Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
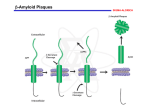
Dennis J. Selkoe Towards a remembrance of things past The question of the dualism of mind and brain has gripped the attention of philosophers and scientists for centuries. In recent decades, the debate about dualism has been influenced by enormous progress in the fields of neurobiology and psychology. Emerging knowledge about how the brain functions has seemed to strengthen the position of those who argue that mind versus brain is a false dichotomy. From the perspective of the neurobiologist, the functions of the mind – reasoning, abstraction, introspection, creativity and so forth – appear to emerge from the summed properties of millions, or more likely billions, of neurons and their unimaginably complex connections. If it is true, as I believe, that the faculties of the human mind are encoded by the activities of many elaborate sets of neurons, then diseases of the mind should arise directly from altered biochemical function of these neurons. The concept that mental faculties emerged directly from molecular and cellular neurobiology has fascinated me, like so many others, for decades. Indeed, this issue intrigued me long before I became closely familiar with specific disorders of mental function, such a schizophrenia and Alzheimer’s disease. More than any other factor, it was my sense of excitement about the still not fully resolved question of the mind-brain relationship that spurred me into the field of neurology. Among the myriad disorders that can wreak havoc with mental function, the example of Alzheimer’s disease is of special interest to neuroscientists. This is not only because it is the most common of the brain degenerative diseases, but also because it begins with a remarkably pure impairment of cognitive function. Alzheimer’s disease robs its victims of their most human qualities: memory, insight, reasoning, language and emotional homeostasis. Alzheimer patients enter upon their tragic journey by losing their ability to encode new memories in the brain, first of trivial and then important details of life. The insidious dissolution of a patient’s ability to learn new information, to store that to which one has been exposed for later retrieval, evolves in an individual whose motor and sensory functions are entirely preserved and who is otherwise neurologically intact. Over time, both declarative and non-declarative memory become profoundly impaired, and the capacities for other complex mental functions such as abstraction and language slip away. But the subtlety and variability of the earliest amnestic symptoms, occurring in the absence of any other clinical evidence of brain dysfunction, suggest that something is discretely, perhaps intermittently, interrupting the function of synapses which help encode new declarative memories. Research that my colleagues and I have conducted over more than two decades suggests that this “something” is the amyloid ß-protein (Aß), a hydrophobic 42-amino acid peptide with an ominous tendency to assemble into long-lived aggregates in brain regions important for memory and thinking. My own interest in the Alzheimer process arose during my training in both neurology (a clinical discipline) and neurochemistry (a basic biomedical discipline). I first encountered patients with Alzheimer’s disease as a medical student and then, more intensively, as a neurology resident. But my fascination with the disease arose from laboratory studies I performed on microtubules and neurofilaments in cultured neural cells during my postdoctoral training in biochemistry and neuronal cell biology. Microtubules are critical for the life of the cell and the organism, and I was assigned a project to examine aspects of how tubulin, their subunit protein, polymerizes into intact microtubules. As I began studying this subject and interfacing it with my clinical interest in neurology, I became increasingly aware that microtubules and their constituent proteins could potentially be implicated in one of the two classical brain lesions of Alzheimer’s disease -- the neurofibrillary tangle. Because of my strong desire to contribute somehow to the alleviation of neurological disease, I experienced a 180° change in scientific 1 direction, from the study of microtubules during the early development of the neuron to researching their role in the degeneration of neurons at the end of life. As it turned out some years later, the microtubule subunit, tubulin, was itself not directly implicated in the formation of neurofibrillary tangles. However, this apparent connection was enough to encourage me to begin to isolate neurofibrillary tangles from the cerebral cortex of patients dying with Alzheimer’s disease and attempt to decipher their biochemical composition. Before describing how these early experiments led me into a career-long quest for the causes and treatments of Alzheimer’s disease, I should mention