Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Heart failure wikipedia , lookup
Saturated fat and cardiovascular disease wikipedia , lookup
Cardiac contractility modulation wikipedia , lookup
Management of acute coronary syndrome wikipedia , lookup
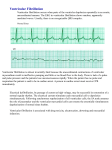
Electrocardiography wikipedia , lookup
Cardiovascular disease wikipedia , lookup
Quantium Medical Cardiac Output wikipedia , lookup
Coronary artery disease wikipedia , lookup
Hypertrophic cardiomyopathy wikipedia , lookup
Antihypertensive drug wikipedia , lookup
Ventricular fibrillation wikipedia , lookup
Arrhythmogenic right ventricular dysplasia wikipedia , lookup
Comprehensive Summaries of Uppsala Dissertations from the Faculty of Medicine 992 _____________________________ _____________________________ Left Ventricular Hypertrophy and the Insulin Resistance Syndrome BY JOHAN SUNDSTRÖM ACTA UNIVERSITATIS UPSALIENSIS UPPSALA 2001 Dissertation for the Degree of Doctor of Philosophy (Faculty of Medicine) in Geriatrics presented at Uppsala University in 2001 Abstract Sundström, J. 2001. Left ventricular hypertrophy and the insulin resistance syndrome. Acta Universitatis Upsaliensis. Comprehensive Summaries of Uppsala Dissertations from the Faculty of Medicine 992. 68 pp. Uppsala. ISBN 91-554-4919-0. Left ventricular hypertrophy (LVH) and the insulin resistance syndrome are common conditions associated with a markedly increased cardiovascular risk. In a fairly large prospective longitudinal study of men from the general population, we found that an unfavorable serum fatty acid profile and components of the insulin resistance syndrome such as dyslipidemia, obesity and hypertension at age 50 predicted the prevalence of LVH at age 70. In cross-sectional analyses at age 70, several components of the insulin resistance syndrome were significantly related to left ventricular relative wall thickness and concentric remodeling, but less to LVH. Left ventricular relative wall thickness was inversely related to insulin sensitivity in skeletal muscle and borderline significantly directly related to insulin sensitivity in the myocardium in a healthy, normotensive sample of the cohort investigated with positron emission tomography, whereas left ventricular mass index was not related to myocardial or skeletal muscle insulin sensitivity. At age 70, echocardiographic LVH was related to a variety of common electrocardiographic diagnoses. In a prospective mortality analysis with baseline at age 70 and a median follow-up time of five years, echocardiographic and electrocardiographic LVH predicted mortality independently of each other and of other cardiovascular risk factors, implying that echocardiographic and electrocardiographic LVH in part carry different prognostic information. In summary, components of the insulin resistance syndrome predicted LVH twenty years later, but were cross-sectionally more related to increased left ventricular relative wall thickness and concentric remodeling. Echocardiographic and electrocardiographic LVH predicted mortality independently of each other and of components of the insulin resistance syndrome. Key words: Left ventricular hypertrophy, insulin, glucose, lipids, mortality. Johan Sundström, Department of Public Health & Caring Sciences, Section for Geriatrics, Box 609, SE-751 25 Uppsala, Sweden © Johan Sundström 2001 ISSN 0282-7476 ISBN 91-554-4919-0 Printed in Sweden by Uppsala University, Tryck & Medier, Uppsala 2001 to Anna Left ventricular hypertrophy and the insulin resistance syndrome This thesis is based on the following investigations, which will be referred to by their Roman numerals: I. Sundström J, Lind L, Vessby B, Andrén B, Aro A, Lithell H. Dyslipidemia and an unfavorable fatty acid profile predict left ventricular hypertrophy 20 years later. Circulation 2001; in press. II. Sundström J, Lind L, Nyström N, Zethelius B, Andrén B, Hales CN, Lithell H. Left ventricular concentric remodeling rather than left ventricular hypertrophy is related to the insulin resistance syndrome in elderly men. Circulation 2000;101:2595-600. III. Sundström J, Lind L, Valind S, Holmäng A, Björntorp P, Andrén B, Waldenström A, Lithell H. Myocardial insulin-mediated glucose uptake and left ventricular geometry. Blood Pressure 2001; in press. IV. Sundström J, Lind L, Andrén B, Lithell H. Left ventricular geometry and function are related to electrocardiographic characteristics and diagnoses. Clin Physiol 1998;18:463-470. V. Sundström J, Lind L, Ärnlöv J, Zethelius B, Andrén B, Lithell H. Echocardiographic and electrocardiographic left ventricular hypertrophy predict mortality independently of each other in a population of elderly men. Submitted for publication. The original articles were reprinted with permission from the publishers. Contents ABBREVIATIONS _________________________________________________________________________ 6 INTRODUCTION _________________________________________________________________________ 7 THE ETIOLOGY OF LEFT VENTRICULAR HYPERTROPHY ______________________________________________ 8 LEFT VENTRICULAR HYPERTROPHY AND THE INSULIN RESISTANCE SYNDROME ____________________________ 9 ELECTROPHYSIOLOGIC CO-MORBIDITY OF LEFT VENTRICULAR HYPERTROPHY __________________________ 12 PROGNOSTIC SIGNIFICANCE OF LEFT VENTRICULAR HYPERTROPHY __________________________________ 12 AIMS OF THE STUDY_____________________________________________________________________ 13 METHODS______________________________________________________________________________ 14 THE COHORT __________________________________________________________________________ 14 Study populations _________________________________________________________________ 15 INVESTIGATIONS AT AGE 50 _______________________________________________________________ 15 INVESTIGATIONS AT AGE 70 _______________________________________________________________ 16 Echocardiography ________________________________________________________________ 16 Electrocardiography ______________________________________________________________ 17 Blood pressure measurements _____________________________________________________ 19 Hyperinsulinemic euglycemic clamp _______________________________________________ 19 Oral glucose tolerance test ________________________________________________________ 20 Lipid measurements_______________________________________________________________ 20 Positron emission tomography _____________________________________________________ 20 FOLLOW-UP AFTER AGE 70________________________________________________________________ 21 STATISTICAL ANALYSIS ____________________________________________________________________ 21 RESULTS _______________________________________________________________________________ 23 STUDY I ______________________________________________________________________________ 23 STUDY II ______________________________________________________________________________ 25 STUDY III______________________________________________________________________________ 26 STUDY IV _____________________________________________________________________________ 28 STUDY V______________________________________________________________________________ 30 DISCUSSION ___________________________________________________________________________ 34 LEFT VENTRICULAR HYPERTROPHY AND THE INSULIN RESISTANCE SYNDROME ___________________________ 34 Components of the insulin resistance syndrome are risk factors for later left ventricular hypertrophy ______________________________________________________________________ 34 Fatty acids and left ventricular hypertrophy ________________________________________ 34 Relative wall thickness is related to components of the insulin resistance syndrome ___ 36 Relative wall thickness is related to skeletal muscle insulin resistance _________________ 38 INSULIN RESISTANCE AND LEFT VENTRICULAR HYPERTROPHY: CAUSE OR CONSEQUENCE? ________________ 39 Potential mechanisms whereby insulin resistance or hyperinsulinemia may cause left ventricular hypertrophy____________________________________________________________ 39 Insulin resistance and left ventricular hypertrophy as parallel phenomena ____________ 42 Left ventricular hypertrophy as a causal factor for hypertension and insulin resistance_ 42 THE CLINICAL IMPORTANCE OF LEFT VENTRICULAR HYPERTROPHY ___________________________________ 43 Electrophysiologic co-morbidity of left ventricular hypertrophy_______________________ 43 Prognostic significance of echocardiographic and electrocardiographic left ventricular hypertrophy ______________________________________________________________________ 44 STRENGTHS AND LIMITATIONS OF THE STUDY ___________________________________________________ 45 CONCLUSIONS_________________________________________________________________________ 47 FUTURE PERSPECTIVES ___________________________________________________________________ 48 ACKNOWLEDGEMENTS _________________________________________________________________ 51 SUMMARY IN SWEDISH / SAMMANFATTNING PÅ SVENSKA _________________________________ 52 REFERENCES ___________________________________________________________________________ 53 Abbreviations LVH ECG RWT LVMI SD CE PET OGTT CV ICD HDL LDL LA IVS PW LVEDD LVESD ASE LVEDV LVESV LVOT FVI IVRT SBP DBP AUC NEFA 18FDG ROI PYAR ANOVA CI HR ROC BMI PPAR IGF-I GLUT PKC ET-1 ACE AT1 left ventricular hypertrophy electrocardiography left ventricular relative wall thickness left ventricular mass index standard deviation cholesteryl ester positron emission tomography oral glucose tolerance test intra-individual coefficient of variation International Classification of Diseases high-density lipoprotein low-density lipoprotein left atrial diameter interventricular septal thickness posterior wall thickness left ventricular end-diastolic diameter left ventricular end-systolic diameter American Society of Echocardiography left ventricular end-diastolic volume left ventricular end-systolic volume left ventricular outflow tract flow velocity integral isovolumetric relaxation time systolic blood pressure diastolic blood pressure area under the curve nonesterified fatty acids 2-[18F]-fluoro-deoxyglucose region of interest person-years at risk analysis of variance confidence interval Cox proportional hazard ratio receiver operating characteristic body mass index peroxisome proliferator-activated receptor insulin-like growth factor-I glucose transporter protein kinase C endothelin-1 angiotensin-converting enzyme angiotensin II type-1 receptor 6 Introduction Under some circumstances, of which we today only know a handful, the mass of the left ventricle of the heart increases. A too large left ventricular mass is known as left ventricular hypertrophy (LVH). LVH is a very common condition. The prevalence of echocardiographic LVH in the general population ranges from 10-20% in young and middle-aged subjects to 30-50% in elderly subjects1-3, and LVH is also more common in the settings of obesity, hypertension, valvular disease or previous myocardial infarction3,4. Irrespective of the underlying cause of LVH and diagnostic method, LVH imposes a great risk for cardiovascular and all-cause mortality and morbidity5-21. The only way of accurately determining the weight of the left ventricle is by autopsy, but several diagnostic methods give reasonable assessments of left ventricular mass in living subjects. Electrocardiography (ECG) was one of the first methods, and is still the most extensively used because of its low cost, widespread accessibility and proven prognostic value17-21. In the last decades, the introduction of cardiac ultrasound (echocardiography) has made direct assessment of cardiac geometry and calculation of left ventricular mass possible. Other aspects of the left ventricle than the mass, such as the ratio between the thickness of the left ventricular walls and the diameter of the left ventricular cavity (the relative wall thickness, RWT), have been possible to assess. The grouping of subjects into four left ventricular geometric groups according to left ventricular mass indexed for body size (left ventricular mass index, LVMI) and RWT (figure 1) has been shown to be relevant and to give prognostic information7,11-13. A hypertrophic left ventricle (increased LVMI) is denoted eccentric LVH if RWT is normal, and concentric LVH if RWT is increased. A normal LVMI with increased RWT is denoted concentric remodeling22. Recently, measuring left ventricular mass by nuclear magnetic resonance tomography has been found to have a high precision23, but is yet expensive and time-consuming. There are several ways of determining the clinical importance of an exposure, such as LVMI or RWT, and establishing limits for what should be regarded as healthy and unhealthy levels of this exposure. One way is to investigate the levels of the exposure in a healthy population, and simply regard the highest 5% of the values of the exposure (or >2 standard deviations (SD) above the mean exposure) as pathological. This is the way the definitions of echocardiographic LVH were originally derived24. Another way is to investigate if the exposure is cross-sectionally related to other pathological conditions, and if that is the case, determine at which level of the exposure the relation to pathological conditions appears. A third, and more appropriate, way is to investigate at which level of the exposure an increased risk for subsequent morbid or mortal events appears. In this study, a combination of these approaches were used in the investigation of left ventricular mass and geometry. Evaluation of the impact of prospectively determined risk factors on subsequent LVH, as well as evaluation of echocardiographic and ECG-LVH as predictors of later total and cardiovascular mortality and morbidity was made. Cross-sectional relations between left ventricular geometry and ECG abnormalities and glucose metabolism in the heart, skeletal muscle and the whole body were also investigated. 7 Fig. 1: Distribution of left ventricular geometric patterns in the study population Relative wall thickness Concentric remodeling, n=79 Concentric LVH, n= 40 Normal, n= 262 Eccentric LVH, n= 94 0.44 LVH = left ventricular hypertrophy 150 g/m2 Left ventricular mass index The etiology of left ventricular hypertrophy An increased mass of the left ventricle has generally been thought to be the consequence of a compensation by the ventricle for a hemodynamic stimulus, such as an increased demand for cardiac work. If the heart is challenged with a higher afterload, such as an aortic valve stenosis or an increased peripheral arterial resistance with arterial hypertension, the left ventricle is believed to respond with thickening of the ventricular walls and, ultimately, concentric LVH. If the heart is challenged with a higher preload, as in the case of a larger circulating blood volume in obesity or a regurgitating valve, the left ventricle is believed to respond with dilatation and, ultimately, eccentric LVH. Both wall thickening and dilatation may initially serve as relevant compensation for the increased hemodynamic stress and improve cardiac function, but also increase the weight of the heart. This increased mass is due to cardiomyocyte hypertrophy, rather than hyperplasia, because cardiomyocytes are probably terminally differentiated shortly after birth25. The myocyte hypertrophy is accompanied by a variable degree of non-myocyte proliferation and fibrosis26. Eventually, the structure of the myocardium is altered and the risk of subsequent morbidity and mortality increases, and the hypertrophy is no longer considered compensatory or physiologic, but pathologic27. However, the repertoire of stimuli that may cause LVH stretches far beyond hemodynamic ones, and is yet largely unknown. 