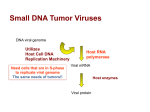
Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
West Nile fever wikipedia , lookup
Oesophagostomum wikipedia , lookup
Biological warfare wikipedia , lookup
Eradication of infectious diseases wikipedia , lookup
Hepatitis B wikipedia , lookup
Middle East respiratory syndrome wikipedia , lookup
Bioterrorism wikipedia , lookup
Ebola virus disease wikipedia , lookup
Marburg virus disease wikipedia , lookup
Orthohantavirus wikipedia , lookup
Herpes simplex virus wikipedia , lookup
Cross-species transmission wikipedia , lookup
Henipavirus wikipedia , lookup
Influenza A virus wikipedia , lookup