some of the possible avenues by which a scientist might have approached a complex problem like Alzheimer’s disease in the late 1970’s, when my work began. Both the clinical and neuropathological features of the disease were analyzed in great detail in the decades after the Bavarian psychiatrist, Alois Alzheimer, described his index case, a 53-year-old woman from Frankfurt, in 1906. Clinically, the disease was marked by an inexorable progression of cognitive failure, usually beginning with subtle impairment of recent memory and proceeding to more severe memory difficulty, disorientation, confusion about matters of every day life, word-finding difficulty, problems with mathematical and geographical concepts, and emotional lability. Pathologically, patients who died with this syndrome after 10 or more years of progressive symptoms showed the two classical brain lesions Alzheimer had called attention to: the senile (neuritic) plaques and the neurofibrillary tangles. These usually occur in large numbers in the limbic and association cortices of the brain and, to some extent, in certain subcortical neuronal populations that project to the cortex. As one approached the question of how to decipher the cause and mechanism of this syndrome, one could imagine several routes. One was to grind up the patient’s brain and analyze it neurochemically in order to learn which normal constituents were changed in amount and whether some such altered constituents had a credible link to mental function. This avenue was pursued effectively by searching for changes in neurotransmitters. By 1977, several scientists had discovered that there was a profound decrease in the enzymes which synthesize and degrade acetylcholine, (choline acetyltransferase and acetyl cholinesterase), in the hippocampus and cerebral cortex. This knowledge suggested that neurons which use acetylcholine as a neurotransmitter were dying out in the disease, but it did not answer what might be killing them. Another potential approach would be to identify genes that caused inherited forms of Alzheimer’s disease. It was apparent that some cases ran strongly in families, but the methods for rigorous genetic linkage analysis and positional cloning were only beginning to emerge in the 1970’s, and there were not yet large collections of Alzheimer families to approach this problem at the time. A third approach, beyond the neurochemical and the genetic, was to pursue biochemical pathology. By this I mean the attempt to analyze the protein composition, and ultimately the molecular origin, of the classical neuropathological lesions that occur in Alzheimer cases, the amyloid plaques and neurofibrillary tangles. This is the approach that I chose to take around 1978 and later worked on intensively with a number of very talented colleagues. The avenue of biochemical pathology was not without its hazards. Experts in Alzheimer’s disease pointed out that the plaques and tangles were very likely to be “tombstones” of the process and would not lead readily to an accurate understanding of the earliest events in the disorder. While this was certainly possible, it seemed to me more likely that the innumerable neuritic plaques and neurofibrillary tangles that were the hallmarks of all cases of the disease represented a biochemical and cellular reaction to the underlying cause that might well provide a direct window on what that cause was. It was this thinking that encouraged me to isolate first neurofibrillary tangles and then amyloid plaques from the brains of patients dying with Alzheimer’s disease. I began the purification of neurofibrillary tangles shortly after completing a project isolating spinal cord neurons of rabbits that I had treated experimentally with aluminum salts 2 during my postdoctoral fellowship in the laboratory of Michael Shelanski at Harvard Medical School. Using methods similar to those Shelanski had recommended to study the aluminuminduced bundles of filaments in spinal neurons, I tried to characterize neurofibrillary tanglebearing neurons from AD cerebral cortex. In repeated attempts to analyze these biochemically and determine the subunit protein of the abnormal paired helical filaments (PHF) comprising the tangles, I was unable to discern any differences in the protein composition of tangle-rich neuronal fractions versus identically prepared fractions from age-matched normal cortex devoid of tangles. Despite the presence of abundant tangles, it appeared that their subunit protein could not be extracted and displayed on denaturing electrophoretic gels. In particular, I could not confirm an earlier report that the subunit of the tangles was a ~50,000 MW protein. One possible explanation for my inability to detect the proteins comprising the tangles on the gels was that the tangles were highly insoluble and thus not broken down by the denaturing agents [sodium dodecyl sulfate (SDS) and ß-mercaptoethanol (BME)] used in protein gel electrophoresis. Based