8 Previous cross-sectional studies of correlates of LVH3,4,28-31 have shown age (although not in healthy elderly32), hypertension, obesity, certain valvular diseases, alcohol use, heredity, blood viscosity and previous myocardial infarction to be related to LVH or left ventricular mass. In one prospective study33, average blood pressure over 30 years was associated with LVMI at follow-up. Hypertension is generally regarded as the worst culprit, indicated by the large number of clinical trials on the subject (overviews in references34-36) showing that antihypertensive therapy lowers left ventricular mass and RWT. However, the correlation between degree of blood pressure lowering and degree of LVH reduction in previous studies is not always impressive34, and the variation in 24-h blood pressure explains only 2530% of the variation in left ventricular mass37, although findings are conflicting38. Thus, other factors must be of importance in the etiology of LVH. Body size is a powerful determinant of left ventricular mass39, and may explain part of the gender difference regarding left ventricular mass40. It is therefore common to index left ventricular mass for a measurement of body size, such as lean body mass, body surface area, height or height2.7, to obtain LVMI. Weight reduction has been shown to decrease RWT, left ventricular mass41 and LVMI in overweight hypertensive patients even more than pharmacological antihypertensive treatment42. Lifestyle intervention aimed at reducing weight, dietary salt intake, alcohol consumption and increasing physical activity in sedentary subjects has been found to be as effective in decreasing left ventricular mass by itself as in combination with antihypertensive medication43, suggesting that lifestyle factors may account for part of the hitherto unexplained LVH. Numerous intervention studies aiming at determining causal factors or possible treatments for LVH exist, but very few longitudinal studies of predictors of LVH33 have been made. Therefore, we have undertaken such a study aimed at identifying hemodynamic, metabolic, dietary and psychosocial predictors in men aged 50 for the prevalence of echocardiographic LVH and left ventricular geometric subtypes twenty years later (study I). As a proxy for dietary fat quality, we investigated the fatty acid composition of serum cholesteryl esters (CE), which mainly reflects dietary fat quality over the past couple of weeks44,45. Serum CE fatty acid composition has been shown to predict myocardial infarction46, but relation to LVH is not known. Neither have relations between smoking or other psychosocial factors and later LVH been investigated. Left ventricular hypertrophy and the insulin resistance syndrome It is well known that important cardiovascular risk factors, such as hypertension, glucose intolerance, hyperinsulinemia, dyslipidemia, and obesity, often cluster in the same individuals. The existence of a syndrome involving some of these disorders was proposed nearly 80 years ago by the Swede Eskil Kylin47, among others. Insulin resistance to glucose uptake, as assessed by the hyperinsulinemic euglycemic clamp48, has been proposed to be of pathogenetic importance in the syndrome, particularly by Gerald Reaven49-51. Consequently, the term ”insulin resistance syndrome” has, among other terms50,52,53, been proposed49. There is no internationally agreed definition of the insulin resistance syndrome at present, but 9 the most accepted definitions54,55 include the presence of insulin resistance or glucose intolerance together with two or more other components of the syndrome: insulin resistance, glucose intolerance, hypertension, abdominal obesity, raised plasma triglycerides, decreased HDL-cholesterol or microalbuminuria. The insulin resistance syndrome is a highly prevalent condition, present in 12.5% of the present cohort at age 50 and 18.8% at age 7056, in 20% of another general Swedish population57 and in 22-27% of a general American population58. In the present study, the mentioned definitions of the insulin resistance syndrome have not been used, but the relations between individual components of the insulin resistance syndrome and LVH have been investigated. The associations between LVH and components of the insulin resistance syndrome, mainly insulin levels, have recently gained interest (table 1). The left ventricular geometric correlates of insulin resistance are not yet clear, since some studies have found relations between insulin resistance variables and RWT59-61 whereas others have not62, and some studies have found relations between insulin resistance variables and the sum of left ventricular wall thickness62-64, LVH or left ventricular mass (with or without indexation for body size by various methods), whereas others have not. We used a fairly large sample of elderly men from the general population for a study of the relations between several important variables of the insulin resistance syndrome and RWT, LVMI and left ventricular geometric subgroups (study II). Pathogenetic processes linking the insulin resistance syndrome and LVH or increased RWT also remain to be elucidated. Although relations between wholebody insulin resistance and LVH or increased RWT have been investigated previously, the relations between insulin sensitivity in the myocardium and RWT or LVMI are not yet fully understood. Insulin-mediated glucose uptake, as measured with 18F-fluorodeoxyglucose and positron emission tomography (PET), has been found to be increased in the myocardium of hypertensives in one study65, but reduced in the myocardium in subjects with LVH in other studies66,67. As the increased work imposed on the left ventricle by hypertension is a powerful denominator of myocardial glucose uptake65, we investigated the relations between myocardial and skeletal muscle insulin sensitivity and RWT and LVMI in a homogenous sample of healthy, normotensive men with wide ranges of RWT and LVMI (study III). 10 Table 1: Previous studies of relations between left ventricular geometric properties and insulin resistance Study Study population LVM indexation Findings 229 (111 ♂), 107 DM type-2 Insulin resistance method OGTT Uusitupa, 198768 /BSA Sharp, 199269 102 (51 ♂), 51 HT, 2 LVH f-Ins /height Sasson, 199370 40 (18 ♂) healthy obese NT IVGTT /height Marcus, 19941 851 (439 ♂) healthy general population, age 18-42 89 (42 ♂) general population, age 36-37 f-Ins /BSA f-Ins /height /weight 50 HT clamp, f-Ins - in ♀: LVMI ➸ f-Ins, OGTT-Ins in ♂: LVMI ✗ f-Ins, OGTT-Ins in HT: LVMI ➸ f-Ins, f-Glu in NT: LVMI ✗ f-Ins, f-Glu LVMI ➸ IVGTT-f-Ins, -90min-Ins, -AUC-Ins and kvalue LVMI ➸ f-Glu, TG, stress-NE and -E in men: LVH ➸ f-Ins➹ ➹, TG➹ ➹, stress-NE and –E➹ ➹ LVM ➸ f-Ins LVM/height ➸ f-Ins LVM/weight ✗ f-Ins LVWT ➸ f-Ins (-)➸ ➸WBGD 36, 26 HT, 14 LVH ? Kupari, 199471 Lind, 199563 Paolisso, 199572 Paolisso, 199573 37 (19 ♂) HT, 25 LVH Souza, 199574 14 HT, 6 LVH clamp, OGTT, IC clamp, OGTT ITT ? LVH ➸ KITT➷ 29 (14 ♂) healthy lean HT OGTT /BSA LVMI ✗ OGTT-peak-Ins, IRI OGTT Kamide, 199676 390 ♂, 180 HT, work-site population 40 (17 ♂) healthy lean HT /BSA /height2.7 /BSA LVMI ✗ f-Ins, OGTT-Ins, f-Glu, OGTT-Glu RWT ➸ OGTT-Ins, f-Glu, OGTT-Glu ✗ f-Ins LVMI (-)➸ ➸ WBGD Rabkin, 199677 23 (13 ♂) DM type-2 LVMI ✗ f-Ins, IGF-I, IGF-II, GH Paolisso, 199762 26 ♂ healthy HT /BSA /height /height Avignon, 199778 24 ♀ healthy obese NT Tomiyama, 52 ♂ healthy HT Vetta, 199879 Jelenc, 199880 49 ♂ healthy NT, 29 obese, mean age 69 30 ♂ HT, 21 dippers Phillips, 199864 29 (21 ♂) healthy lean HT Chen, 199881 Verdecchia, Costa, 199575 Ohya, 199659 199760 199961 Watanabe, 199982 Rheeder, 199983 Galvan, 200084 clamp, OGTT f-Ins, IGF-I, IGF-II, GH clamp, IC ? in HT: LVMI ➸ f-Ins (-)➸ ➸WBGD, NOGM LVH ➸ WBGD➷ ➷, NOGM➷ ➷, f-Ins➹ ➹ LVH ➸ f-Ins➹ ➹, WBGD➷ ➷ LVMI (-)➸ ➸WBGD, NOGM LVWT ➸ f-Ins, (-)➸ ➸WBGD, NOGM RWT ✗ f-Ins, WBGD, NOGM LVM ➸basal metabolism, f-Glu, TG LVM ✗ SI frequently sampled IVGTT, IC 24h-u-Cpeptide - /height2.7 LVMI, RWT ➸ 24h-u-C-peptide OGTT /height2.7 LVMI ➸ f-Ins, OGTT-AUC-Ins, (-)➸ ➸Glu/Ins-ratio /height2 in dippers: LVM ➸ f-Glu, f-s-C-peptide ✗ f-Ins in non-dippers: LVM ✗ f-Glu, f-C-peptide, f-Ins LVMI, LVWT (-)➸ ➸ SI 1315 (698 ♂) healthy OGTT, s-Cpeptide frequently sampled IVGTT s-C-peptide - LVM ➸ f-s-C-peptide 101 (58 ♂) healthy HT OGTT - LVM ➸ f-Ins, OGTT-2h-Ins, HOMAIR, IGF-I RWT ➸ IGF-I 83 (47 ♂) healthy, 65 HT IST, OGTT /BSA LVMI ➸ f-Ins, SSPG, HbA1c 1655 (749 ♂) general population, mean age 65 50 (32 ♂) healthy, 21 HT OGTT /BSA LVMI ➸ OGTT-2h-Ins clamp, /height2.7 LVMI, PW, IVS ✗ f-Ins, OGTT-Ins, AUC-Ins, WBGD OGTT LVM = left ventricular mass; ♂ = male; HT = hypertensive; LVH = left ventricular hypertrophy; LVMI = left ventricular mass index; ➸ = relation; Ins = insulin; Glu = glucose; NT = normotensive; ✗ = no relation; IVGTT = intravenous glucose tolerance test; AUC = area under curve; BSA = body surface area; TG = triglycerides; NE = norepinephrine; E = epinephrine; ➹ = increased; clamp = hyperinsulinemic euglycemic clamp; LVWT = sum of left ventricular wall thicknesses; (-)➸ ➸ = inverse relation; WBGD = clamp whole-body glucose disposal; OGTT = oral glucose tolerance test; NOGM = non-oxidative glucose metabolism; IC = indirect calorimetry; ➷ = decreased; ITT = intravenous insulin tolerance test; KITT = glucose disappearance rate at ITT; IRI = insulin resistance index (OGTT peak plasma insulin * corresponding plasma glucose * 10-4); RWT = relative wall thickness; IGF-I, IGF-II = insulin-like growth factor-I and –II; GH = growth hormone; ♀ = female; SI = insulin sensitivity by frequently sampled IVGTT and the Bergman minimal model; HOMAIR = f-Ins * f-Glu / 22.5; IST = insulin suppression test; SSPG = steady-state plasma glucose at 2h-IST; PW = left ventricular posterior wall thickness; IVS = intraventricular septum thickness 11 Electrophysiologic co-morbidity of left ventricular hypertrophy Cross-sectional analyses cannot determine causal relations between variables, but are relevant for the study of co-existence of conditions or diseases. In this study, we have investigated the co-existence of ECG aberrations and echocardiographic LVH or left ventricular geometric and functional abnormalities, in order to assess the associations between these two ways (ECG and echocardiography) of measuring cardiac functions (study IV). Previously, LVH has been shown to be related to premature ventricular beats85, and to a wide variety of ECG aberrations, including Q-waves, S-T segment and T-wave aberrations, in patients with hypertrophic cardiomyopathy86. Prognostic significance of left ventricular hypertrophy LVH has previously been shown to be a strong risk factor for mortality and morbidity whether diagnosed by echocardiography5-16 or ECG17-21. Echocardiographically determined LVH has been shown to be a major risk factor for cardiovascular morbidity and mortality (including cerebrovascular stroke9 and sudden death14) in the general population5,6 and in patients with hypertension15, coronary artery disease8,10 or left ventricular dysfunction16. In some studies7,11-13, an increased RWT has also been shown to be of adverse prognostic value, with a worse prognosis in subjects with concentric LVH than in subjects with eccentric LVH, and a worse prognosis in subjects with concentric remodeling than in subjects with a normal left ventricular geometry (table 2). However, in one of these studies11, the geometric pattern gave little additional information when taking left ventricular mass into account. Although many studies of the prognostic value of LVH and geometric subtypes have been made, it is not known if subjects with echocardiographic LVH who later suffer morbid events or die also have ECG-LVH or vice versa, or if the methods provide complementary prognostic information. It is not fully known if the increased cardiovascular risk associated with echocardiographic or ECG-LVH or an increased RWT is independent of associated metabolic disturbances (such as insulin resistance measured with the clamp technique) and hypertension. We therefore studied these prognostic aspects using a fairly large cohort of elderly men from the general population, followed for a maximum of 6.4 years (study V). Table 2: Previous studies of the prognostic importance of left ventricular geometric patterns Rate (endpoints / 100 patient-years) Study Study population 253 (167 ♂) HT Krumholz, 199511 3216 (1400 ♂) general population, age >40 694 (305 ♂) HT 274 (195 ♂) HT Koren, 19917 Verdecchia, 199512 Verdecchia, 199613 Follow-up duration (years) 10.2 Endpoint CV mortality CV morbidity total mortality CV morbidity Normal geometry 0 1.1 0.7 1.2 Concentric remodeling 0.3 1.5 1.6 2.0 Concentric LVH 2.1 3.1 2.7 3.6 Eccentric LVH 1.0 2.3 1.4 2.1 2.7 CV morbidity 1.1 2.4 - - 3.2 CV morbidity - - 3.3 2.2 7.7 LVH = left ventricular hypertrophy; ♂ = male; HT = hypertensive; CV = cardiovascular. Adapted from reference87 with permission. 12 Aims of the study I. To investigate prospectively determined hemodynamic, metabolic, dietary and psychosocial factors at age 50 as predictors for the prevalence of echocardiographic LVH and left ventricular geometric subtypes at age 70, by using a fairly large, regionally determined sample of men from the general population, followed-up for twenty years. II. To cross-sectionally investigate the relations between echocardiographic left ventricular mass and geometry and components of the insulin resistance syndrome. III. To investigate the relations between myocardial and skeletal muscle insulin sensitivity and RWT and LVMI, by measuring glucose uptake in myocardium and skeletal muscle during hyperinsulinemic euglycemic clamp using 18Ffluorodeoxyglucose and PET in a homogenous sample of healthy, normotensive men with wide ranges of RWT and LVMI. IV. To cross-sectionally investigate the relations between echocardiographic indices of left ventricular geometry and function and ECG characteristics and diagnoses, with special emphasis to the role of echocardiographically determined LVH. V. To investigate if prospectively determined echocardiographic and ECG-LVH predict subsequent total and cardiovascular mortality and morbidity independently of each other and of components of the insulin resistance syndrome, and if any additional prognostic information was provided by an echocardiographic examination if the subject’s ECG-LVH and hypertension status was known. 