Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Expression vector wikipedia , lookup
Drug design wikipedia , lookup
Magnesium transporter wikipedia , lookup
Gene expression wikipedia , lookup
G protein–coupled receptor wikipedia , lookup
Interactome wikipedia , lookup
Point mutation wikipedia , lookup
Ancestral sequence reconstruction wikipedia , lookup
Genetic code wikipedia , lookup
Western blot wikipedia , lookup
Biosynthesis wikipedia , lookup
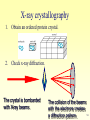
Metalloprotein wikipedia , lookup
Protein–protein interaction wikipedia , lookup
Two-hybrid screening wikipedia , lookup
Amino acid synthesis wikipedia , lookup
Structural alignment wikipedia , lookup
Structural Bioinformatics Proteins 2 Specific databases of protein sequences and structures Swissprot PIR TREMBL (translated from DNA) PDB (Three Dimensional Structures) 3 Myoglobin – the first high resolution protein structure Solved in 1958 by Max Perutz John Kendrew of Cambridge University. Won the 1962 and Nobel Prize in Chemistry. “ Perhaps the most remarkable features of the molecule are its complexity and its lack of symmetry. The arrangement seems to be almost totally lacking in the kind of regularities which one instinctively anticipates.” 4 Why Proteins Structure ? Proteins are fundamental components of all living cells, performing a variety of biological tasks. Each protein has a particular 3D structure that determines its function. Protein structure is more conserved than protein sequence , and more closely related to function. 5 There Are Four Levels of Protein Structure Primary: amino acid linear sequence. Tertiary: the 3D shape of the fully folded polypeptide chain Secondary: -helices, β-sheets and loops. Quaternary: arrangement of several polypeptide chains. 6 Symbols for the 20 amino acids A ala alanine C cys cysteine D asp aspartic acid E glu glutamic acid F phe phenylalanine G gly glycine H his histidine I ile isoleucine K lys lysine L leu leucine M met N asn P pro Q gln R arg S ser T thr V val W trp Y tyr methionine aspargine proline glutamine arginine serine threonine valine tryptophane tyrosine 7 Secondary Structure Secondary structure is usually divided into three categories: Alpha helix Beta strand (sheet) Anything else – turn/loop 8 Alpha Helix: Pauling (1951) • A consecutive stretch of 5-40 amino acids (average 10). • A right-handed spiral conformation. • 3.6 amino acids per turn. 3.6 residues 5.6 Å • Stabilized by H-bonds in the backbone between C=O of residue n, and NH of residue n+4. • Side-chains point out. 9 Beta Strand: Pauling and Corey (1951) • Different polypeptide chains run alongside each other and are linked together by hydrogen bonds. • Each section is called β -strand, and consists of 5-10 amino acids. β -strand 10 3.47Å Beta Sheet 4.6Å (a)Antiparallel (b)Parallel 3.25Å The strands become adjacent to each other, forming beta-sheet. 4.6Å 11 Loops • Connect the secondary structure elements. • Have various length and shapes. • Located at the surface of the folded protein and therefore may have important role in biological recognition processes. • Proteins that are evolutionary related have the same helices & sheets but may vary in loop structures. 12 How is the 3D Structure Determined ? 1. Experimental methods (Best approach): • X-rays crystallography. • NMR. • Others. 2. In-silico methods (partial solutions based on similarity):. • Threading - needs a 3D structure, combinatorial complexity. • Ab-initio structure prediction - not always successful. 13 X-ray crystallography 1. Obtain an ordered protein crystal. 2. Check x-ray diffraction. The crystal is bombarded with X-ray beams. The collision of the beams with the electrons creates 14 a diffraction pattern. X-ray crystallography 3. Analyze diffraction pattern and produce an electron density map. 4. Thread the known protein sequence into the density map. 15 X-ray crystallography • The molecules must be very pure in order to produce perfect and stable crystals. • The method is time-consuming and difficult. 16 NMR - Nuclear Magnetic Resonance (since 1945) • A sample is immersed in a magnetic field and bombarded with radio waves. • The molecule’s nucleus resonate (spin). This motion is determined and is specific for each molecule type. 17 Principles of NMR 18 NMR - Nuclear Magnetic Resonance • The NMR technique is very time consuming and expensive, and the sample has to be in a concentrated solution, and is limited to small and soluble molecules. 19 PDB: Protein Data Bank • Holds 3D models of biological macromolecules (protein, RNA, DNA). • All data are available to the public. • Obtained by X-Ray crystallography (84%) or NMR spectroscopy (16%). • Submitted by biologists and biochemists from around the world. 