on this hypothesis, I painstakingly examined the tangle-rich fractions from AD cortex under the electron microscope, both before and after their extraction in the SDS –BME denaturing buffer. I was excited to find that the PHF of the tangles were still present after boiling the sample in the denaturants, and they now appeared enriched and “cleaner” than in the same fraction prior to SDS-BME treatment. During this time, the first scientist to join my formative laboratory from overseas, Yasuo Ihara of Tokyo University, worked closely with my research technician, Fernando Salazar, and me to establish unequivocally that most neurofibrillary tangles in AD brain were resistant to these harsh solvents and remained as insoluble polymers. This explained why we were unable to identify particular protein differences between the tangle-rich and tangle-free brain fractions. The paper that we published (Selkoe, Ihara and Salazar, Science, 1982) provided perhaps the first example of the remarkable insolubility, and thus stability, that pathological aggregates of neuronal proteins have in neurodegenerative diseases. Thereafter, Ihara, research technician Carmela Abraham and I pursued our studies of the unusual chemical properties of neurofibrillary tangles. We developed the first antibodies to such tangles and showed that they did not react with numerous normal brain proteins we probed. This result suggested that the tangles were composed of conformationally altered epitopes of some unidentified brain protein. After Ihara returned to his own laboratory at Tokyo University, this work was continued with postdoctoral fellow Kenneth Kosik. We were later observed that the anti-tangle antibodies reacted with an altered form of the microtubule-associated protein, tau. This result and similar observations from other laboratories indicated that tau was the major antigenic component of the paired helical filaments making up the tangles. Subsequent biochemical analyses showed that the tangles were largely if not solely composed of insoluble forms of the tau protein. This work on the biochemical pathology of the neurofibrillary tangle helped me appreciate the remarkable chemical alterations of normal brain proteins that could occur during neurodegeneration. Years later, it became apparent that other age-related brain disorders such as Huntington’s and Parkinson’s were also characterized by the progressive insolubilization and accumulation of normally soluble neuronal proteins. Indeed, one of the most exciting lessons that has emerged from the study of these previously obscure diseases is that they seem to share a general mechanism of protein misfolding and aggregation. Insoluble aggregates of proteins can accumulate in neurons in many neurological diseases, although the individual proteins comprising the aggregates are distinct in disorders like Parkinson’s, Alzheimer’s, Huntington’s and ALS. By 1983, I had become increasingly aware that neurofibrillary tangles, while a hallmark of Alzheimer-type brain pathology, were by no means unique to this disorder. Indeed, tangles occurred in a wide range of etiologically distinct neurodegenerative disorders, including for example, subacute sclerosing panencephalitis (SSPE, a late post-measles neurodegeneration) and Kuf’s disease (a lipid storage disorder). This knowledge implied that neurofibrillary tangles could be responsible for injuring neurons but they might arise, in turn, from different kinds of 3 initial insults. During the course of Alzheimer’s disease, an insult arose that was presumably distinct from the causes of other tangle-bearing syndromes, such as SSPE and Kuf’s disease. Based on such considerations, I concluded that the amyloid plaques of Alzheimer’s disease might be a more specific – and perhaps earlier -- feature of the disorder than the tangles. Among human neurological disorders, amyloid plaques were known to accumulate primarily in Alzheimer’s disease and Down’s syndrome. Patients with trisomy 21 (Down’s syndrome) invariably develop both the neuritic plaques and neurofibrillary tangles of Alzheimer’s disease, but they generally do so already in their 30’s and 40’s, and this can be accompanied by further intellectual deterioration in these individuals, who are already retarded from birth. The only other circumstance in which substantial numbers of cortical amyloid plaques occur is very late in life in both humans and lower primates. On this basis, Carmela Abraham and I began to develop methods for isolating and purifying the amyloid that made up the cores of the innumerable senile plaques found in limbic and association cortices of AD brains. We found that the amyloid plaques, like the tangles we had studied earlier, were highly insoluble and remained largely intact after treatment in harsh denaturants such as SDS and guanidine hydrochloride. We took advantage of their insolubility and their roughly spherical shape to use fluorescent-activated cell sorting to purify