hydrocarbons are oxidized with poor regioselectivities MMO M. Capsulatus 12 min Coon Biochem. Biophys. Res. Comm. 1974 (57) 1011. Munck PNAS 1997 (94) 2981. + OH OH 1.3 : 1 Lipscomb J. Biol. Chem. 1992 (267) 17588. A beginning... The Shilov system: Cl Cl CH4 + H 2O PtII Cl (K+) 2 Cl cat. K2Pt(IV)Cl6 oxidant 120oC Shilov Zh. Fiz. Khim. (Engl. Trans.) 1972 (46) 785. regioselectivities: Bercaw JACS 1990 (112) 5628. CH3OH + CH3Cl In 1972 Shilov and coworkers demonstrated that a combination of chloroplatinum(II) and (IV) salts in aqueous solutions at elevated temperatures effects the oxidation of alkanes to mixtures of alcohols and alkyl chlorides. The regio- and chemoselectivity of the Shilov system reflects those of other organometallic systems in that the stronger 1o methyl hydrogens of propane and even ethanol are more reactive than the methylene hydrogens. Unfortunately only modest selectivites are observed. Some overoxidized products and regioisomeric mixtures of alcohols are observed because the product alcohols are more soluble in the aqueous reaction media than the hydrocarbon. M.C. White, Chem 153 C-H Activation -269- Week of November 4, 2002 MMO ·Hydroxylase Active Site of MMO OH 2 O O O E114 Fe H147 O N OH 2 O O O Fe(II) (II) N E243 O E243 O N E144 N E114 O H246 H147 O O H2 O E209 Fe (III) O N N O H O Fe (III) O N E144 N E209 O H246 MMOHox MMOHred o Based on crystallographic studies of M. capsulatus(-160 C) Proposed mechanism (thought to be operating for POM as well): - 2e (II) Fe H O (II) Fe ·O O· H (III) O (III) Fe Fe H2O2 "peroxide shunt" -H+ ROH H O (IV) (III) Fe Fe OH R· H O (IV) (IV) Fe Fe O H (III) O (III) Fe Fe O O µ-1,2 peroxo adduct P H+ H O (III) (III) Fe Fe O O+ H Key piece of evidence supporting substrate radical intermediate: CH3 CH3 CH3 MMO + D D D HO OH T H T T (R)-ethane (S)-ethanol RH (IV)Fe O· Q Fe(III) H+ -H2O The second iron in MMO transiently stabilzes intermediate Q by supplying an e- to fill the oxygen atom's octet. This avoids energetically unfavorable Fe(V) intermediates. (R)-ethanol 35% Lipscomb Chem. Reviews 1996 (96) 2625. Attempts to mimic Nature's solution have failed. The key to chemo- and regioselectivity in these radical systems may be MMO and POM's protein suprastructure which thus far have not been mimicked in solution. O N N H O Lippard Nature 1993 (366) 537. Fe III N 2+ (ClO4-) 2 O O Cl Cl Fe III N N O OH N cat. H2O2, CH3CN, air note: the same yields and selectivities were observed when the reactions were run under an inert atmosphere (Ar) or in air. This indicates that free radicals, propagated with O2, are not acting as the oxidant. Nishida Chem. Lett. 1995 885. + 4 tn 2 tn M.C. White, Chem 153 C-H Activation -270- Week of November 4, 2002 The Shilov System/C-H activation via late, electrophilic complexes H σ−donation>> π-backbonding M M heterolytic cleavage + C H+ C σ-complex C-H activation processes that occur via heterolytic cleavage result in no oxidation state change at the metal. Generally, electrophilic metal complexes are used that incorporate metals in their highest stable oxidation states. Unlike the Bergman nucleophilic complexes, electrophilic complexes are compatable with oxidants and provide a route to oxidative functionalization of hydrocarbons (the most desirable form of functionalization). The Shilov system: 2 (K+) 2 Cl Cl PtII Cl Cl cat. + H 2O CH4 