Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Rutherford backscattering spectrometry wikipedia , lookup
Atomic theory wikipedia , lookup
G protein–coupled receptor wikipedia , lookup
Biochemistry wikipedia , lookup
Double layer forces wikipedia , lookup
Peptide synthesis wikipedia , lookup
History of molecular biology wikipedia , lookup
Self-assembling peptide wikipedia , lookup
Interactome wikipedia , lookup
Mass spectrometry wikipedia , lookup
Gas chromatography–mass spectrometry wikipedia , lookup
Thermal shift assay wikipedia , lookup
Western blot wikipedia , lookup
Bottromycin wikipedia , lookup
Metalloprotein wikipedia , lookup
Two-hybrid screening wikipedia , lookup
Chemical biology wikipedia , lookup
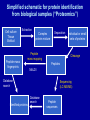
Lecture 5 Condensed phase ionisation techniques: spray methods At the end of this lecture you should be able to: • describe the ion formation models in ESI-MS • calculate molecular weights and charge states from low- and high-resolution ESI-MS spectra Ionisation Techniques: Overview Gas-Phase Methods • Electron Impact (EI) • Chemical Ionization (CI) Desorption Methods • Secondary Ion MS (SIMS) and Liquid SIMS • Fast Atom Bombardment (FAB) • Laser Desorption/Ionization (LDI) • Matrix-Assisted Laser Desorption/Ionization (MALDI) Spray Methods • Atmospheric Pressure Chemical Ionization (APCI) • Electrospray (ESI) Condensed phase ionisation techniques (2): spray methods Overview • Thermospray • APCI: Atmospheric pressure chemical ionisation • APPI: Atmospheric pressure photoionisation • Electrospray ionisation Atmospheric pressure chemical ionisation (APCI) • For non-polar and thermally stable compounds < 1500 Da • Useful to combine with liquid chromatography Spray needle/ Capillary Ions Spray To mass spectrometer Flow Cone Nebuliser gas Skimmers Corona discharge Atmospheric pressure Vacuum www.chm.bris.ac.uk/ms/theory/apci-ionisation.html Electrospray Ionisation (ESI) • • • • Nobel Prize to Fenn in 2002 Also atmospheric pressure ionisation Very versatile Also works for (very) large (bio)molecules, including proteins, nucleic acids, carbohydrates • Softest ionisation technique of all Spray needle/Capillary Spray To mass spectrometer Flow 2-10 ml/min 3-4 keV Cone Skimmers Atmospheric pressure Vacuum http://www.chm.bris.ac.uk/ms/theory/esi-ionisation.html Electrospray Ionisation (ESI) • Flowrates: 2 to 10 ml/min: Best interface for LC/MS • Can be combined with almost any mass analyser Common: TOF, Ion Trap, Quadrupole, FT-ICR • Uses: Mass detection, structure elucidation, protein folding, H/D exchange, protein sequencing…. Sample characteristics • Common solvents: mixtures of water with acetonitrile or methanol • Usually with added acid (acetic, formic), < 1% • Can’t tolerate (non-volatile) salt or buffers • Can do positive or negative electrospray: selected by capillary voltage What it looks like Ionisation models Ion evaporation model 1. Spray generates multiply charged droplets 2. Solvent evaporation leads to increasing charge density 3. When charge density too high: Coulombic explosion Spray needle tip Multiply charged droplet Analyte molecule Rayleigh limit is reached Multiply charged droplet www.chm.bris.ac.uk/ms/theory/esi-ionisation.html Charged residue model Charged amino acids Usually negatively charged at pH 7 O- 4.24 O O 3.65 O N Acidic N Aspartate Glutamate O 6.00 N Positively/uncharged at pH 7 O O- N NH Histidine Basic Always positively charged 12.5 +N + O H3N 10.8 Lysine N O N N Arginine N Charges on surface of proteins Red: Negatively charged Blue: Positively charged White: No charge; hydrophobic Examples • ESI of large molecules usually produces multiply charged ions • e.g. proteins: 12+ 11+ 10+ 9+ 13+ 8+ Charge state series 7+ 6+ 750 1000 1250 1500 1750 2000 2250 2500 2750 m/z • Each peak corresponds to the same protein, but with different number of protons attached: Observed ions are [M+nH]n+ How to determine the molecular mass of a protein from an ESI-MS spectrum • Observed ions have composition [M+nH]n+ • Let m1,m2,…, mn : m/z values of the different peaks 848.7 808.3 771.6 893.3 • mn = 738.1 942.8 [M+ nH] n • Its neighbouring peak to the left: 707.4 998.1 1060.5 • mn+1 = [M+ (n+1)H] n+1 • Solving both equations for n and M: m/z n must be an integer mn+1 H • n= mn mn+1 • M = n(mn – H) • e.g. m2 = 998.1 and m1 = 1060.5 • n = 16, and M = 16952 Da Deconvolution of ESI mass spectra Deconvoluted spectrum Charge states M = n(mn – H) 3000 30000 Mass (Da) Quick reminder: average and monoisotopic mass