them from other brain constituents. As we were pursuing this method, I became aware of a paper from George Glenner at the University of California, San Diego, who had taken the approach of isolating not the amyloid plaques but rather the meningovascular amyloid deposits known to occur in most Alzheimer brains. Glenner’s work revealed the subunit protein of the vascular amyloid, which he named the ß-protein, now referred to as amyloid ß-protein (Aß). In my view, Glenner’s original isolation of Aß in 1984 has turned out to be the seminal event in the unraveling of the pathogenesis of Alzheimer’s disease. Our analyses indicated that the amino acid composition of purified plaque cores was very similar to that which Glenner had described for the meningovascular amyloid. In 1985, Colin Masters and Konrad Beyreuther published an analysis of their isolated plaque core fractions that came to just this conclusion: amyloid plaque cores solubilized in formic acid yielded a protein fragment that was similar if not identical to the ß-protein Glenner had described. We published our purification method shortly thereafter, together with further biochemical and immunochemical characterization of the amyloid plaque cores. A collaboration with Daniel Kirschner at Harvard allowed us to examine the biophysical characteristics of the purified amyloid plaques by X-ray fiber diffraction analyses. These revealed a cross ß-pleated sheet conformation typical of classical amyloid deposits in systemic organs. Shortly thereafter, Kirschner, Abraham and I showed that synthetic peptides made to the partial sequence of Aß could likewise assume stable, cross ß-pleated sheet conformations, providing an initial in vitro model of the Alzheimer amyloid fibril. By the time that we began publishing our analyses of both natural, purified amyloid plaque cores and synthetic Aß assemblies, it had become clear to me that the accumulation of the amyloid protein as an insoluble aggregate represented a reasonable candidate for the etiologic agent of Alzheimer’s disease. In other words, the characteristics that Glenner, Masters, our lab and others were describing for the cerebral amyloid of Alzheimer’s disease were highly reminiscent of those found in various systemic amyloid deposits that occur in distinct human amyloidosis. Because some of these disorders were known to be caused by mutations in the genes encoding the respective amyloid subunit proteins, it appeared reasonable that both familial and sporadic forms of Alzheimer’s disease could begin with accumulation of the amyloid ßprotein in either mutant or wild-type form. The identification of the gene encoding the precursor protein of Aß (amyloid ß-protein precursor, or APP) by 4 laboratories in 1987 led to several exciting insights. One that particularly intrigued me was the fact that the 40-42 amino acid Aß region was a fragment of the large precursor molecule predicted to derive in part from the single transmembrane domain of this receptor-like molecule (Fig. 1). This was highly curious, because it suggested that one of the two putative proteolytic cleavages that released Aß from its precursor must occur within the transmembrane region of the molecule. However, there was no known 4 mechanism by which a proteolytic cleavage within the membrane could occur, as proteolysis of a peptide bond requires water molecules, and these would not be found in the hydrophobic environment of a lipid membrane. This apparent conundrum would occupy much of our attention in the next decade and a half. The other exciting insight that emerged from the cloning of APP was that its gene was found on the long arm of human chromosome 21. This recognition provided an immediate (and as it turns out, correct) hypothesis for why patients with trisomy 21 invariably develop Alzheimer-type pathology early in life. Some years later, Dr. Cynthia Lemere, a graduate student in my lab, found that early Aß deposits (so-called “diffuse” or “pre-amyloid” plaques) could occur in large numbers in the cerebral cortex of Down’s patients as early as age 12, long before mature, amyloid-bearing neuritic plaques or neurofibrillary tangles arose. This work and similar studies in other laboratories strongly suggested that accumulation of the amyloid ßprotein in such early deposits preceded the development of other neuropathological features of AD by years or decades, at least in patients with trisomy 21. My laboratory continued to focus strongly on the biology of APP and the way in which Aß arose and accumulated in the brain. We provided the first biochemical description of the heterogeneous polypeptides that comprise the APP gene products in the brain and noted that APP underwent a proteolytic cleavage to yield a ~10 kDa C-terminal fragment (later referred to as C83). We also found that the amyloid deposits that occurred with advanced age in lower primates (e.g., monkeys) and certain other mammals (e.g., dogs) were composed of the same