13 Methods The cohort In 1970-73, all men born in 1920-24 and resident in the municipality of Uppsala were invited to a health survey aimed at identifying risk factors for cardiovascular disease in ≈50 year-old men and selecting high-risk individuals for treatment88. Of the invited subjects, 2322 participated (82%). The cohort was reinvestigated 20 years later (in 1991-95, at age ≈70) with echocardiographic and Doppler examinations, ambulatory blood pressure monitoring, hyperinsulinemic euglycemic clamp and oral glucose tolerance test (OGTT) in addition to the previous study protocol. All investigations in the same subject were performed within 1 month. In the latter investigation, 1221 subjects participated. A reproducibility study was made of all investigations in 22 subjects ≈1 month after the original investigations. The intraindividual coefficients of variation (CVs) presented are from this reproducibility study. Echocardiographic examinations were performed in the first 583 consecutive men in the investigation at age 702, and determination of left ventricular geometry was possible in 475 of these (figure 2). Fig. 2: The Uppsala Longitudinal Study of Adult Men 1970-73 422 died 219 moved 2841 men born 1920-24 (≈ 50 year old) and living in Uppsala municipality were invited, 2322 (82%) participated 1991-95 44 died 1681 eligible participants of the 1970-73 study (≈ 70 year old) were invited, 1221 (73%) participated 583 first consecutive men had echocardiography 475 men had left ventricular geometry determinations PET = positron emission tomography 14 9 in PET study Study populations The population of studies I, II and V consisted of 475 of the first 583 consecutive men of the investigation at age 70 in which determination of left ventricular geometry was possible2. The men included in the study did not differ significantly from the excluded men in any of the investigated variables. Fifty-four subjects had been hospitalized due to ischemic heart disease (ICD-9 codes 410-414) between the investigations at 50 and 70 years of age. At age 70, 167 subjects were regularly using antihypertensive medication, of which 6 were using α-receptor blockers, 60 calcium antagonists, 24 angiotensin-converting enzyme (ACE) inhibitors, 57 diuretics, and 88 β-receptor blockers, as monotherapy or in combination. At age 70, 38 subjects used lipid lowering drugs, of which 16 used statins, 15 fibrates, 6 resins and 2 used nicotinic acid, as monotherapy or in combination. Only 17 subjects had significant echocardiographic valvular disease (aortic or mitral stenosis or regurgitation grades 3 or 4). According to the criteria proposed by the World Health Organization in 198589, 66 subjects had diabetes mellitus type-2 and 115 had impaired glucose tolerance at age 70. In study I, all analyses were also made on a subset (n=421) without ischemic heart disease during follow-up, and in study II, all analyses were also made on a subset (n=458) without valvular disease. The population of study III consisted of 9 subjects, chosen from the investigation at age 70 by echocardiographic left ventricular geometry to have wide ranges of RWT and LVMI. The subjects all had supine office blood pressures <160/95 mm Hg at a single office visit, were free from regular medication, history of heart disease, significant echocardiographic valvular disease and signs of coronary heart disease on resting ECG, exercise testing and 24-h ambulatory ECG monitoring. The population of study IV consisted of 540 of the first 583 consecutive men of the investigation at age 70 who had both an ECG and an echocardiographic examination. Investigations at age 50 These investigations were used in study I and have been described extensively previously88. Blood pressure in the recumbent position was measured with a mercury manometer and radial pulse rate was counted. All blood samples were drawn in the morning after an overnight fast. Blood glucose, serum insulin, cholesterol, triglycerides and HDL-cholesterol were measured, and LDL-cholesterol was calculated using Friedewald’s formula: LDL = serum cholesterol – HDL – (0.45 * serum triglycerides). The serum CE proportions of several fatty acids (14:0, 16:0, 16:1, 18:0, 18:1, 18:2 ω6, 18:3 ω6, 18:3 ω3, 20:3 ω6, 20:4 ω6, 20:5 ω3, 22:6 ω3) were determined by gas chromatography46. No dietary records were obtained, but in other population studies in middle-aged men in the 1970s in Sweden, intake of fat corresponded to about 40%, carbohydrates 45-50% and protein 13-15% of energy intake. The estimated intake of saturated fats corresponded to 17-18% of energy intake90. 15 A questionnaire covered level of physical activity (four categories) and education (five categories). Coding of smoking (smoker, non-smoker, ex-smoker), civil status and socioeconomic status (three social classes, Central Bureau of Statistics) was based on interview reports88. Investigations at age 70 Seven-day dietary records in 444 of the 475 subjects in studies I, II and V showed an intake of fat corresponding to 35%, carbohydrates 48% and protein 16% of energy intake, which was comparable to other contemporary Swedish populations of the same age90. Coding of smoking (smoker, non-smoker) was based on an interview question also at age 70. Echocardiography A comprehensive two-dimensional and Doppler echocardiography was performed with a Hewlett Packard Sonos 1500 cardiac ultrasound unit (Hewlett Packard, Andover, MA) as described previously2. A 2.5 MHz transducer was used for the majority of the examinations. Dimensions were measured in M-mode using the leading-edge to leading-edge convention. The measurements included left atrial diameter (LA), interventricular septal thickness (IVS), posterior wall thickness (PW) and left ventricular diameter at the end of diastole and the end of systole (LVEDD and LVESD) (figure 3). Left ventricular mass was determined using the Troy formula according to the recommendations of the American Society of Echocardiography (ASE)24: left ventricular mass (grams) = 1.05 * ((LVEDD + IVS + PW)3 – LVEDD3). To correct for differences in body constitution, left ventricular mass was divided with body surface area to obtain LVMI. LVH was defined as LVMI ≥150 g/m2 according to data from the Framingham Heart Study24, corresponding to 131 g/m2 for LVMI measured with the Penn convention and the modified cube formula24. Left ventricular mass by both conventions correlate well with necropsy left ventricular mass91, but slightly overestimate it (6% by the Penn convention and 25% by the ASE convention). Thus, LVMI measured with the leading-edge to leading-edge convention and the Troy formula can easily be transformed to reflect anatomic measurements91: left ventricular mass = 0.80 * (ASE mass) + 0.6g. RWT was calculated as (IVS + PW) / LVEDD, and a partition value of 0.44 was used. Thus, left ventricular geometry was considered normal if RWT was <0.44 and LVMI <150 g/m2. A normal LVMI with increased RWT was denoted concentric remodeling, and a hypertrophic left ventricle was denoted eccentric if RWT was normal and concentric if RWT was increased (figure 1)22. Left ventricular volumes (LVEDV, LVESV) were calculated according to the Teichholz formula: Vol = 7 * D3 / (2.4 + D). From these volumes ejection fraction was calculated. Stroke volume was calculated from Doppler measurements of left ventricular outflow tract diameter (LVOT) and the flow velocity integral (FVI) as π * LVOT2 / 4 * FVI and was divided by body surface area to obtain stroke index. The left ventricular filling pattern in diastole was examined by measuring mitral blood 16 flow velocities with the transducer in the apical position. The peak velocities of the early rapid filling phase (E-wave) and the atrial systolic phase (A-wave) were recorded and E/A ratio calculated. Left ventricular isovolumetric relaxation time (IVRT) was measured as the time between the aortic valve closure and the start of the mitral flow using the Doppler signal from the area between the LVOT and mitral flow. All examinations were performed with the subjects in the standard left lateral position in expiratory apnea or quiet breathing. The best five cardiac beats were chosen and the average of these was calculated. All examinations and readings of the images were done by one experienced physician (Bertil Andrén) who was unaware of other data of the subjects. The CVs were for dimensional measurements LA 6.5%, IVS 8.8%, PW 6.7%, LVEDD 3.5%, LVMI 12.5% and RWT 6.9% and for Doppler measurements IVRT 6.8%, E-wave 10.0%, A-wave 9.4% and E/A-ratio 16.9%. Fig. 3: Echocardiographic left ventricular geometric measurements IVS LVEDD PW Relative wall thickness = (IVS+PW)/LVEDD Left ventricular mass = 1.05((IVS+LVEDD+PW )3-LVEDD3) Electrocardiography A standard 12-lead ECG was recorded at 50 mm/s and 10 mm/mV and evaluated according to the Minnesota code92 by one experienced physician (Lars Lind) who was unaware of other data of the subjects. In study IV, the analyzed diagnoses were: Q-wave (Minnesota codes 1-1 or 1-2), S-T segment depression (4-1 or 4-2), T-wave items (5-1, 5-2 or 5-3), left bundle-branch block (7-1), frequent premature beats (10% 17 or more of recorded beats, 8-1), atrial flutter or fibrillation (8-3), bradycardia (less than 50 beats per minute, 8-8), atrioventricular block type 1 (6-3) and left anterior hemiblock (7-7). All analyses of S-T segment depression, T-wave items and left bundle-branch block were carried out in a subset without Q-wave. All analyses of frequent premature beats, atrial flutter or fibrillation, bradycardia, atrioventricular block type 1 and left anterior hemiblock were carried out in a subset without Q-, S-Tor T-wave abnormalities or left bundle-branch block (figure 4). In study IV, ECGs of a subset of 107 consecutive subjects without ECG abnormalities defined in the Minnesota code system were also analyzed for linear relations between echocardiographic and ECG measurements. The Sokolow-Lyon Voltage was determined as the amplitude of SV1 + RV5 or RV6, the Cornell Voltage as SV3 + RaVL and the Cornell Product as SV3 + RaVL * QRS duration. The direction of the QRS axis in the frontal plane was calculated from the QRS amplitudes in the extremity leads and expressed as degrees in a 360° circle, with 0° defined as the direction of extremity lead I, increasing clockwise. The early diastolic phase at ECG was defined as the time between the end of the T-wave and the beginning of the P-wave. All intervals were corrected for heart rate according to Bazette93. Fig. 4: Distribution of electrocardiographic diagnoses in the study population Total n=540 No Q-, ST- or T-wave abnormalities or left bundle-branch block, n=364 Frequent premature beats n=9 Q-wave n=77 Normal electrocardiogram n=273 No Q-wave n=463 Left bundlebranch block n=9 Atrial flutter / fibrillation n=12 n=22 Bradycardia n=18 Atrioventricular block type 1 n=12 18 T-wave items n=58 ST-segment depression n=54 In study V, we analyzed the predictive value of several ECG criteria for LVH: Sokolow-Lyon Voltage (SV1 + RV5 or RV6) ≥3.5 mV, Cornell Voltage (SV3 + RaVL) >2.8 mV, left ventricular strain (down-sloping ST-segment depression >0.1 mV with Twave flattening or inversion in leads V4-V5) (which have previously been validated in prospective studies17,19,21) and Cornell Product (SV3 + RaVL * QRS duration) >244 µVs (not validated in a prospective study before, but used as inclusion criterion in the LIFE study94). Sensitivities/specificities (%) for detection of echocardiographic LVH were 27/88 for Sokolow-Lyon Voltage ≥3.5 mV; 17/91 for Cornell Voltage >2.8 mV; 28/88 for Cornell Product >244 µVs; and 21/92 for left ventricular strain, in the present population. Blood pressure measurements The ambulatory blood pressure measuring device Accutracker II (Suntech Medical Instruments) was attached to the subject’s nondominant arm. Systolic and diastolic blood pressures (SBP, DBP) and heart rate were measured over 24 hours, every 30 minutes during daytime (6 AM to 11 PM) and every hour during nighttime. Data were edited to a limited extent, omitting all readings of 0, all heart rate readings <30 beats per minute, DBP readings >170 mm Hg, SBP readings >270 and <80 mm Hg, and all readings for which the difference between SBP and DBP was <10 mm Hg. The CV for 24-h mean arterial blood pressure (DBP + (SBP - DBP) / 3) was 5.5%. Office supine SBP, DBP and heart rate were also measured. Blood pressures were measured twice in the right arm after 10 minutes rest, and means were calculated. The rate-pressure product (an index of cardiac work) was calculated as office SBP*office heart rate. Hyperinsulinemic euglycemic clamp Whole-body glucose uptake and insulin sensitivity index were determined with the hyperinsulinemic euglycemic clamp, performed according to the method of DeFronzo et al48 with a slight modification. After a priming dose of insulin given during 10 minutes, insulin (Actrapid Human®, Novo) was infused at a constant rate of 56 (instead of 4048) mU/(min * m2 body surface area). Such a high concentration of insulin has been shown to inhibit hepatic glucose output by 88-95% also in diabetics95. The arterialized plasma glucose concentration was determined every fifth minute (Beckman Glucose Analyzer II®, Beckman instruments) and the rate of a 20% dextrose solution was thereafter adjusted every fifth minute to keep plasma glucose at the target level of 5.1 mmol/l. The whole-body glucose uptake was calculated as the amount of glucose infused during the last 60 minutes of the 2-h clamp (mg/kg body weight/min), and the insulin sensitivity index (studies II and V) was calculated by dividing whole-body glucose uptake by the mean plasma insulin concentration times 100 (mU/L) during the last 60 minutes of the 2-h clamp. In study III, wholebody glucose uptake was calculated immediately before the PET measurements were done. The CV for whole-body glucose uptake was 9.2%, and for insulin sensitivity index, it was 13.9%. 19 Oral glucose tolerance test Blood samples for determining fasting concentrations were drawn in the morning after an overnight fast. An OGTT was performed by measuring the concentrations of plasma glucose and ”immunoreactive insulin” immediately before and 30, 60, 90, and 120 minutes after 75 g anhydrous dextrose was ingested. Fasting, 2-h levels and the incremental areas under the curves (AUC) of glucose and immunoreactive insulin were analyzed. Glucose was measured by the glucose dehydrogenase method (GlucDH, Merck), and immunoreactive insulin was analyzed by use of an enzymaticimmunological assay (Enzymun, Boehringer Mannheim) performed in an ES300 automatic analyzer (Boehringer Mannheim). Fasting specific insulin and 32-33 split and intact proinsulin concentrations were measured with a specific 2-site immunoradiometric assay technique96 in Cambridge, UK, on a Nuclear Enterprises 1600 gamma counter of 125I. The CV for fasting plasma glucose was 5.8%, and for immunoreactive insulin, it was 15.4%. Lipid measurements HDL was separated by precipitation with magnesium chloride/phosphotungstate. Cholesterol and triglyceride concentrations in serum and HDL were assayed by enzymatic techniques (Instrumentation Laboratories) in a Monarch 2000 centrifugal analyzer. LDL-cholesterol was calculated with Friedewald’s formula. Serum nonesterified fatty acids (NEFA) were measured by an enzymatic colorimetric method (Wako Chemical GmbH) applied for use in the Monarch 2000. The CVs were for serum total cholesterol 5.7%, HDL-cholesterol 11.1%, LDL-cholesterol 6.6%, serum triglycerides 14.8%, and NEFA 24.2%. Positron emission tomography In study III, myocardial and skeletal muscle glucose uptake were determined by means of the radiolabelled tracer 2-[18F]-fluoro-deoxyglucose (18FDG) and PET. PET investigations were performed during hyperinsulinemic euglycemic clamp with the subject in supine position, using a GEMS 4096-15WB scanner (General Electric Medical Systems) with a spatial resolution of 6 mm (full width at half maximum) covering 100 mm (15 tomographic slices) with a 6.5 mm slice spacing. Attenuation of photons was corrected for using a 10 minutes transmission scan obtained with a rotating 68Ga/68Ge pin radiation source. Emission tomograms were reconstructed to a 128*128 matrix with 2 mm pixel size using a 4.2 mm Hanning filter. 