CH3OH K 2Pt(IV)Cl6 oxidant Because Pt is a late "soft" metal, the relatively diffuse alkane C-H bond is able to intermolecularly compete with the hard oxygen lone pair of H2O for binding to the metal. + CH3Cl 120oC Proposed mechanism: 2 Cl - 2 H2O PtII Cl II Pt CH4 OH 2 OH 2 OH 2 Cl 2 Cl Cl H Cl (K+)2 Cl Cl Cl OH 2 PtII H MeOH H 2O Inversion of stereochemistry at the platinum bound C using deuteruim labeled substrates provided strong evidence for S N2 functionalization pathway Bercaw ACIEE 1998 (37) 2180. Cl PtIV OH 2 CH3 HCl soft deprotonation CH3 IV Cl K+ Cl Pt Cl Cl or Cl- CH3 K+ Cl Cl- Cl Cl H 2O PtIV Cl OH 2 CH3 Cl Cl Cl PtII OH 2 CH3 K+ note: no oxidation state change to the metal K2 Pt(IV)Cl6 K2 Pt(II)Cl4 Pt(II) catalyst is regenerated M.C. White, Chem 153 C-H Activation -271- Week of November 4, 2002 C-H activation via late, electrophilic complexes in highly acid media Hg(II)(OSO 3H) 2 cat. N N CH4 + 2 H2SO4 Cl note that the product cannot undergo further oxidation. PtII N 200oC Periana Science 1993 (259) 340 Cl N 500 tn CH4 + 2 H2SO4 CH3OSO3H 50% yield (based on CH4) H2SO4 (ox/solv) H 2SO4 (ox/solv) 200oC Periana Science 1998 (280) 560. CH4 + CF3CO2H CH3OSO3H 70% methyl bisulfate (90% conversion/ 80% selectivity) based on methane. Pd(OAc)2 stoic. CH3O2CF3 + Pd (0) CF3CO2H (solv) Sen JACS 1987 (109) 8109 Heterolytic cleavage directly from the σ-complex is clearly operating for Pd(II) and Hg(II) systems where the M(n+2) oxidation state of the alkyl(hydrido)metal intermediate is prohibitively high in energy. Proposed mechanism: 2+ + - N N N OSO3 H N PtII N N N ( OSO3 H) PtII OSO3 H OSO3 H N OSO3 H (-OSO3 H)2 II Pt N N 14 e- complex CH3OSO3H N H H N OSO3 H The ligand may become protonated under the reaction conditions. Protonation will withdraw electron density from the Pt through the σ-bonding framework of the bidiazine ligand thereby enhancing its electrophilicity. CH4 -OSO3 H + OSO3 H N N N OSO3 H N PtIV N PtII H CH 3 N OSO3 H N N OSO3 H CH 3 + (-OSO3 H) -OSO3 H N N H OSO3 H (-OSO3 H) PtIV or N N CH 3 heterolytic cleavage oxidation N SO2 + H2O N OSO3 H PtII SO3 + 2 H2SO4 N N CH 3 Although the Periana Pt system is unparalleled with respect to its efficiency at oxidative functionalization of methane, the high cost associated with platinum coupled to the operational difficulty in seperating the product from the solvent renders this route to methanol non-competitive with traditional reforming. M.C. White Chem 153 C-H Activation -272- Week of November 4, 2002 Substrate-directed vinyl alkylation via electrophilic C-H activation N N H 2+ H3 CCN H3 CCN Pd II NCCH3 NCCH3 1.PdCl 2(CH 3CN) 2/AgBF 4 NEt 3, CH3CN 2. NaBH 4 40-45% N N H (BF4 -)2 generated via in situ metathesis NaBH 4 NCCH3 2+ + - (BF4 )2 H3 CCN (BF4 -)2 Pd II N LnPd N H N H recall that Pd(IV) is a prohibitively high energy oxidation state + NEt3 N (BF4 -)2 Ln Pd II NEt3 migratory insertion N H Trost JACS 1978 (100) 3930. MeO2 C CO 2Me O N N H Corey JACS 2002 (124) 7904. N H OMe NCO2 Me Pd(OAc)2 (1 eq) NaOAc (1 eq) AcOH: H 2O (1:1) 25oC, 24h 31% N H model system for key cyclization step in (+)-Austamide synthesis M.C. White, Chem 153 C-H Activation -273- Week of November 4, 2002 Substrate-directed alkane arylation via electrophilic C-H activation OMe OMe OMe N PdOAc 2 (4 mol%) OMe Cu(OAc)2 (2 eq.) benzoquinone (4 mol%) N 100oC Ph 2Si(OH)Me (2 eq) or S R S