amyloid ß-protein as accumulated in AD. Since these otherwise healthy older animals could not all be considered to have a disease per se, we reasoned that Aß must be produced and accumulate in normal brain tissue over time. Our attention was particularly drawn to the enigma mentioned earlier of how Aß occurred as a free subunit protein in the amyloid deposits when it was initially a portion of the membraneanchored region of APP. This location of the Aß region was generally interpreted as indicating that some injury to the membrane of a cell (e.g., a neuron in the cerebral cortex) was needed before an unidentified protease could access the Aß region and clip it out of the precursor to release it. If this interpretation were correct, then Aß formation and accumulation represented, at the very least, a secondary event in the pathogenesis of AD, that is, it would follow some prior membrane injury. But my colleagues and I felt that the accumulation of Aß deposits in normal elderly humans, primates and other lower mammals suggested that Aß might be formed as a normal metabolic event over time. To this end, we began an intensive search for evidence that Aß could occur in normal biological fluids, for example, in plasma, as was the case for the protein subunits of systemic amyloid deposits (e.g., transthyretin in senile cardiac amyloidosis). This search led to the discovery of normal Aß production by cells throughout life, an observation made by postdoctoral fellows Michael Schlossmacher and Christian Haass in the laboratory. The extensive experiments that Haass, Schlossmacher and other co-workers carried out in late 1991 and early 1992 led us to report the unexpected finding that Aß was a normal product of cellular metabolism and occurred in the extracellular fluids of healthy cells. Similar results were published at the same time by the research groups of Steven Younkin and Dale Schenk, respectively. The discovery of the normal production of soluble Aß throughout life had three immediate implications. First, it would now be possible to study the conversion of APP to Aß dynamically in cultured cells and in animals. Prior to this, Aß had only been observed after painstaking purification of insoluble amyloid deposits from postmortem human brain tissue. But now, it was clear that essentially any cultured cell being studied in the laboratory could be analyzed for its secretion of Aß. Second, the presence of Aß in normal biological fluids, such as cerebrospinal fluid and plasma, suggested that one might ultimately be able to quantify Aß and follow changes in its amount over time and during treatment of Alzheimer’s disease. Indeed, the 5 measurement of Aß42 has subsequently been shown to be useful as a biomarker of Alzheimer’s disease. Third, and perhaps most important, the finding of normal Aß secretion by cells provided cell culture assay systems that could be used to screen large libraries of chemical compounds in order to find those that could lower Aß production without injuring the cell. Each of these three implications of the discovery of soluble Aß has been extensively exploited in the decade since the initial reports. In my own laboratory, postdoctoral fellow Martin Citron found that an APP mutation discovered by geneticists to cause a rare form of early-onset AD in a Swedish family increased the cellular production of Aß about 5-8 fold. This observation and a similar finding from Steve Younkin and colleagues provided the first genotype-to-phenotype relationship in familial Alzheimer’s disease. Our laboratory subsequently examined the mechanisms of other APP mutations, including in skin fibroblasts cultured directly from affected patients. After presenilin (PS) was discovered as an AD-causing gene by Peter St. George Hyslop in 1995, we and others analyzed the effects of various natural mutations in PS1 and PS2 on APP processing. In all cases, we found that genetic mutations causing early-onset familial AD led to an increase in the production of Aß, in particular, the hydrophobic and highly self-aggregating Aß42 species. This evidence for a direct conversion from Alzheimer-causing genotypes to the phenotype of cerebral Aß accumulation remains one of the strongest supports for the “amyloid” hypothesis (more correctly the Aß hypothesis) of Alzheimer’s disease. Among the exciting aspects of researching the mechanisms of a disease such as Alzheimer’s are the implications one’s discoveries may have for fundamental biology. For my colleagues and me, this has been particularly well-illustrated by work on the presenilin protein and its connection to the unknown protease referred to as -secretase. The finding of normal Aß production mentioned above meant that two proteolytic activities, nicknamed ß-secretase and secretase, must cleave APP at the N- and C-termini of the Aß region, respectively, to liberate small peptides of 39-43 amino acids into intraluminal and extracellular fluids. Because mutations in presenilin