18FDG is a glucose analogue, transported across the cell membrane by the same transport proteins as glucose and phosphorylated by hexokinase. However, 18FDG is not further metabolized, but the phosphorylated moiety is trapped in the cytosol. Starting at the time of an intravenous bolus injection of some 6 MBq/kg body weight of 18FDG, the accumulation of tracer in the studied tissue was followed for 60 minutes. Venous blood, arterialized by heating of the hand, was collected from a vein on the back of the hand for measurements of the plasma concentration of radioactivity and of plasma glucose levels. Altogether 15 samples were taken to 20 outline the variation of 18FDG in plasma during the study. The glucose uptake (metabolic rate of glucose), expressed in µmol/min/100cm3 tissue, was calculated by the graphical method97, using a value of the ”lumped constant” equal to 1.0 for myocardium98, and 1.2 for skeletal muscle. Partial volume or spill-over effects in 18FDG time-activity curves were corrected for corresponding 15O-water measurements from assessments of myocardial perfusion. Images acquired during peak myocardial concentration of 18F (40-60 minutes after injection of the tracer) were used for the definition of regions of interest (ROI) in the short axis view. ROIs defining the left ventricular wall were 10 mm in the radial direction, symmetrically placed from the endo- to the epicardial surface in sections 10 mm apart, covering the left ventricular wall from apex to base. All ROIs were lumped together to provide a volume, representing the entire left ventricular myocardium. ROIs representing skeletal muscle were defined in the soft tissues of the left upper arm in six adjacent image slices which were joined together. Bone was identified in the transmission measurement and excluded from the ROIs. Follow-up after age 70 The subjects had a median follow-up time of 5.2 years (range 0.7-6.4 years) after the investigation at age 70, contributing to 2415.6 person-years at risk (PYAR). Endpoints were defined using the Swedish national cause-of-death and hospital discharge registers. During follow-up, 44 subjects died (rate 1.82/100 PYAR), 18 from cardiovascular disease (ICD codes 390-459, rate 0.75/100 PYAR) (of which 7 died from acute myocardial infarction and 4 from stroke). Morbidity was defined as first hospitalization or death from cardiovascular disease or any cause, and was evaluated only for subjects who had not previously been hospitalized for cardiovascular disease or any cause, respectively. During follow-up, 48/122 (39%, rate 9.50/100 PYAR) had a morbid event of any cause, and 64/338 (19%, rate 4.09/100 PYAR) had a cardiovascular morbid event. Statistical analysis Variables with a skewed distribution (fasting serum insulin, triglycerides, LDL/HDL-cholesterol, CE proportion of palmitoleic, stearic, γ-linolenic, α-linolenic, eicosapentaenoic and docosahexaenoic acids at age 50; and fasting plasma glucose, immunoreactive insulin, specific insulin, proinsulin, 32-33 split proinsulin, serum triglycerides and NEFA, 2-h glucose and immunoreactive insulin levels, and the AUC of immunoreactive insulin at the OGTT at age 70) were logarithmically transformed to achieve normal distribution, and these transformed variables were used in all analyses. One-way and multi-way ANOVA (adjusted for possible confounders) was used to calculate differences in means between subgroups. Posthoc comparisons between subgroups were only performed if overall ANOVA was significant. Chi2-test was used to evaluate differences in nominal variables between subgroups. Pearson’s correlation coefficients and partial correlation coefficients (adjusted for possible confounders) were used to evaluate relations between pairs of 21 continuous variables. Two-tailed 95% confidence intervals (CI) and significance values were given, with P<0.05 regarded as significant. In study I, logistic regression was used with LVH or increased RWT at age 70 as outcome variables and standardized continuous variables (mean=0, SD=1) or indicator variables for multilevel nominal variables at age 50 as explanatory variables. Multiple logistic regression was used to adjust for possible confounders, treated as dichotomous variables. Squared terms and interaction terms were tested in all regressions. In study II, relations to LVMI but not to RWT were adjusted for use of antihypertensive medication, because subjects using antihypertensive medication had significantly higher LVMIs but did not differ in RWT. Squared variables and interaction terms between independent variables were tested in all regression models. Scatter plots were visually examined for other nonlinear associations. In study V, the prognostic value of transfer from one level of a dichotomous variable to another, or a one SD increase in a continuous variable, was investigated with Cox proportional hazard ratios (HR). Proportionality of hazards was confirmed with Kaplan-Meier plots. Multiple Cox proportional hazards analyses were used to adjust for either LVMI, previous ischemic heart disease or nine cardiovascular risk factors. Other cut-off levels for LVMI than 150 g/m2 were sought using histograms of quartiles of LVMI for cardiovascular and total mortality, and by performing logistic regression and receiver operating characteristic (ROC)-curves. JMP 3.2 (SAS Institute Inc.) and Stata 6.0 (Stata Corporation) software were used. 22 Results Prospectively defined predictors at age 50 for left ventricular hypertrophy or increased relative wall thickness at age 70 (I) The prevalence of LVH at age 70 was 28% in the present cohort. Among the variables associated with the insulin resistance syndrome, a one SD increase in any of body mass index (BMI), SBP, DBP, fasting LDL/HDL-cholesterol or serum triglycerides at age 50 resulted in a 27-41% increased odds of having LVH at age 70 (figure 5). A one SD increase in the serum CE proportion of any of myristic (14:0), palmitic (16:0), stearic (18:0), oleic (18:1) or eicosapentaenoic acid (20:5 ω3) at age 50 increased the odds of having LVH at age 70 by 29-37%, whereas a one SD increase in the proportion of linoleic acid (18:2 ω6) at age 50 reduced the odds of having LVH at age 70 by 24%. No studied psychosocial factors at age 50, crude or adjusted, predicted later LVH. The prevalence of an increased RWT at age 70 was 25% in the present cohort. A one SD increase in serum CE proportion of oleic acid (18:1), γ-linolenic acid (18:3 ω6) or dihomo-γ-linolenic acid (20:3 ω6) at age 50 increased the odds of having increased RWT at age 70 by 28-32%, whereas a one SD increase in the proportion of linoleic acid (18:2 ω6) at age 50 reduced the odds of having increased RWT at age 70 by 26%. The studied psychosocial issues were not associated with an increased RWT. Fig. 5: Odds-ratios and 95% confidence intervals for having left ventricular hypertrophy at age 70 for a one standard deviation increase in a variable at age 50 Body mass index Systolic blood pressure Diastolic blood pressure Pulse rate Total cholesterol LDL/HDL-cholesterol Triglycerides Glucose Insulin Myristic acid (14:0) Palmitic acid (16:0) Palmitoleic acid (16:1) Stearic acid (18:0) Oleic acid (18:1) Linoleic acid (18:2ω6) 0.6 0.8 1.0 1.2 23 1.4 1.6 Approximately the same results were obtained when: 1, adjusting all the above analyses for ischemic heart disease (ICD-9 codes 410 to 414) during follow-up and valvular disease and use of antihypertensive medication at age 70; 2, adjusting for these three variables and BMI at age 50; 3, adjusting for the first three variables and use of lipid lowering drugs at age 70; and 4, analyzing a subsample of 421 subjects without ischemic heart disease during follow-up, adjusted for valvular disease and use of antihypertensive medication at age 70. Characteristics at age 50 according to left ventricular geometric patterns at age 70 (I) The levels of several variables associated with the insulin resistance syndrome and serum CE proportions of several fatty acids determined at age 50 varied significantly between the four left ventricular geometry groups determined at age 70 (figure 6). In the concentric remodeling group, serum CE proportions of oleic (18:1) and γ-linolenic acid (18:3 ω6) were significantly higher, and linoleic acid (18:2 ω6) significantly lower than in the normal geometry group. In the concentric LVH group, DBP and serum CE proportions of myristic (14:0), palmitic (16:0), oleic (18:1) and eicosapentaenoic acid (20:5 ω3) were significantly higher, and linoleic acid (18:2 ω6) significantly lower than in the normal geometry group. In the eccentric LVH group, BMI, SBP, DBP, fasting serum triglycerides and CE proportions of oleic (18:1) and eicosapentaenoic acid (20:5 ω3) were significantly higher, and linoleic acid (18:2 ω6) significantly lower than in the normal geometry group. Adjusting for ischemic heart disease during follow-up and valvular disease and use of antihypertensive medication at age 70, or adjusting for these three variables and BMI at age 50, gave similar results. Fig. 6: Significant predictors at age 50 for left ventricular geometric subtypes at age 70 18:1 ↑ 18:2 ω6 ↓ DBP ↑ 14:0 ↑ 16:0 ↑ 18:1 ↑ 18:2 ω6 ↓ Concentric remodeling LVH = left ventricular hypertrophy 14:0 = myristic acid 16:0 = palmitic acid 18:1 = oleic acid 18:2 ω6 = linoleic acid BMI = body mass index SBP = systolic blood pressure DBP = diastolic blood pressure TG = triglycerides Normal Concentric LVH Eccentric LVH 24 BMI ↑ SBP ↑ DBP ↑ TG ↑ 18:1 ↑ 18:2 ω6 ↓ Relations between components of the insulin resistance syndrome and left ventricular geometric parameters at age 70 (II) Several components of the insulin resistance syndrome were significantly and directly related to RWT, such as 24-h SBP and DBP, 24-h heart rate, 2-h glucose level and the AUC of glucose at the OGTT, fasting specific insulin and 32-33 split proinsulin, waist-to-hip ratio, BMI, serum triglycerides, and NEFA, whereas clamp insulin sensitivity index was inversely related to RWT (figure 7). On the other hand, of the measured variables, only 24-h SBP (directly), 24-h heart rate, and 2-h immunoreactive insulin level at the OGTT (inversely) were significantly related to LVMI. In a subsample of 458 subjects without valvular disease, controlling for previous ischemic heart disease, results were essentially the same as in the original analyses. There was no relation between RWT and LVMI. Fig. 7: Significant correlations between left ventricular geometric determinants and components of the insulin resistance syndrome at age 70 Left ventricular mass index Relative wall thickness 24-h SBP 24-h DBP 24-h heart rate Insulin sensitivity index OGTT 2-h glucose OGTT AUC glucose Fasting specific insulin Fasting 32-33 split proinsulin W aist-to-hip ratio Body mass index Serum triglycerides Serum NEFA SBP = systolic blood pressure DBP = diastolic blood pressure OGTT = oral glucose tolerance test AUC = area under the curve NEFA = non-esterified fatty acids -0.1 0 Correlation coefficient 0.1 0.2 Metabolic and other characteristics of subjects with various left ventricular geometries (II) The levels of several components of the insulin resistance syndrome — 24-h SBP and DBP, 24-h heart rate, clamp insulin sensitivity index, 2-h glucose level and the AUC of glucose at the OGTT, waist-to-hip ratio, and BMI — differed significantly between the 4 left ventricular geometric groups (figure 8). The values of 24-h heart rate, waistto-hip ratio, 2-h glucose level, and the AUC of glucose at the OGTT were significantly 25 higher and clamp insulin sensitivity index was significantly lower in the concentric remodeling geometry group compared with the group with normal left ventricular geometry. The 24-h SBP and DBP were significantly higher in the concentric LVH group compared with the group with normal left ventricular geometry. The difference in 24-h heart rate between the groups (higher in the concentric remodeling geometry group and lower in the eccentric LVH group than in the normal geometry group) remained significant when adjusting simultaneously for the possible confounders ischemic heart disease, stroke index and use of β-receptor blockers and any other antihypertensive medication. In a subsample without valvular disease, controlling for previous ischemic heart disease, differences between the groups were essentially the same as in the original analyses. Fig. 8: Metabolic and other characteristics of left ventricular geometric subtypes at age 70 24-h HR ↑ Insulin sensitivity index ↓ OGTT glucose ↑ Waist-to-hip ratio ↑ 24-h SBP ↑ 24-h DBP ↑ Concentric remodeling Concentric LVH 24-h HR ↓ LVH = left ventricular hypertrophy HR = heart rate OGTT = oral glucose tolerance test SBP = systolic blood pressure DBP = diastolic blood pressure Normal Eccentric LVH Myocardial and skeletal muscle insulin-mediated glucose uptake and left ventricular geometry (III) In a homogenous group of 9 subjects, insulin-mediated glucose uptake in the skeletal muscle was inversely correlated to RWT (figure 9) whereas there was a borderline significant positive correlation between insulin-mediated myocardial glucose uptake and RWT (figure 10). The myocardial to skeletal muscle glucose uptake ratio showed a positive correlation to RWT (figure 11). We found no relations between LVMI and insulin-mediated glucose uptake in the myocardium, skeletal muscle or the ratio between myocardial and skeletal muscle glucose uptake. 