Si(OH)Me2 Ph R = Ph, 73% R= PhCH=CH, 64% OMe OMe 2 CuOAc N O S Pd II O O OMe O + (OAc -) OMe OMe 2 CuOAc2 N Pd(0)Ln Ph OMe OMe OMe N AcO Pd II N S H Pd II S pka ~ 50 - Ph OAc OMe OMe base-assisted heterolytic cleavage Sames JACS 2002 (124) 13372. transmetalation N Pd II S AcO Ph 2SiOHMe S M.C. White/M.W. Kanan Chem 153 C-H Activation -274- Week of November 4, 2002 Intermolecular arene vinylation via electrophilic C-H activation H Pd(OAc)2 1mol% + CO2Et O CF3CO2H/CH2Cl 2 (4:1) 25oC O O O H HO O O + O O O O CF 3 ( O2CCF3) O CO 2Et O O protonolysis HO H O PdII Reactions run in CF3CO2D yielded products with vinyl deuterium incorporation α to the ester. + CH3 O2CCF3 Pd II O 61% (-O2CCF3 ) CF 3 Reactions run in acetic acid failed. TFA is thought to be necessary for the formation of cationic Pd(II) species. Reactions run with Pd(0) sources gave only trace amounts of product (<20%). PdII CO2Et O PdII O CF3 O trans migratory insertion O O O O CO2Et CO 2Et CF3 O ? Fujiwara Science 2000 (287) 1992. Reaction exhibits excellent functional group tolerance with unprotected OH, CO2Et Br, and acetals tolerated in the arene. Coupling to activated alkenes (vinyl esters) was also effected in high yields (65-96%). O CF 3 M.C. White, Chem 153 Q&A -275- Week of November 4, 2002 Silylformylation Provide a detailed mechanism for the following transformation reported in the literature. O O Co 2(CO) 8 2 mol% HSiEt 2Me (1.2 eq), CO (50 atm) 3-5 eq. MeEt2SiO H 53% Reaction works for 3,4, and 5 membered cyclic ethers. Ring strain may promote the nucleophilic ring opening reaction. Neither tetrahydropyran or diethyl ether react under these conditions. The high affinity of silicon for oxygen is used to account for the fact that R3SiCo(CO)4 and not HCo(CO)4 interacts with the cyclic ether. H (OC)4Co Co(CO)4 Co(CO)4 HSiR3 O O R3Si MeEt2SiO Co(CO)4 H O SiR 3 MeEt2SiO Co(CO)3 HSiR3 H O SiR 3 O Co(CO)4 MeEt2SiO Co(CO)3 migratory insertion Murai ACIEE 1977 (16) 789. Murai ACIEE 1979 (18) 837. Co(CO)4 MeEt2SiO nucleophilic ring opening M.C. White, Chem 153 Q&A -276- Week of November 4, 2002 Carbonylation PdLnI PdLnI cat. PdLn cat. PdLn I I CO CO PdLnI PdLnI O O When a competition exists between cyclic acylpalladation and cyclic carbopalladation, the preferred outcome is different for alkenyl and alkynyl substrates. For alkenyl substrates, cyclic acylpalladation is favored over cyclic carbopalladation, and for alkynyl substrates, this preference is reversed. Given these empirical observations, predict the products of the following transformations: n-Bu OH E I E I Me OH I Me 5% Cl2Pd(PPh3)2 NEt3 ( 2eq), CO (1 atm) 73% MeOH, 70oC E E 5% Cl2Pd(PPh3)2 NEt3 ( 2eq), CO (1 atm) 66% MeOH, 70oC O O n-Bu O E O E = CO2Me 5% Cl2Pd(PPh3)2 NEt3 ( 2eq), CO (1 atm) MeOH, 70oC O E E E Negishi JACS 1994(116) 7923. Negishi JACS 1985(107) 8289. M.C. White/ M.W. Kanan, Chem 153 TESO Q&A -277- Week of November 4, 2002 O TESO O Pd(PPh 3)4 800 psi CO O or R TESO R R O OTf OH OH O i-Pr 2NEt, PhCN 65 to 110°C OTf Leighton Org. Lett. 2000 (2) 2905. Provide a mechanism for this one-pot transformation. Explain why both the E- and Z-tetrasubstituted enol triflates react to form the desired product in comperable yields. TESO TESO TESO PdLn O R R TfO OH OH + CO O O R - CO 4 Pd(OTf)Ln OH O Pd(OTf)Ln TESO R TESO TESO O Pd(OTf)Ln O R O R H OH 5 OH OH + CO Ln(OTf)Pd Pd(OTf)Ln - CO TESO R O