cause the most aggressive known form of AD by increasing the generation of Aß42 peptide compared to that of the Aß40 peptide, it appeared that presenilin modified the -secretase reaction in some way. In other words, presenilin mutations allowed secretase to cleave more APP molecules after the 42nd rather than the 40th residue in the Aß sequence, leading to more Aß42, which has a stronger tendency to aggregate into oligomers and polymers. To approach how presenilin alters -secretase processing of APP, postdoctoral fellow Weiming Xia asked whether the two proteins interact directly. He obtained evidence that APP and PS1 could be co-immunoprecipitated from cells, even at endogenous levels of protein expression. We also performed various subcellular fractionation experiments that convinced us that APP and presenilin were often found in the same vesicular fractions of cells, and they could again be co-precipitated from such vesicles. Indeed, presenilin and APP contacted each other within subcellular vesicles in which we could demonstrate the de novo production of Aß upon incubation at 37°C. My close collaborator, Michael Wolfe, a medicinal chemist by training, pioneered a separate line of investigation. Dr. Wolfe decided to design and synthesize peptidomimetic inhibitors of -secretase that mimicked the Aß40-45 region within APP. By installing difluoro ketone or difluoro alcohol moieties in the P1/P1’ positions of these substrate-based molecules and then collaborating with my lab to show that they inhibited cellular Aß production, he was able to conclude that whatever -secretase was, it was very likely to be an aspartyl protease, rather than a member of one off the three other classes of protein-cleaving enzymes. As my colleagues and I were obtaining the various lines of evidence just reviewed, Dr. Bart DeStrooper at the Catholic University of Leuven, Belgium, reported the striking observation that deleting the presenilin 1 gene in a mouse caused a sharp decrease (~70%) in Aß production by its neurons. Although this finding was generally interpreted to suggest that PS served as a 6 critical cofactor for the unknown -secretase, my colleagues and I felt that another plausible interpretation was that presenilin was the actual protease. Much of the evidence we had obtained to date (above) was consistent with this provocative hypothesis, and we pursued it. A close inspection of the presenilin amino acid sequence led Dr. Wolfe to observe two aspartate residues in adjacent transmembrane domains of both presenilin 1 and presenilin 2 (the two human homologues). This exciting finding led immediately to our speculating that the two aspartates might serve as the active site of an unprecedented intramembranous aspartyl protease. Subsequent mutagenesis of the aspartates in PS1 led to a dramatic drop in cellular Aß production, about to the same extent as knocking out the entire PS1 gene. Moreover, mutating either intramembrane aspartate abrogated the normal endoproteolysis of presenilin into its biologically active heterodimeric form. We interpreted these results to indicate that the two aspartates were the active site of -secretase and could activate the enzyme by autoproteolysis. It was of great interest that all homologues of presenilin in lower animals (e.g., in C. elegans and Drosophila) also contain these two aspartates. Upon our publication of these data in early 1999, several laboratories confirmed and extended the findings, showing that the aspartate mutations acted in a dominant-negative fashion to markedly decrease the function of presenilin in intact cells. Nonetheless, the hypothesis that presenilin was -secretase remained controversial. As my colleagues and I pursued our identification of -secretase, it became increasingly apparent from the work of Iva Greenwald, Raphael Kopan, Bart DeStrooper and others that this unusual protease had a crucial role in the normal development of all multicellular organisms – as a mediator of the Notch signaling pathway. Work in these laboratories showed that C. elegans, Drosophila and mammals all required their presenilin homologues for proper function of Notch. More specifically, a cleavage within the Notch transmembrane domain first described by Kopan and colleagues at Washington University, liberates the Notch cytoplasmic domain to signal in the nucleus, and presenilins are required for this processing. It collaborative studies led by Bradley Hyman at the Massachusetts General Hospital, we found that the aspartate mutations in PS1, as well as the APP substrate-based -secretase inhibitors mentioned earlier, could markedly interfere with Notch nuclear entry and thus decrease Notch signaling. This realization meant that PS was necessary not only for generating the Aß peptide of Alzheimer’s disease, but also for the proper function of Notch in cell fate decisions throughout metazoan development. If we wished to inhibit -secretase (aka presenilin) with compounds such as those