26 Fig. 9: Insulin-mediated skeletal muscle glucose uptake in relation to left ventricular relative wall thickness Skeletal muscle glucose uptake (µmol/min/100cm3) 10 8 6 4 2 0.35 0.40 0.45 0.50 Relative wall thickness Fig. 10: Insulin-mediated myocardial glucose uptake in relation to left ventricular relative wall thickness 120 Myocardial glucose uptake (µmol/min/100cm3) 100 80 60 40 20 0.35 0.40 0.45 Relative wall thickness 27 0.50 Myocardial / skeletal muscle glucose uptake (µmol/min/100cm3) Fig. 11: Ratio between insulin-mediated myocardial and skeletal muscle glucose uptake in relation to left ventricular relative wall thickness 40 30 20 10 0 0.35 0.40 0.45 0.50 Relative wall thickness Insulin-mediated whole-body glucose uptake (measured with hyperinsulinemic euglycemic clamp) was directly related to the insulin-mediated glucose uptake in the skeletal muscle, and tended to be inversely related to the insulin-mediated myocardial glucose uptake (the latter two measured with 18FDG- PET). There was no significant relation between insulin-mediated myocardial and skeletal muscle glucose uptake or between insulin-mediated myocardial muscle glucose uptake and fasting plasma glucose, 24-h systolic, diastolic or mean arterial blood pressure or the rate-pressure product. Echocardiographic characteristics of various electrocardiographic diagnoses at age 70 (IV) For distribution of ECG diagnoses in the study population at age 70, see figure 4. An increased LVMI was the most common echocardiographic finding in the analyzed ECG diagnoses (figure 12). Subjects with a Q-wave had significantly higher LVEDD and LVMI and lower ejection fraction than subjects without a Q-wave. Subjects with ST- or T-wave abnormalities had significantly larger LA, IVS, PW and LVMI than subjects without ST- or T-wave abnormalities. Subjects with left bundle-branch block had significantly higher LVEDD and LVMI and lower ejection fraction than subjects without left bundle-branch block. Subjects with atrial flutter or fibrillation had a significantly larger LA than subjects without atrial flutter/fibrillation. Subjects with atrioventricular block type 1 had significantly larger IVS, PW, IVRT and LVMI than subjects without atrioventricular block type 1. Subjects with frequent premature 28 beats had significantly larger LA, IVS, PW and LVMI than subjects without premature beats. Subjects with bradycardia had a significantly higher E/A ratio than subjects without bradycardia. The prevalences of several ECG diagnoses differed significantly between the four echocardiographically determined left ventricular geometric groups (figure 13); the prevalence of Q-waves was highest in the eccentric LVH group, whereas the prevalences of ST- or T-wave abnormalities and atrioventricular block type 1 were highest in the concentric LVH group. Fig. 12: Left ventricular mass index in various electrocardiographic diagnoses Atrioventricular block type 1 Frequent premature beats Left bundle-branch block ST- or T-wave abnormalities Q-wave Normal electrocardiogram 100 110 120 130 140 150 160 Left ventricular mass index (g/m2) 170 Fig. 13: Prevalences (%) of various electrocardiographic diagnoses in the left ventricular geometric subgroups Any electrocardiographic abnormality 70 ST- or T-wave abnormalities Q-wave 60 Atrioventricular block type 1 Left bundle-branch block 50 40 30 20 10 0 Normal Concentric remodeling LVH = left ventricular hypertrophy 29 Concentric LVH Eccentric LVH Relations between echocardiographic and electrocardiographic measurements (IV) Left ventricular systolic function, as measured by the ejection fraction, was inversely related to the Sokolow-Lyon Voltage. Left ventricular diastolic function, as measured by the E/A ratio, was related to the duration of the early diastolic phase at the ECG and inversely related to the Sokolow-Lyon Voltage. The IVRT was inversely related to the clockwise deviation of the QRS axis from the direction of extremity lead I. Sokolow-Lyon Voltage was also significantly related to LVMI. Prognostic value of prospectively determined echocardiographic left ventricular hypertrophy (V) Echocardiographic LVH was a significant predictor of cardiovascular morbidity and mortality. Dividing the population into geometric subgroups provided some additional information (figure 14): concentric remodeling and eccentric LVH were significant predictors of cardiovascular mortality compared to subjects with a normal geometry. Concentric LVH had a similar but non-significant hazard ratio as concentric remodeling. Also LVMI as a continuous variable was a significant predictor of morbidity and mortality from cardiovascular disease and total mortality. Other cut-off levels for LVMI than the proposed24 150 g/m2 were sought using quartiles of LVMI and logistic regressions and ROC-curves with total and cardiovascular mortality as dependent variables. Total mortality seemed to increase rather linearly with increasing LVMI, and no obvious cut-off level for prediction of total mortality was found. In contrast, the risk for cardiovascular mortality was markedly increased only in the fourth quartile of LVMI (figure 15). This information combined with the ROC-curve for LVMI regarding cardiovascular mortality (figure 16) indicated that 150 g/m2 was a cut-off level that provided a reasonable combination of sensitivity and specificity for detection of this increased risk. Nine cardiovascular risk factors (clamp insulin sensitivity index, proinsulin, total and HDL-cholesterol, triglycerides, waist circumference, smoking, hypertension and previous ischemic heart disease) were also evaluated as risk factors for mortality and morbidity. Smoking or a 1 SD increase in proinsulin increased the risk, and a 1 SD increase in HDL-cholesterol decreased the risk for mortality from cardiovascular disease. Smoking also predicted total morbidity. When adjusting for these nine cardiovascular risk factors, concentric remodeling and eccentric LVH remained significant predictors of cardiovascular mortality, and the predictive value of LVMI as a continuous variable for total and cardiovascular mortality remained significant. 30 Fig. 14: Cox proportional hazard ratios for cardiovascular mortality in various categories of left ventricular geometry Hazard ratio for cardiovascular mortality P=0.002 9 8 7 6 5 4 3 2 1 0 P=0.04 LVMI ≥ 150 g/m2 LVMI < 150 g/m2 RWT ≥ 0.44 RWT < 0.44 RW T = relative wall thickness LVMI = left ventricular mass index Hazard in the normal geometry group was set to 1.0 A B 34 61 Total mortality (%) Cardiovascular mortality (%) Fig. 15: Total (A) and cardiovascular (B) mortality in quartiles of left ventricular mass index 0 0 71 <117 117 <135 135 <154 71 <117 154 250 117 <135 135 <154 154 250 Left ventricular mass index (g/m2) Left ventricular mass index (g/m2) 31 Fig. 16: Receiver operating characteristics-curve for left ventricular mass index (g/m2) regarding cardiovascular mortality 100 130 120 110 140 Sensitivity (%) 75 150 160 50 170 25 0 25 50 75 100 100 - Specificity (%) Area under left ventricular mass index curve = 76 % 0 Prognostic value of prospectively determined electrocardiographic left ventricular hypertrophy (V) LVH as defined by Sokolow-Lyon Voltage ≥3.5 mV was a significant predictor of total mortality. LVH as defined by Cornell Voltage >2.8 mV was a significant predictor of total mortality, also after adjustment for LVMI. LVH as defined by Cornell Product >244 µVs was a strong predictor of both cardiovascular and total mortality, also after adjustment for nine other cardiovascular risk factors. Adjustment for LVMI made prediction of cardiovascular, but not total mortality, lose in significance. The left ventricular strain pattern only predicted morbidity from cardiovascular disease. Comparison between the prognostic values of echocardiographic and electrocardiographic left ventricular hypertrophy (V) In multivariate Cox proportional hazards analyses with LVMI, Cornell Product >244 µVs, Sokolow-Lyon Voltage ≥3.5 mV and nine other cardiovascular risk factors as independent variables, LVMI and Cornell Product >244 µVs were significant predictors of total mortality. LVMI, previous ischemic heart disease, HDLcholesterol, smoking and proinsulin were significant predictors of cardiovascular mortality. 32 By assessing echocardiographic LVH if ECG Cornell Product ≤244 µVs, another 7 of the 44 total deaths (another 6 of the 18 cardiovascular deaths) could be predicted. By assessing ECG Cornell Product if echocardiography showed no LVH, another 8 of the 44 total deaths (another 2 of the 18 cardiovascular deaths) could be predicted (figure 17). We also stratified for hypertension. In 263 normotensive subjects, no more total or cardiovascular deaths could be predicted by assessing echocardiographic LVH if ECG Cornell Product ≤244 µVs. In 212 hypertensive subjects, another 7 of the 19 total deaths (another 6 of the 12 cardiovascular deaths) could be predicted by assessing echocardiographic LVH if ECG Cornell Product ≤244 µVs. Fig. 17: Total (A) and cardiovascular (B) mortality rates and Kaplan-Meier survival plots in groups with/without LVH as defined by Cornell Product and echocardiography B 100 6 5 4 3 50 Echo+ 2 1 0 EchoCP+ CP - PYAR = person-years at risk CP+/- = Cornell Product >244 µVs +/Echo+/- = left ventricular mass index ≥150 g/m2 +/LVH = left ventricular hypertrophy 33 Analysis time (years) 0 2 4 6 3 2 75 Echo+ 1 0 100 Survival (%) Analysis time (years) 2 4 6 Survival (%) Total mortality rate (/100 PYAR) 0 Cardiovascular mortality rate (/100 PYAR) A EchoCP+ CP - Discussion In this prospective longitudinal study, we found that an unfavorable serum fatty acid profile and components of the insulin resistance syndrome at age 50 predicted the prevalence of LVH at age 70 in a twenty-year follow-up of a fairly large, regionally determined sample of men from the general population. In cross-sectional analyses at age 70, several components of the insulin resistance syndrome were significantly related to RWT and concentric remodeling, but less to LVH. More specifically, RWT was inversely related to insulin sensitivity in skeletal muscle and borderline significantly directly related to insulin sensitivity in the myocardium in a small, healthy and normotensive sample of the cohort, whereas LVMI was not related to myocardial or skeletal muscle insulin sensitivity. At age 70, echocardiographic LVH was related to a variety of common ECG diagnoses. In a prospective mortality study with baseline at age 70, echocardiographic and ECG-LVH were found to predict mortality independently of each other and of other cardiovascular risk factors, implying that echocardiographic and ECG-LVH in part carry different prognostic information. Left ventricular hypertrophy and the insulin resistance syndrome Components of the insulin resistance syndrome are risk factors for later left ventricular hypertrophy In the twenty-year prospective study (I), dyslipidemia and a serum CE fatty acid composition with a high proportion of saturated fatty acids and low proportion of linoleic acid at age 50 predicted the prevalence of LVH at age 70 to a similar degree as hypertension and obesity. The impact of obesity, dyslipidemia and an unfavorable fatty acid profile on LVH was independent of history of ischemic heart disease, valvular disease and use of antihypertensive medication at age 70. The relations between dyslipidemia and an unfavorable fatty acid profile and later LVH were also independent of obesity at age 50. Hypertension has previously been related to the development of LVH in a prospective study33, but the predictive value of obesity, dyslipidemia and an unfavorable fatty acid profile for LVH has not been shown before. No psychosocial variables were associated with later development of LVH, in accordance with a cross-sectional population study1 where exercise physical activity and smoking were not related to LVH. Fatty acids and left ventricular hypertrophy The relation in study I between an unfavorable serum CE fatty acid composition and later LVH is important since the fatty acid composition mainly reflects dietary fat quality over the past couple of weeks44,45 and thus is a modifiable risk factor. A diet with a high proportion of saturated fatty acids has recently been shown to impair 34 insulin sensitivity in humans99, supported by the experimental finding that nonesterified palmitate has been shown to decrease insulin-mediated as well as basal glucose uptake100. The effects of dietary fat quality on insulin sensitivity may in part be explained by the observation that long-chain unsaturated fatty acids have a higher affinity than shorter saturated fatty acids for peroxisome proliferator-activated receptors (PPARs)101, which have an important role in regulating glucose and fatty acid metabolism101,102. Previous studies of the present cohort have shown an unfavorable fatty acid profile to be related to insulin resistance103 and to predict myocardial infarction over 19 years46. Also the impact of other traditional risk factors on myocardial infarction46 was similar to their impact on LVH in the present study, which suggests that LVH might be regarded as an intermediary risk factor on the pathway between the described risk factor profile and myocardial infarction. The apparently opposite effects of linoleic acid (which comprises more than 50% of all serum CE fatty acids) and the saturated fatty acids may be due to passive redistribution of the less abundant acids following changes in linoleic acid proportion, but may also reflect true pathophysiological effects of the saturated fatty acids. Oleic acid is a major component in olive oil and the cardioprotective Mediterranean diet, but in the present study, a high serum CE oleic acid proportion was related to later LVH and increased RWT. The serum CE oleic acid proportion is a poor marker for dietary oleic acid intake45, but the serum CE linoleic-to-oleic acid ratio has been shown to be a good indicator of dietary polyunsaturated-to-saturated fatty acids ratio104. In men of the present study, the dietary source of oleic acid was not olive oil (which was not used by middle-aged men in Sweden in the early 1970s) but food groups containing a high proportion of saturated fatty acids, such as dairy products, solid margarines and meat products. The low impact of ω3polyunsaturated fatty acids, which are generally regarded as favorable, and the at a first glance surprising relation between eicosapentaenoic acid (20:5 ω3) levels and later LVH may be due to the fact that a high intake of linoleic-acid-rich vegetable fats reduces ω3-acid levels in serum44 by a decreased conversion of α-linolenic acid (18:3 ω3) to eicosapentaenoic acid, through competition for the same enzyme systems. Furthermore, of the two major ω3 fish fatty acids eicosapentaenoic acid and docosahexaenoic acid (22:6 ω3), the latter seems to have the most beneficial effects on blood pressure and heart rate105. Other factors than dietary fat intake may affect serum CE fatty acid profile, but the relations between LDL/HDL-cholesterol and triglycerides and later LVH support the adverse role of a diet rich in saturated fats. Fatty acids are the major oxidative fuel for the heart under fasting circumstances106, and myocardial substrate uptake is determined by the glucose-free fatty acid cycle107. Cardiac fatty acid β-oxidation is under the control of PPAR-α101,108, in such a way that high levels of fatty acids activate PPAR-α, which up-regulates transcription of genes coding for proteins involved in cardiac fatty acid transport and metabolism108. During development of LVH, cardiac substrate utilization changes towards more use of exogenous glucose109, which is preferentially metabolized through glycolysis110,111. Furthermore, cardiac expression of PPAR-α and fatty acid βoxidation falls112, which may lead to myocardial fibrosis113, contractile dysfunction and rhythm disturbances112. 