The distance between isotopic peaks reveals charge state mix of 6 proteins LCT prot_mix_0724a 651 (10.856) Sm (SG, 2x6.00); Cm (648:651) protein_modeling 505.3506 TOF MS ES+ 783 505.3506 100 +1 % 1.00 506.3584 mix of 6 proteins LCT 915.4818 prot_mix_0724a 350 (5.837) Sm (SG, 2x6.00); Cm (343:374) 100 506.3584 protein_modeling 915.7363 TOF MS ES+ 1.86e3 915.7363 507.3566 915.4818 507.3566 915.9765 +4 % 915.9765 915.2247 0 500 m/z 501 502 503 504 505 506 507 508 509 510 511 512 916.2311 915.2274 916.2311 916.4857 0.25 916.4857 916.7402 0 915 916 917 m/z mix of 6 proteins 918 prot_mix_0724a 655 (10.923) Sm (SG, 2x6.00); Cm (645:675) LCT protein_modeling 1086.5515 TOF MS ES+ 454 1086.5515 100 1086.0433 1086.0433 0.51 1086.0444 +2 % 1087.0444 1087.5529 1087.5529 1088.0460 Jonathan A. Karty 1088.0460 0 m/z 1084 1085 1086 1087 1088 1089 1090 Charge States and distance between isotopic peaks a.i. 1.5 0.1 +10 1.0 0.5 0.0 1695.7 1696.2 1696.7 m/z Example beyond molecular mass: Protein folding • Calbindin: Calcium binding induces protein folding: increase in charge states with higher m/z (=lower charge, more folded) No Ca2+ excess Ca2+ raw data deconvoluted Recent developments: ambient mass spectrometry: DESI and DART • • • • DESI: Desorption electrospray ionisation DART: Direct analysis in real time Applicable to solids, liquids, and gases No prior sample treatment ! Ionsiation techniques: Summary Ionisation Vola- Thermal Size tile Amount Examples EI Yes Stable Small 1-2 mg organics CI Yes Stable Small 1-2 mg organics FAB No Stable Medium 0.5-1 mg Polar/ionic organics, organometallics, peptides, biomolecules FD No Labile Medium 1-2 mg Non-polar organics, organometallics MALDI No Labile Large 250 fmol500 pmol Peptides, proteins, polymers ESI No labile Large 1-300 pmol/ml Polar/ionic organics, peptides, proteins, biomolecules, organometallics, polymers Table adapted from http://www.scs.uiuc.edu/~msweb/SLM530.pdf Summary: Application ranges of various techniques masspec.scripps.edu/MSHistory/whatisms.php Self-assessment questions • Q1 Describe the two ion formation models in ESI • Q2 A positive-ion ESI spectrum shows the following adjacent signals at m/z 4348.8, 4546.5, 4762.9, 5001.0 5264.2. Calculate the molecular mass of the molecule. • Q3 The following m/z (979,1040,1109,1189,1280,1387,1512,1664) were obtained by electrospray ionisation of a protein from an aqueous solution. – Calculate the molecular mass of the protein within 10 Da. – Describe how the mass spectrum would have looked if the same protein had been ionised by MALDI. • Q4 What distance between isotopic peaks would you expect for a +12 charge state ? • Q5 The following peaks arose from the different isotopes contributing to the ESI mass spectrum resulting from the protonation of a species of relative molecular mass m to reach a charge z.:m/z=848.40, 848.45, 848.50, 848.55, 848.60, 848.65, 848.70. – What is the value z and what is the mass of the species giving rise to the peak at m/z 848.55 Lecture 6 Tandem MS Peptide/protein identification by MS At the end of this session you should be able to • explain how structural information can be obtained by Tandem MS and MALDI-TOF/PSD • explain how mass spectrometry data can be used to identify known and unknown proteins Tandem MS (also termed MS2 and MSn) • Used for: – Identify and quantify compounds in complex mixtures – Structure elucidation of unknown compounds • Applied in: – – – – Proteomics Metabolomics Biomarker discovery De novo protein sequencing Tandem MS • Multistage technique: mass selection of precursor ion Ion source MS-1 Activation and fragmentation Mass analysis of product ions MS-2 Normal spectrum MS/MS spectrum Tandem MS Fragmentation techniques • Collision-induced dissociation (CID): – most common – Possible with ESI coupled to triple-quad, Ion trap, FTICR, and MALDI-TOF • Electron Capture Dissociation (ECD): – only for multiply charged biopolymers – Primarily with FT-ICR • Electron-Transfer Dissociation (ETD) • Absorption of electromagnetic radiation • Not Tandem MS, but useful fragmentation technique: Post-source decay (PSD) combined with MALDI reflectron TOF Reflectron-TOF and Post-Source decay for MALDI-TOF in d u c e s p o s t Field-free region s o u rc e d e c a y D e te c to r r e fle c tr o n • (Some) parent ions fragment in field-free drift region • Parent and product ions arrive at reflectron simultaneously (same velocity) • Product ions leave Reflectron earlier (smaller Ekin) Example: MALDI-PSD TOF spectrum of a neuropeptide Parent Ion