that Dr. Wolfe had made, we risked interfering with the normal function of Notch. This function is not restricted to early development but also in cell fate decisions in adulthood, such as during hematopoeisis and turnover of epithelial cells. At this writing, it is not yet clear whether one could safely inhibit presenilin/-secretase with a small molecule in a way that chronically lowers Aß perhaps 30% or so but does not cause side effects on Notch processing. Moreover, several labs, including ours, have discovered numerous additional substrates of PS/-secretase, raising questions about other pathways that could be impacted by inhibitors. Nevertheless, it may still be possible to modulate the secretase complex in a way that subtly lowers Aß production without serious side effects. This is particularly true in view of recent evidence we obtained that presenilin is the active site member of a four-protein complex which includes 3 other membrane proteins, nicastrin, Aph1 and Pen2. While this finding indicates the complexity of -secretase, it also provides additional potential therapeutic targets. My colleagues and I are also very interested in the consequences of the lifelong Aß production reviewed above. We have been studying for some years the proteases that might normally degrade Aß. Just as heightened production of Aß (e.g., as a result of a presenilin mutation) could cause the chronic elevation of Aß42 that I believe precipitates AD, so could decreased degradation of the peptide. Wei Qiao Qui, a postdoctoral fellow in the lab, screened cell lines for potential Aß-degrading proteases and found that the known thiol metalloendopeptidase, insulin degrading enzyme (IDE), is responsible for a substantial portion of 7 Aß degradation by microglia and other cells. On this basis, we hypothesized that mutations in the IDE gene that decrease its function might underlie some forms of AD in which the cause of chronic Aß elevation is yet unknown. My colleague at Harvard, Dr. Rudy Tanzi, has recently obtained intriguing genetic evidence that suggests this could be the case. However, much work is needed before we will know whether dysfunction of IDE, or another Aß-degrading protease in the brain, can actually lead to the Alzheimer syndrome. Among numerous other aspects of this fascinating riddle in human neurobiology that my laboratory is investigating, growing evidence that small oligomers of Aß, rather than the large amyloid fibrils of the senile plaques, may be the principal neurotoxic form of the peptide intrigues us. In this regard, work led by Dominic Walsh in my lab, in collaboration with Michael Rowan and coworkers at Trinity College, Dublin, has shown that soluble oligomers of Aß -- first discovered in cell culture by postdoctoral fellow Marcia Podlisny -- can block long-term potentiation (LTP) when injected into the hippocampus of living rats. These experiments therefore suggest that diffusible oligomers of Aß might be responsible for some of the alterations in hippocampal neuronal activity in that presumably underlie memory impairment in AD patients. Finally, like others, I am intrigued by the emerging parallels between the work on the pathobiology of AD summarized here and the attempt to implicate oligomers of misfolded proteins in the mechanisms of other neurodegenerative diseases. In this regard, postdoctoral fellows Michael Schlossmacher and Hideki Shimura, in collaboration with Kenneth Kosik and myself, have recently observed a special interaction of the two key gene products implicated in familial Parkinson’s disease, -synuclein and parkin. It appears that parkin, an E3 ubiquitin ligase, can ubiquitinate an O-glycosylated form of a-synuclein that we discovered in human brain, and this process may contribute to the progressive accumulation and aggregation of synculein in dopaminergic neurons in Parkinson’s disease. Much additional research is needed before we can learn whether this parkin--synuclein interaction carries a similar import for Parkinson’s disease that the presenilin-APP interaction described earlier does for Alzheimer’s disease. The application of molecular and cell biological methods to the elucidation of Alzheimer’s disease, with special attention to inherited forms of the disorder, has allowed us to develop and refine a hypothetical model describing how the disease begins, unfolds and eventually produces progressive dementia (Fig. 2). This schema remains a hypothesis, one that we believe has strong experimental support but that can ultimately be validated only in the clinic. The ideas and findings reviewed in this lecture have helped bring us to the verge of human trials of several Aß-lowering therapeutic agents. If one or more of these turn out to delay the development or slow the course of progressive cognitive failure, then Alzheimer’s disease may turn out to exemplify the power of reductionist science applied to the most complex of biological systems, the human cerebral cortex. 8