35 Intervention trials in animals have shown that administration of ω3-polyunsaturated fatty acids affects cardiac lipid profile114 and reduces or prevents LVH115, whereas inhibition of long-chain fatty acid β-oxidation increases left ventricular mass116. In controlled trials in humans with recent myocardial infarction, treatment with ω3polyunsaturated fatty acids reduced left ventricular enlargement117 and cardiovascular events118. Long-chain unsaturated fatty acids have been suggested to be the natural ligands for the PPARs101. A possible interpretation of these findings may be that the beneficial effect of long-chain unsaturated fatty acids in LVH may, at least in part, be mediated through an improvement of the disturbed PPAR-α activity seen in LVH. Relative wall thickness is related to components of the insulin resistance syndrome In study II, several components of the insulin resistance syndrome were found to be related to an increased left ventricular RWT and left ventricular concentric remodeling but less to LVH, at age 70. Several attempts have been made to investigate the relations between components of the insulin resistance syndrome and echocardiographic left ventricular mass, mass index, sum of wall thicknesses or relative wall thickness (table 1). The previous studies show a wide variety of results, and are difficult to compare because of different methods of assessing left ventricular parameters and the insulin resistance syndrome and the use of different populations. The method of determining left ventricular parameters seems to influence the results, as investigators who have not indexed left ventricular mass for body size, or have not clearly stated the method of indexation, have more often found significant relations between left ventricular mass and signs of insulin resistance than investigators who have indexed left ventricular mass for height or height raised to any power, followed by investigators who have indexed left ventricular mass for body surface area. The characteristics of the investigated populations also seem to influence the results, as more significant relations between left ventricular mass and signs of insulin resistance were found in hypertensive populations than in the general population, followed by normotensive populations. Most of the previous studies in table 1 have comprised a limited number of middle-aged, hypertensive subjects. Because hypertension is a part of the insulin resistance syndrome, the relations found in hypertensive populations between components of the insulin resistance syndrome and left ventricular mass and geometry may not easily be generalized to normotensive or general populations. We therefore used a sample of men from the general population in study II. When investigating relations between LVH and components of the insulin resistance syndrome, it is important to take into account that both phenomena are related to an increased body size. Therefore, simple correlations between left ventricular mass and for instance insulin are, not surprisingly, often positive. Indexation for body size of some kind should be made for this kind of investigation to be meaningful. The best body size determinant of left ventricular mass is probably lean body mass119, which is most closely matched by the surrogate measurement body surface area120. Indexation methods based solely on height tend to overestimate the influence of obesity on left ventricular mass, whereas indexation for body surface area (which has 36 been criticized for disregarding the effect of obesity on left ventricular mass3,39) gives relations between left ventricular mass and obesity of similar magnitude as indexation for lean body mass119. This suggests that relations found between components of the insulin resistance syndrome and left ventricular mass indexed for body surface area are relatively independent of obesity. In study II, waist-to-hip ratio and BMI were related to RWT and not to LVMI and were highest in the concentric remodeling group. In another population study, left ventricular mass indexed to body surface area was not correlated to BMI in men1, suggesting that indexation for body surface area effectively takes care of the relation between left ventricular mass and obesity. In study II, insulin sensitivity index derived from the hyperinsulinemic euglycemic clamp, glucose tolerance at an OGTT, and fasting levels of specific insulin and 32-33 split proinsulin were all related to left ventricular RWT but not to LVMI in the present study, and subjects with left ventricular concentric remodeling had lower insulin sensitivity index and impaired glucose tolerance compared with subjects with normal left ventricular geometry. These findings are in accordance with another population study of men59 in which the insulin levels tended to be higher in subjects with concentric remodeling and concentric LVH and glucose and insulin levels correlated with RWT but not with LVMI. The values of several insulin sensitivity variables were similar for concentric remodeling and concentric LVH, although significant only for the former. This may be due to a small group of men with concentric LVH, but may also reflect a real difference. Heart rate was significantly increased in the concentric remodeling group, in accordance with previous research59. Tachycardia is proposed to be a reliable marker for an increased sympathetic activity in population studies121. The inverse relation between LVMI and heart rate reflects the increased heart rate in the concentric remodeling group and decreased heart rate in the eccentric LVH group, the latter probably made up of both subjects with heart failure and subjects with a physiological LVH caused by exercise, with low sympathetic activity and normal metabolic status. An inverse relation between LVMI and heart rate in men has been found in another population-based study1. In study I, lipid derangements, unfavorable fatty acid profile and obesity seemed to be powerful longitudinal predictors of LVH development, in contrast to study II, in which the main finding was that several measures of glucose intolerance and insulin resistance were related to an increased RWT and concentric remodeling, but less to LVH. The reason for these differences is not clear, but the metabolic associations may be affected by the normal age-related increase in RWT32. A possible sequence of events may be that the insulin resistance syndrome is cross-sectionally related to an increased RWT, which later develops to LVH. This may be a reason for the finding that factors of the insulin resistance syndrome at 50 are related to later LVH, but that (other) factors of the insulin resistance syndrome at 70 are cross-sectionally related to increased RWT, but not to LVH. 37 Relative wall thickness is related to skeletal muscle insulin resistance In study III, left ventricular RWT was inversely related to insulin sensitivity in skeletal muscle and borderline significantly directly related to insulin sensitivity in the myocardium. LVMI was not related to insulin sensitivity in the myocardium or skeletal muscle. Because skeletal muscle is the most important determinant of wholebody glucose uptake, this correlates well with the findings of study II, in which RWT, but not LVH, was related to whole-body insulin resistance. The relation between RWT and the ratio between myocardial and skeletal muscle glucose uptake indicates that myocardial and skeletal muscle insulin sensitivity are separate qualities, in accordance with some previous studies65,67 but in contrast to other122,123. Thus, the term ”insulin sensitivity”, frequently used to describe insulinmediated glucose uptake in skeletal muscle, might have different determinants and implications in the heart. For instance, cardiac work load is one of the most powerful denominators of myocardial glucose uptake65,67. Comparisons between study III and a recent similar study by Paternostro et al66 are difficult, since LVH patients of that study were more whole-body insulin resistant, older and had more pharmacotherapy than controls. Whether changes in myocardial glucose uptake are involved in the pathogenesis of the growth of left ventricular walls or merely reflect adaptive changes in oxygen demand has to be further evaluated. Supporting the former theory is the finding that induced hyperinsulinemia in the rat was followed by marked ventricular hypertrophy without hypertension124. In favor of the latter theory is the fact that oxidizing glucose yields more ATP per oxygen atom than oxidizing palmitate, which may be important in a situation with a decreased coronary reserve such as LVH. Induction of LVH in rats has been found to be associated with a shift in myocardial myosin isoenzymes from a faster to a slower form together with a decrease in ATPase activity and oxygen consumption125, although this mechanism is possibly of less importance in humans126. Increased myocardial glucose uptake and increased RWT may also be non-causally related parallel findings. The insulin resistance syndrome may be causally related to increased RWT due to other mechanisms, and the observed increased myocardial glucose uptake may be due to an increased availability of circulating glucose associated with the insulin resistance syndrome, since cardiac substrate utilization is largely dependent of substrate availability. In study III, we defined the study population from a thoroughly investigated cohort study, which enabled us to make the sample homogenous in all variables except the left ventricular geometry variables under study. As a result, too few subjects with LVH were eligible to make a meaningful division of the population into two groups, with and without LVH. Furthermore, no medication-free normotensive subjects with concentric LVH could be found in the health survey population from which the present study population was drawn, so only subjects with eccentric LVH were included. Although eccentric LVH is proposed to be associated with a lower risk for coronary heart disease than concentric LVH (table 2), the risk is still higher than that of normal left ventricular geometry. The findings may not have been the same if subjects with concentric LVH were included. 38 Insulin resistance and left ventricular hypertrophy: Cause or consequence? Potential mechanisms whereby insulin resistance or hyperinsulinemia may cause left ventricular hypertrophy Several lines of evidence indicate that insulin resistance or hyperinsulinemia may be causal factors in the development of thick left ventricular walls and LVH: i. Insulin at high concentrations is capable of inducing hypertrophic effects via the insulin-like growth factor (IGF)-I receptor (which are abundant in the heart), but also at low concentrations via the insulin receptor127,128. Stimulation of the IGF-I receptor by IGF-I causes myocyte hypertrophy in vitro129, and the effect on cardiac hypertrophy in vivo is probably mainly due to local rather than circulating IGF-I130,131. Cardiomyocytes produce IGF-I during development of hypertrophy132-134, a process suggested to be independent of the systemic reninangiotensin system132. Administration of growth hormone to humans (which mainly exerts its cardiac effect via cardiac IGF-I) leads to left ventricular wall thickening and an increased utilization of carbohydrates and lower oxygen consumption135. Long-term exposition to increased cardiac IGF-I levels result in myocardial fibrosis130. ii. Glucose uptake may in itself have an effect on LVH. Type-2 diabetic American Indians have been shown to have an increased RWT and LVMI136, and changes in blood glucose levels in type-2 diabetics during one year of follow-up have been shown to correlate to changes in LVMI137. The main glucose transporters (GLUT) in the heart are GLUT-1 and GLUT-4, of which GLUT-1 is the most important for basal glucose uptake138. Several findings indicate that increased basal cardiac glucose uptake may lead to cardiac hypertrophy. Conventionally used therapeutic doses of the thiazolidinedione troglitazone lead to a 3-4-fold increase in cardiac GLUT-1 and a <2-fold increase in GLUT-4138, and much higher doses lead to LVH139. Selective knock-out of the cardiac GLUT-4-receptor in mice leads to increased GLUT-1 levels and a 3-fold increase in cardiac basal glucose uptake and LVH140. In a study in humans66, cardiac GLUT-1 levels were increased in patients with LVH and whole-body insulin resistance. Hyperglycemia induces activation of protein kinase C (PKC)-β in the heart141, which is capable of enhancing angiotensin effects and macrovascular contractility, among other things142. Transgenic mice which overexpress PKC-β2 in the heart develop LVH and myocardial fibrosis143. iii. Hyperinsulinemia leads to renal sodium and water retention, an increased blood volume144,145 and a higher preload, which is a known trigger of LVH. iv. Hyperinsulinemia inhibits myocardial protein degradation in insulin resistant subjects. Myocardial protein undergoes continual turnover with a half-life of approximately 10 days, and acute insulin administration has been shown to decrease myocardial protein degradation by 80%146, which might lead to LVH. 39 v. Insulin resistance and hyperinsulinemia have been shown to cause systemic147,148 and cardiac149 sympathetic activation, which has been related to cardiac hypertrophy in experimental studies150,151. In dogs, repeated pressor episodes with elevated plasma norepinephrine levels152 or chronic infusion of norepinephrine153 resulted in LVH, but did not induce a sustained elevation of blood pressure. In humans, raised plasma catecholamine levels1,72 and an increased cardiac norepinephrine release154 have been found in subjects with LVH, and in the present study II and one other study59, heart rate (a marker for an increased sympathetic activity 121) was related to left ventricular concentric remodeling. In other studies, sympathetic activity has not been convincingly shown to be related to LVH151. Heart rate variability, an index of cardiac autonomic function, has been shown to be abnormal in subjects with the insulin resistance syndrome155,156 and in subjects with LVH157,158, suggesting that abnormal cardiac autonomic function may be a link between the insulin resistance syndrome and LVH. vi. Hyperinsulinemia has been shown to be a potent inducer of endothelin (ET)-1 release, more so in subjects with the insulin resistance syndrome than in normal subjects or subjects with insulinoma159, and insulin resistance is associated with an altered balance between the vasoconstricting effects of ET-1 and the vasodilating effects of NO160. Cardiomyocytes produce prepro-ET-1 during development of hypertrophy133,134, and prepro-ET-1 production has been related to cardiomyocyte protein synthesis161 and RWT133. vii. Insulin resistance is related to an increased pressor response to angiotensin II162. Fructose-fed rats (a model of insulin resistance) also exhibit an increased pressor response to angiotensin II163 and LVH, which is preventable with an angiotensin II type-1 receptor (AT1) blocker, indicating that angiotensin II may be responsible for the LVH in this model of insulin resistance164. Angiotensin II has been proposed to play a major role in the development of LVH, and angiotensinogen is produced in cardiomyocytes during hypertrophy development133,134. In a transgenic mouse model, increased cardiomyocyte angiotensinogen production was related to LVH and increased RWT, an effect that was reversible by ACE inhibition or AT1 blockade165. However, the hypertrophic effect of angiotensin II seems to be mediated by cardiac ET-1161, and cardiac hypertrophy develops also in aortic-banded AT1A knock-out mice166,167 and in pressure overload animal models after treatment with an ACE inhibitor or AT1 blocker168,169, suggesting that hypertrophy is a complex process that cannot be explained solely by the