Necla Birgül, Christoph Weise, Hans-Jürgen Kreienkamp and Dietmar Richter , The EMBO Journal (1999) 18, 5892–5900 Simplified schematic for protein identification from biological samples (“Proteomics”) Cell culture Tissue Biofluid Peptide-mass fingerprints Extraction Complex protein mixture Separation Peptide mass mapping Cleavage Peptides MALDI Database search Identified proteins Individual or small sets of proteins Sequencing (LC-MS/MS) Database search Peptide sequences Peptide mass mapping/ fingerprinting • Makes use of specific cleavage agents – Chemical cleavage: e.g. CNBr – Digestion with endoproteases (proteolytic enzymes): Trypsin, pepsin, chymotrypsin etc. – See exercises Peptide mass mapping/fingerprinting Peptides Mass Spectrum Abundance Protein Digest 600 900 1200 1500 1800 m/z Compare QNICPRVNRIVTPCVAYGLG RAPIAPCCRALNDLRFVNTR NLRRAACRCLVGVVNRNPGL RRNPRFQNIPRDCRNTFVRP FWWRPRIQCGRIN Peptide sequences Theoretical Digest NTFVRPFWWRPR IVTPCVAYGLGR CLVGVVNR APIAPCCR FQNIP ... Theoretical Mass Spectrum Abundance Protein Sequence (in database) 600 900 1200 m/z 1500 1800 De novo protein discovery • Mass fingerprinting only practicable if protein is already in a database • If previously undiscovered protein: Need to sequence • Can be done by sequencing peptides Peptide sequencing: fragmentation rules R1 R2 R3 R4 +NH ―CH―CO―NH―CH―CO―NH―CH―CO―NH―CH―CO ― 3 2 N-terminus • • • • Three types of bonds along backbone amino alkyl bond alkyl-carbonyl bond amide bond C-terminus Peptide sequencing: fragmentation rules a1 b1 c1 R1 R2 R3 R4 NH2―CH―CO―NH―CH―CO―NH―CH―CO―NH―CH―CO2H x3 y3 z3 • Each bond can be broken by fragmentation Six possible product ions • But: peptide bond most likely to break in low energy CID (and MALDI/PSD): Mostly b and y fragments Peptide fragments generated by low energy CID or PSD b1 b2 b3 R3 R2 R1 R4 NH2―CH―CO―NH―CH―CO―NH―CH―CO―NH―CH―CO2H N-terminus C-terminus y3 y2 y1 For example: R1 R3 R2 + b3: NH2―CH―CO―NH―CH―CO―NH―CH―C=O R3 y2: R4 +NH ―CH―CO―NH―CH―CO H 3 2 m/z values of b ions: residue masses + 1 (+H+) m/z values of y ions: residue masses +17 (OH-) + 2 (2H+) = 19 Example: Peptide analysed by MALDI TOF reflectron and PSD y7 y8 y6 y5 y4 y1 Arg y2 Leu/ Ile/Asn Leu/Ile: 113.08 Asn: 114.04 Ala y3 Phe Leu/ Ile/Asn His Thr His Proposed sequence: HTH[LIN]FA[LIN]R Self-assessment questions • Q1 How are MALDI and ESI used for the identification of proteins ? • Q2 Describe how Peptide Fingerprinting works • Q3 A MALDI-PSD spectrum of a peptide shows the following y-peaks: 174.8 / 288.3 / 359.0 / 506.1 / 619.6 / 756.0 / 857.6 / 995.2 Find out the masses for amino acids (e.g. at Wikipedia). Calculate the differences between peaks in this spectrum and suggest possible sequences for this peptide Exercises • Exercise 1: Protein cleavage/digestion 1. Go to http://www.expasy.ch/tools/peptidecutter/ 2. In the box, enter ALBU_HUMAN (this is the swissprot name of human serum albumin) - you can also choose a different protein if you like. Sequences and swissprot codes can for example be found in the swissprot database (at www.expasy.ch). 3. Scroll down, and tick the box “only the following selection of enzymes and chemicals”, and then select one chemical or enzyme, which you want to use for cleaving the albumin protein, from the list 4. Scroll back up, and click “Perform” 5. Inspect the output. How many times is albumin cleaved by your chosen cleavage agent ? Find out what the specificity of your cleavage agent is. • Exercise 2: Calculation of molecular masses of proteins and peptides – 1. Copy a peptide fragment from the output of Exercise 1 (or make one up yourself), go to http://www.expasy.ch/tools/protparam.html – and paste your sequence into the appropriate box (the large one). – 2. Click “Compute Parameters”. – 3. Inspect the output. What is the molecular formula of the peptide ? How many positively and negatively charged side-chains does your peptide have ? What charge would it have at pH 7 ? • Exercise 3. Average and monoisotopic masses – 1. Copy the molecular formula (or the one-letter code sequence) of the peptide from exercise 2, and go to http://education.expasy.org/student_projects/isotopident/ htdocs/ – 2. Paste the formula or sequence in the appropriate box. Make sure that you have selected the correct “Type of composition” (e.g. “chemical formula”) in the respective pulldown menu. – 3. Click “Submit query”. – 4. Inspect the output. How many isotopic peaks are there? Which is the most abundant peak ? What are the monoisotopic and the average masses ?