TESO Pd(OTf)Ln O O TESO O R O O 3,3-sigmatropic rearrangement O O R OH Oxidative addition into the Z-tetrasubstituted enol triflate leads directly to intermediate 5 which can continue on the path to the desired product. Oxidative addition into the E-tetrasubstituted enol triflate leads to intermediate 4 which can isomerize to 5 via a -allenyl intermediate. M.C. White, Chem 153 Q&A -278- Week of November 4, 2002 Carbonylation O O R + H 3C H N H N H CH3 O CO, 0.25% PdBr2(PPh3)2 30% LiBr, 1% H2SO4 R N CH3 N H 3C O Substituted hydantoins can be prepared by the ureidocarbonylation of an aldehyde in presence of LiBr and H2SO4. Propose a mechanism for this transformation. O H 3C H 3C Br R N O N H CH3 H 3C R N N R + H H 3C CH3 CO H R Br PdL2 CH3 Br oxidative addition H O O N H O LiBr, H 2SO 4 N O H 3C R OC PdL2 N N H H N CH3 HBr migratory insertion reductive elimination O H 3C L 2Pd(H)Br N N H R PdL2Br O O R Beller ACIEE 1999 (38) 1454. H 3C N N CH3 O CH3 CH3 N H PdLn Br M.C. White/Q. Chen, Chem 153 Q&A -279- Week of November 4, 2002 Hydrophosphinylation n-Hex H O PPh2 5% Pd(PPh3) 4 + Ph 2P(O)H n-Hex 1a) Hydrophosphinylation of internal and terminal alkynes with Ph2P(O)H can be catalyzed by a variety of palladium sources. Pd(PPh3)4 is selective for the anti-Markovnikov product. Propose a mechanism for the hydrophosphinylation of 1-octyne below. O n-Hex PPh2 O Ph 2PH Pd(PPh3)2 reductive elimination oxidative addition R Ph3P Ph3P Pd Ph3P H Pd Ph3P PPh2 PPh 2 O O hydropalladation Ph3P Ph3P R Tanaka, M. ACIEE 1998, 37, 94-96. Tanaka, M. OM 1996, 15, 3259-3261. R H Pd PPh2 O H H M.C. White/Q. Chen Chem 153 Q&A -280- Week of November 4, 2002 Hydrophosphinylation n-Hex Ph2P(O)H + H O PPh2 5% PdMe 2(dmpe) 5% Ph2P(O)OH n-Hex b) The regioselectivity of the hydrophosphinylation can be completely reversed by using PdMe2(dmpe) (dmpe = dimethylphosphinoethane) and the phosphinic acid, Ph 2P(O)OH. Propose a mechanism for the formation of the Markovnikov product under these conditions. O Ph 2POH Me Me P Me Pd P Me Me Me To explain the observed reversal in regioselectivity in the presence of diphenylphosphinic acid, Tanaka and coworkers propose the formation of a new catalytically active species under these conditions. O Ph2PH O PPh2 P n-Hex Pd P O O OPPh 2 R H PPh2 O "protonolysis" Ph 2P H O OPPh 2 P P Pd Pd P P Ph2P R R O Tanaka, M. ACIEE 1998, 37, 94-96. Tanaka, M. OM 1996, 15, 3259-3261. phosphinylpalladation O OPPh2 PPh2 O H M.W. Kanan/M.C. White, Chem 153 Q&A -281- Week of November 4, 2002 Carbometallation i Pr Since very little CH3D is observed, oxidative addition of a C-H from the ligand isopropyl group in intermediate A must be much faster than oxidative addition of C6D6 to A. i Pr-d7 i Me Me Pr N Me Pt Me N i Me N C 6D 6 150°C 10 min. + C2H6, CH4 Pt N i Pr Me Pr-d7 D D D Me i CD3 i Pr Pr-d7 1 2-d27 Provide a mechanism for the formation of 2-d27. Note that very little CH3D is formed under the reaction conditions. i Goldberg JACS 2002 (124) 6804. i Pr Me N Pt Me i H Pr N Me Pt N iPr iPr CD3 Me iPr-d 7 D DD Pt N i CD3 Pr-d7 i Pr and/or and/or iPr Pt i D Pr N C6D5 Pt N N iPr N iPr 2 H N ox. add'n. Pt N N iPr iPr iPr iPr N Pr-d7 7 - RH ....... iPr A H HH Pt Pr - C 2H 6 iPr iPr-d i 1 Red. Elim. N iPr Me N i Pr-d7 iPr iPr Me iPr - CH4 N Pt Red. Elim. C6D6 ox. add'n. D N Pt N iPr iPr iPr C6D5 iPr Red. Elim. N Pt N N iPr i Pr C6D5 iPr-d 1