effects of angiotensin II170. viii. Insulin resistance to glucose uptake is often related to a decreased vasodilatory response to insulin171,172. Insulin resistance has been associated with a deranged microcirculation173 as well as a defect in the ability of insulin to increase aortic compliance174. In a recent study175, a decreased insulin-stimulated femoral artery blood flow was related to increased left ventricular wall thickness. After ACE inhibitor treatment, but not after β-receptor blocker treatment, an increased 40 insulin-stimulated blood flow was related to a reduced left ventricular wall thickness. Increased vascular stiffness and peripheral resistance induce an increased afterload, which is of pathogenetic importance for left ventricular concentric remodeling22 and LVH. ix. The development from physiologic to pathologic LVH with increased myocardial fibrosis may also be accelerated by insulin. An altered echocardiographic myocardial texture indicative of increased fibrosis has been found in hypertensive subjects with LVH, but not in athletes with the same degree of LVH176. This texture alteration has also been found to correlate to hyperinsulinemia during an OGTT177, supported by the finding of increased myocardial fibrosis in an animal model of the insulin resistance syndrome and diabetes mellitus type-2178. Hyperinsulinemia may increase serum aldosterone levels in obese, insulin resistant individuals179, and elevated aldosterone levels play an important role in the development of myocardial fibrosis180. Further support for the hypothesis that insulin resistance or hyperinsulinemia may play a causal role in the development of LVH comes from an elegant study in which induced chronic moderate hyperinsulinemia in rats, with control of hormones with effects opposing insulin, was followed by pronounced cardiac hypertrophy, without elevated blood pressure124. LVH is also present in endocrine disorders with insulin resistance and hyperinsulinemia such as acromegaly181 and hypothyroidism182. The abundance of proposed mechanisms behind LVH and the difficulty to fit the results from various experimental studies into the same hypothesis indicate that there are probably several LVH phenotypes, with different molecular background170. In view of our findings, it seems reasonable to hypothesize that the growthstimulating effects of insulin in humans mainly affect RWT, whereas hemodynamic load mainly affects LVMI. If the primary hypertrophic stimulus is an increase in a growth-stimulating hormone such as insulin, it is possible that the cardiomyocytes respond with an increased cross-sectional area leading to an increased RWT, rather than elongation or any other adaptation leading to an enlarged left ventricular cavity. These geometric parameters are however not independent of each other, and LaPlace’s law (figure 18) indicates that a possible consequence of an increased wall thickness is a dilatation of the left ventricle, in order to keep wall tension constant. It is also possible that metabolic and hemodynamic factors act in a synergistic manner and potentiate the effects of one another on left ventricular mass and geometry. An augmenting effect of insulin on the pressor responses of norepinephrine183 and angiotensin II162,163 has been found. Further support for a synergistic effect is seen in table 1, in which more significant relations between left ventricular mass and signs of insulin resistance have been found in hypertensive populations than in normotensive populations or the general population. 41 Insulin resistance and left ventricular hypertrophy as parallel phenomena LVH and the insulin resistance syndrome may be parallel consequences of one or more common etiologic factors, such as an increased activity in the sympathetic nervous system, vascular alterations or aging. Left ventricular wall thickness and the prevalence of left ventricular concentric remodeling increase with age32. This is mainly due to cardiac myocyte hypertrophy, for which an increased stroke work resulting from increased arterial stiffness has been proposed to be an etiological factor184. Because insulin sensitivity also decreases with age, the described association between components of the insulin resistance syndrome and the growth of left ventricular walls could in part be a consequence of aging, a process proceeding faster in some subjects prone to both insulin resistance and cardiac remodeling. Fig. 18: LaPlace’s law P* r W all tension = W all thickness P = intraventricular blood pressure r = left ventricular cavity radius Left ventricular hypertrophy as a causal factor for hypertension and insulin resistance LVH has been shown to precede and has been proposed to contribute to the development of hypertension185,186. LVH has also been proposed to be a possible cause for increased sympathetic nervous activity. Subjects with LVH have been shown to have a reduced cardiac sensitivity to the effectors of the sympathetic nervous system, which may lead to a compensatory systemic sympathetic hyperactivity151. An increased sympathetic activity has also been suggested as a potential cause for the insulin resistance syndrome187. LaPlace’s law (figure 18) indicates the relation between blood pressure, left ventricular wall tension, wall thickness and chamber diameter. Hitherto, this law has mostly been used to legitimize the theory that an increased blood pressure leads to increased left ventricular wall thickness in order to keep wall tension constant. However, this law also indicates that an alternative response to an increased blood pressure could be a diminished chamber diameter, with no change in wall thickness (concentric remodeling). LaPlace’s law does not indicate a certain chain of events, or the reason for the initiating event, but rather the consequences of the initiating event. This also means that if an increased wall thickness (possibly due to growth stimulation by insulin) were the initiating event, this would result in an increased blood pressure or a dilatation of the left ventricle, or a combination of these. 42 The clinical importance of left ventricular hypertrophy Electrophysiologic co-morbidity of left ventricular hypertrophy The most consistent finding in study IV was that almost all major ECG abnormalities were related to an increased LVMI. Furthermore, both left ventricular systolic and diastolic dysfunction were related to an increased Sokolow-Lyon Voltage, a commonly used ECG sign of LVH. Subjects having suffered from a myocardial infarction (definite abnormal Q-wave at the ECG) showed an elevated LVMI, and the prevalence of Q-waves was highest in the eccentric LVH group. Whether or not the increased LVMI is the cause or the consequence of the myocardial infarction in this group of patients can not be told from a cross-sectional study like this. It seems likely that both of these associations between an abnormal Q-wave at the ECG and an increased LVMI exist in the present sample, as structural changes of the left ventricle frequently occur following a myocardial infarction, changes most likely to result in an enlarged left ventricle188 and eccentric LVH. The situation is probably the same for left bundle-branch block, which most often is a consequence of a myocardial infarction189. ST-segment or T-wave changes are non-specific ECG items found in coronary artery disease and several other cardiac disorders, including LVH. The prevalence of ST- or T-wave abnormalities was highest in the concentric LVH group, which might indicate a higher prevalence of angina pectoris and ischemia or the left ventricular strain pattern of LVH in this group. Subjects with frequent premature beats or atrioventricular block type 1 (without Q-, S-T- or T-wave abnormalities) had an increased LVMI and a thick IVS. This resembles the geometry found in patients with hypertrophic cardiomyopathy in whom conduction disturbances and ventricular arrhythmias are commonly found190. None of the subjects in the present study fulfilled the criteria for hypertrophic cardiomyopathy191, but the pathophysiological events linking septal enlargement to ventricular arrhythmias and atrioventricular block may be similar in these groups of subjects. Arrhythmias have previously been shown to be related to LVH192,193, and LVH is a strong risk factor for sudden death14. The most commonly used index of left ventricular diastolic function, the E/A ratio, was not altered in the present sample even in subjects with Q-waves, in whom left ventricular diastolic dysfunction has previously been shown in middle-aged subjects194,195. The possible reason for this is that left ventricular diastolic function and thus the E/A ratio declines with age also in healthy subjects196 and that the effect of aging in itself overrides the effects of the different pathological conditions in the present sample aged 70. This seems also to be the case for IVRT, the echocardiographic index of the earliest left ventricular diastolic phase. 43 In a sub-sample without Minnesota code ECG abnormalities, existing LVH was mainly eccentric, and was better found with the Sokolow-Lyon Voltage than the Cornell assessments. This is in contrast to the findings in the whole cohort, where the sensitivities and specificities for echocardiographic LVH were similar for SokolowLyon Voltage and the Cornell assessments. Sokolow-Lyon Voltage, one of the most commonly used ECG signs of LVH, was not only related to an increased LVMI, but also to impairments in both systolic and diastolic function. Prognostic significance of echocardiographic and electrocardiographic left ventricular hypertrophy In study V, echocardiographic LVMI and the ECG-LVH criterion Cornell Product >244 µVs predicted mortality independently of each other and of other cardiovascular risk factors, which indicates that echocardiographic and ECG-LVH were not identical conditions. It was also shown that many but not all of the subjects whose deaths were predicted by ECG-LVH were also identified by echocardiography and vice versa. The main new finding was that the previously known prognostic value of ECG-LVH to some extent was independent of echocardiographic LVMI and vice versa. We therefore assessed the clinically relevant question of how much additional prognostic information would be gained by referring a subject to an echocardiographic examination if the subject’s ECG-LVH and hypertension status was known. Our conclusion was that the additional prognostic value of an echocardiographic LVH assessment if ECG Cornell Product ≤244 µVs was low in normotensive subjects, whereas in hypertensive subjects or the population as a whole, echocardiography and ECG provided complementary prognostic information. ECG is today generally regarded as merely a less sensitive method than echocardiography for detecting anatomic LVH, but in view of the findings of the present study, the LVH information obtained with ECG should rather be regarded as of equal prognostic importance as echocardiographic LVH information. The implication for the clinician assessing the risk associated with LVH is that the decision of which patient to refer to echocardiography should be based on knowledge of ECG-LVH and hypertension status. A normotensive patient without ECG-LVH may well have echocardiographic LVH, but it is probably of a benign, ”physiologic” nature of low prognostic importance and hardly motivates an echocardiographic examination. Total mortality seemed to increase linearly with increasing LVMI, but the risk for cardiovascular mortality was markedly increased only in the fourth quartile of LVMI, seemingly in accordance with one large previous study6, but in contrast to two studies5,15, in which the relation between LVMI and cardiovascular morbidity was apparently linear. Defining LVH as LVMI ≥150 g/m2, (a cut-off level originally derived from a healthy, middle-aged sample of men living in Framingham, USA24) seemed to limit the information carried in the continuous variable LVMI regarding prediction of all-cause mortality, but was relevant for the prediction of cardiovascular mortality and morbidity in the present population. Altogether, the cut-off level 150 g/m2 seems appropriate for LVMI measured with the leading edge 44 to leading edge convention and the Troy formula, corresponding to 131 g/m2 measured with the Penn convention and the modified cube formula24. Eccentric LVH was the most hazardous of the left ventricular geometric patterns investigated in the present study, followed by concentric remodeling. This is in contrast to findings in previous studies (table 2), in which concentric LVH has been associated with the worst prognosis. The reason for this discrepancy may be that the previous studies were carried out in middle-aged hypertensive subjects and the present study in a general elderly population, with a fairly high prevalence of ischemic heart disease. Eccentric LVH in the elderly is often an end-stage geometry in patients with congestive heart failure due to ischemic heart disease, but it is also found in a substantial proportion of apparently healthy subjects without ischemic heart disease2. In the present study, the predictive power of eccentric LVH was not attenuated when adjusting for previous hospitalization for ischemic heart disease, suggesting that eccentric LVH is not a benign condition in this age-group even in absence of ischemic heart disease. High LVMI was a stronger independent risk factor for cardiovascular disease than most other well known risk factors in this age group investigated (including the novel risk factor proinsulin, which was an independent predictor of cardiovascular mortality in a 27-year follow-up study from age 50 in this cohort (Björn Zethelius, MD, unpublished data, 2000), and no other investigated risk factors than LVMI and Cornell Product were independent predictors of total mortality. In study II, we found that components of the insulin resistance syndrome were closest related to the concentric remodeling geometry. Controlling for factors of the insulin resistance syndrome in study V, the concentric remodeling geometry still implied an increased risk, indicating that concentric remodeling is not only an integrated measurement of risk factors of the insulin resistance syndrome, but in itself adds to the total risk. Strengths and limitations of the study The determination of the main effect variables in the present study have been made with M-mode echocardiography. Measurement of left ventricular mass with this technique is considered as the golden standard, and has been extensively validated against autopsy materials and as a predictor of subsequent morbidity and mortality. It is however investigator-dependent, and sufficient image quality may be difficult to obtain in certain individuals, such as elderly or patients with pulmonary disease. Proper angulation of the M-mode ultrasound may also be difficult. In the present study, only one echocardiographer (Bertil Andrén) made all the analyses, which eliminates one source of measurement error. At the start of the echocardiographic study in 1991, there was no consensus on which formula to use for calculation of left ventricular mass. The procedures used were in accordance with the contemporary recommendation from the ASE. Comparisons have been made between the techniques used in the present study and the techniques more commonly used today91. The methods have been shown to be equally reliable, and the values of left ventricular mass obtained with one technique can easily be converted to corresponding values of the other technique24,91. 