20 PDB – Protein Data Bank http://www.rcsb.org/pdb/ 21 How Many Structures ? PDB Content Growth http://www.rcsb.org/pdb/holdings.html 22 Structure Prediction: Motivation • Hundreds of thousands of gene sequences translated to proteins (genbanbk, SW, PIR) • Only about 28000 solved structures (PDB) Experimental methods are time consuming and not always posible • Goal: Predict protein structure based on sequence information 23 Structure Prediction: Motivation • Understand protein function – Locate binding sites • Broaden homology – Detect similar function where sequence differs • Explain disease – See effect of amino acid changes – Design suitable compensatory drugs 24 Prediction Approaches • Primary (sequence) to secondary structure – Sequence characteristics • Secondary to tertiary structure – Fold recognition – Threading against known structures • Primary to tertiary structure – Ab initio modelling 25 Can we predict the secondary structure from sequence ? -helix b-sheet nonpolar polar polar polar Non-polar Secondary structures have an amphiphilic nature : one face polar and the other non polar 26 Secondary Structure Prediction Methods • Chou-Fasman / GOR Method – Based on amino acid frequencies • Artificial Neural Network (ANN) methods – PHDsec and PSIpred • HMM (Hidden Markov Model) • Best accuracy now ~80% 27 Chou and Fasman (1974) The propensity of an amino acid to be part of a certain secondary structure (e.g. – Proline has a low propensity of being in an alpha helix or beta sheet breaker) Name Alanine Arginine Aspartic Acid Asparagine Cysteine Glutamic Acid Glutamine Glycine Histidine Isoleucine Leucine Lysine Methionine Phenylalanine Proline Serine Threonine Tryptophan Tyrosine Valine P(a) 142 98 101 67 70 151 111 57 100 108 121 114 145 113 57 77 83 108 69 106 P(b) 83 93 54 89 119 037 110 75 87 160 130 74 105 138 55 75 119 137 147 170 P(turn) 66 95 146 156 119 74 98 156 95 47 59 101 60 60 152 143 96 96 114 50 Success rate of 50% 28 Secondary Structure Method Improvements ‘Sliding window’ approach • Most alpha helices are ~12 residues long Most beta strands are ~6 residues long Look at all windows of size 6/12 Calculate a score for each window. If >threshold predict this is an alpha helix/beta sheet TGTAGPOLKCHIQWMLPLKK 29 Improvements in the 1980’s • Adding information from conservation in MSA • Smarter algorithms (e.g. HMM, neural networks). Success -> ~80% 30 PHDsec and PSIpred • PHDsec – Rost & Sander, 1993 – Based on sequence family alignments • PSIpred – Jones, 1999 – Based on Position Specific Scoring Matrix Generated by PSI-BLAST • Both consider long-range interactions 31 HMM • HMM enables us to calculate the probability of assigning a sequence of hidden states to the observation TGTAGPOLKCHIQWML HHHHHHHLLLLBBBBB observation p=? Hidden state 32 Beginning with an αhelix α-helix followed by α-helix The probability of observing Alanine as part of a β-sheet The probability of observing a residue which belongs to an αhelix followed by a residue belonging to a turn = 0.15 Table built according to large database of known secondary structures 33 HMM • The above table enables us to calculate the probability of assigning secondary structure to a protein • Example TGQ HHH p = 0.45 x 0.041 x 0.8 x 0.028 x 0.8 x 0.0635 = 0.0020995 34 SS prediction using ANN A C D E F G H I K L M N P Q R S T V W Y . Amino acid at position Inputs for one position 35 PHDsec Neural Net A C D E F G H I K L M N P Q R S T V W Y . Amino acid at position Inputs for one position Outputs H= helix E= strand C= Coil Confidence 0=low,9=high Hidden layer 36 Secondary structure prediction • • • • • • • • • • • • • • AGADIR - An algorithm to predict the helical content of peptides APSSP - Advanced Protein Secondary Structure Prediction Server GOR - Garnier et al, 1996 HNN - Hierarchical Neural Network method (Guermeur, 1997) Jpred - A consensus method for protein secondary structure prediction at University of Dundee JUFO - Protein secondary structure prediction from sequence (neural network) nnPredict - University of California at San Francisco (UCSF) PredictProtein - PHDsec, PHDacc, PHDhtm, PHDtopology, PHDthreader, MaxHom, EvalSec from Columbia University Prof - Cascaded Multiple Classifiers for Secondary Structure Prediction PSA - BioMolecular Engineering Research Center (BMERC) / Boston PSIpred - Various protein structure prediction methods at Brunel University SOPMA - Geourjon and Delיage, 1995 SSpro - Secondary structure prediction using bidirectional recurrent neural networks at University of California DLP - Domain linker prediction at RIKEN 37