45 One of the main investigations of the study was assessment of insulin sensitivity with the hyperinsulinemic euglycemic clamp technique, in which serum insulin levels are elevated approximately 10-fold from fasting levels for 2 hours. This may seem high, but corresponds to post-prandial insulin levels in insulin resistant individuals. In this setting, the glucose infused is a direct measurement of the total insulin-mediated glucose uptake in the whole body, independent of the insulin-producing capacity of the pancreatic β-cells. The clamp technique is considered the golden standard for measuring insulin sensitivity, because the influences of counter-regulatory hormones can be minimized. An obvious limitation of the study is the lack of women and other ethnic and age groups. This affects the generalizability to other groups, and motivates further studies for confirmation of the findings of the present study. However, the limitation of the population to one sex, age group and ethnic group eliminates the need for adjustment for the influence of these important determinants of LVMI and factors of the insulin resistance syndrome. The present cohort has been closely followed for twenty years and may therefore be healthier than average Swedish 70-year-old men. Thus, associations in the present study may be weaker than in the general population. One limitation of the prospective study I is the lack of dietary records at baseline, and resulting lack of knowledge about any potential dietary certainties in the studied population, which might influence the generalizability of the findings of the present study. Other limitations of the study include absence of echocardiographic data at baseline and possible bias due to loss to follow-up. One of the strengths of study V is that all subjects were of the same age at baseline, which overcomes the problem of age differences between quantiles of LVMI found in other studies5,6,15. Limitations of the study include possible misclassification of endpoints, although the accuracy of the Swedish hospital discharge and cause-ofdeath registers have been shown to be high197. 46 Conclusions Dyslipidemia and a high serum proportion of saturated fatty acids and low proportion of linoleic acid, as well as obesity and hypertension, at age 50 predicted the prevalence of LVH at age 70 in this twenty-year follow-up of a fairly large prospectively determined sample of men from the general population. The impact of obesity, dyslipidemia and an unfavorable serum fatty acid profile on LVH was independent of history of ischemic heart disease, valvular disease and use of antihypertensive medication, indicating that lipids may be important in the etiology of LVH. Cross-sectionally at age 70, components of the insulin resistance syndrome, such as clamp insulin sensitivity index, OGTT glucose levels, triglycerides, waist-to-hip ratio and 24-h blood pressure, were significantly related to left ventricular RWT and concentric remodeling but less to LVH. In a healthy normotensive sample of men, RWT was inversely related to insulin sensitivity in skeletal muscle and borderline significantly directly related to insulin sensitivity in the myocardium in elderly men, whereas LVMI was not related to myocardial or skeletal muscle insulin sensitivity. Echocardiographic signs of LVH were seen both in subjects with ECG signs of myocardial infarction as well as in subjects with several other ECG diagnoses. Furthermore, both systolic and diastolic dysfunction were related to increased QRS amplitudes on the ECG. These findings suggest an important role for LVH in overall cardiac electrophysiological pathology, and the finding of ECG abnormalities in elderly men should raise the suspicion of structural and/or functional left ventricular abnormality. Total and cardiovascular mortality risk increased with increasing echocardiographic LVMI, independently of other cardiovascular risk factors, and cardiovascular risk was fairly well assessed with dichotomized echocardiographic LVH and geometric subgroups. ECG-LVH also predicted total and cardiovascular mortality, especially the Cornell Product criterion, which predicted total mortality independently of LVMI and other risk factors. Thus, echocardiographic and ECG-LVH are not identical conditions, and to fully assess the considerable risk associated with either condition, both an ECG and an echocardiogram should be performed, especially in hypertensive subjects. 47 Future perspectives The results of the present study show that there may be potential roles for dietary fat quality and components of the insulin resistance syndrome, such as insulin resistance, obesity, hypertension and dyslipidemia, in the etiology of LVH and increased RWT. Proof of causality cannot be obtained solely by epidemiological studies, but intervention studies need to be made. Several intervention studies investigating the etiology of LVH have been made, most addressing the role of hypertension35,36, but some investigating other possible etiologic factors, such as obesity41,42, a sedentary lifestyle and an unhealthy diet43. In fact, treating the latter factors has been shown to be as effective as antihypertensive medication in decreasing left ventricular mass41-43. The correlation between degree of blood pressure lowering and degree of LVH reduction in previous studies is not strong34, which may indicate that other properties of the antihypertensive drugs than their antihypertensive effect may influence left ventricular mass and RWT. It was for instance recently proposed that ”available data support the hypothesis that antihypertensive drugs that inhibit the activity of the renin-angiotensin system or, to a lesser extent, the sympathetic nervous system, reduce LVH more consistently than drugs that stimulate these systems”198. Recently, attention has been paid to the metabolic effects of antihypertensive drugs199-209, and it has been shown that these drugs have the capacity to reduce or increase insulin sensitivity by as much as 30%. In view of these data and the results from the present study, it is tempting to propose that the metabolic effects of an antihypertensive treatment may contribute to the capacity of the treatment to reduce LVH and RWT, and may explain some of the variance in the effects of different classes of antihypertensive drugs on LVH. To illustrate these relations, we have constructed graphs (figures 19 and 20) showing changes in left ventricular mass and RWT in relation to changes in blood pressure and insulin sensitivity, using data from several published randomized clinical trials of antihypertensive drugs and weight reduction35,42,199-212 and unpublished data from trials in our clinic with losartan, irbesartan, verapamil and mibefradil (Hans Lithell, MD, unpublished data, 2000). In figure 20, the mean effect of β1-receptor blockers on insulin sensitivity was derived from investigations in approximately 150 subjects, because β1-receptor blockers have been used as reference drugs in many of the insulin sensitivity trials. The mean effects of the other drug classes on insulin sensitivity were derived from studies of two to five different drugs from each class. As can be seen in figures 19 and 20, the degree of reduction of left ventricular mass or RWT was not correlated to the degree of blood pressure reduction. However, there was a clear inverse correlation between the change in RWT and the change in insulin sensitivity (Spearman's rank-order correlation coefficient –0.89, p=0.02). This is in agreement with the findings of our study II, in which RWT was inversely related to insulin sensitivity. The remaining variance in figures 19 and 20 may be due to other properties of the treatments, such as their ability to inhibit the renin-angiotensin system or the sympathetic nervous system. 48 Fig. 19: Average changes in mean arterial pressure in relation to average changes in relative wall thickness (A) and left ventricular mass (B), in several randomized trials B 0 D -5 β1b AIIa Ca ACEi -10 -17 ⌧∆Left ventricular mass(%) ⌧∆Relative wall thickness (%) A W -5 Ca β1b -10 AIIa -15 ACEi -20 -17 -15 -13 -11 ⌧∆Mean arterial pressure (%) D α1b W -15 -13 -11 ⌧∆Mean arterial pressure (%) D = diuretics β1b = β1-receptor blockers AIIa = angiotensin-II1-receptor blockers Ca = calcium antagonists ACEi = angiotensin-converting enzyme inhibitors W = weight loss α1b = α1-receptor blockers Fig. 20: Average changes in whole-body glucose disposal in relation to average changes in relative wall thickness (A) and left ventricular mass (B), in several randomized trials 0 -5 B D ⌧∆Left ventricular mass(%) ⌧∆Relative wall thickness (%) A β1b AIIa Ca -10 -20 ACEi W -5 Ca -10 W BGD = whole-body glucose disposal D = diuretics β1b = β1-receptor blockers AIIa = angiotensin-II1-receptor blockers Ca = calcium antagonists ACEi = angiotensin-converting enzyme inhibitors W = weight loss α1b = α1-receptor blockers 49 α1b AIIa -15 ACEi -20 -20 0 20 ⌧∆W BGD (%) β1b D W 0 20 ⌧∆W BGD (%) In order to establish the role of the insulin resistance syndrome in LVH, intervention studies specifically addressing this issue should be performed. Some attempts have been made to investigate these factors41-43,213, but the results of these studies are difficult to ascribe to the effects on insulin sensitivity per se. In one study213, the effects of two antihypertensive drugs on glucose and insulin metabolism were related to their LVH-reducing effects. The recent emergence of drugs specifically designed to enhance insulin sensitivity makes studies of the role of insulin resistance in LVH possible. One way of studying this issue might include determination of insulin sensitivity with the hyperinsulinemic euglycemic clamp technique, determination of RWT and left ventricular mass with echocardiography or nuclear magnetic resonance tomography, and a comparison between treatments with a drug that improves insulin sensitivity but does not affect blood pressure and a drug that lowers blood pressure but does not affect insulin sensitivity. 50 Acknowledgements I wish to express my sincere gratitude and appreciation to all those who have helped me complete this thesis, with special thanks to: Lars Lind, my principal supervisor, for introducing me to medical research, for his neverending enthusiasm and ideas, and for always having time for stimulating discussions. Hans Lithell, head of the Section for Geriatrics, my supervisor, for his continuous encouraging support and for providing a creative and enjoyable research atmosphere. Bertil Andrén, for providing the echocardiographic examinations on which this thesis is based, and for creative input in the articles. Lars Berglund and Rawya Mohsen, for always having time and patience for any question a PhD-student may have. Bengt Vessby and Margareta Öhrvall, for providing me with the opportunity of developing my clinical skills in the meeting of patients with the insulin resistance syndrome. Johan Ärnlöv, for being a good friend and for valuable support in the fields of career development, jazz music and physical activity (especially office-golf, FIFA 99 and walking or jogging along the Thames, Mississippi, Hudson and Fyris rivers). My friends and colleagues at the Section for Geriatrics — Liisa Byberg, Björn Zethelius, Arvo Hänni, Kristina Björklund, Anu Hedman, Kristina Dunder, Lennart Hansson, Merike Boberg, Per-Erik Andersson, Lena Kilander, Richard Reneland, Margareta Falkeborn, Ann-Cristin Åberg and Claes Risinger — and the Section for Clinical Nutrition Research — Ulf Risérus, Annika Smedman, Eva Södergren, Agneta Andersson, Anette Järvi, Johanna Helmersson, Cecilia Nälsén, Marie Lemcke-Norojärvi, Samar Basu, Brita Karlström, Anders Sjödin and Achraf Daryani — especially those who have participated in the valuable ”doktorandklubben” seminars, for intellectual stimulation, helping hands and good laughs. My co-authors Niklas Nyström, Sven Valind, Antti Aro, Nicholas Hales, Agneta Holmäng, Per Björntorp and Anders Waldenström, for valuable help with the articles and rapid correspondence. Jan Hall, Siv Tengblad, Eva Sejby and Barbro Simu for their excellent laboratory work. The staff at the Metabolic Ward, for their devotion and competence in conducting this demanding longitudinal study, and for being a great team All the brave Uppsala-men who have participated in the study for almost 30 years. Anna, for expert guidance in the art of thesis cover design and the English language, but most of all for patience, support, devotion and love beyond understanding. This study was supported by grants from the Swedish Medical Research Council (grant no. 5446), the Thuréus Foundation, the Swedish National Association against Heart and Lung Disease, the Foundation for Geriatric Research (Stiftelsen för Geriatrisk Forskning), Geriatric Fund (Geriatriska Fonden), the Ernfors Foundation, Trygg Hansa, ”Förenade Liv” Mutual Group Life Insurance Company, and Uppsala University. The cover illustration is a positron emission tomogram from the PET-Center, Uppsala University. Uppsala in December 2000 51 Summary in Swedish Sammanfattning på svenska Vänsterkammarhypertrofi (en förstoring av hjärtats vänstra kammare) och det metabola syndromet (en anhopning av kardiovaskulära riskfaktorer såsom fetma, insulinresistens och förhöjda nivåer av blodtryck, blodfetter och insulin) är mycket vanliga tillstånd som är förknippade med en markant ökad kardiovaskulär risk. Under 1970-73 inbjöds alla 50-åriga män boende i Uppsala till en hälsoundersökning för att kartlägga riskfaktorer för kardiovaskulär sjukdom. Vid en uppföljning 20 år senare upprepades mätningarna, och vänsterkammarens massa och geometri mättes med ultraljud hos 475 män. I denna prospektiva longitudinella studie fann vi att ett ogynnsamt fettsyramönster i blodet samt delar av det metabola syndromet (fetma, blodfettsrubbning och högt blodtryck) vid 50 års ålder ökade sannolikheten för att ha vänsterkammarhypertrofi vid 70 års ålder. I en tvärsnittsstudie vid 70 års ålder var delar av det metabola syndromet (bukfetma, insulinresistens, nedsatt sockertolerans, blodfettsrubbning och högt blodtryck) kopplade till en ökad relativ väggtjocklek i vänster kammare, men inte till vänsterkammarhypertrofi. I ett litet friskt urval av dessa män var en ökad relativ väggtjocklek i vänster kammare kopplad till insulinresistens i skelettmuskel och antytt ökad insulinkänslighet i hjärtmuskel, medan vänsterkammarmassan inte var kopplad till insulinkänslighet i skelett- eller hjärtmuskel. Vid 70 års ålder var vänsterkammarhypertrofi ett genomgående ultraljudsfynd vid ett flertal vanliga EKG-diagnoser. I en prospektiv mortalitetsstudie med början vid 70 års ålder och med fem års uppföljning fann vi att ultraljudsmässig och EKG-mässig vänsterkammarhypertrofi medförde ökad dödlighet, oberoende av varandra och av andra kardiovaskulära riskfaktorer. Detta innebär att ultraljudsmässig och EKG-mässig vänsterkammarhypertrofi ger olika, men lika viktig, prognostisk information. Sammanfattningsvis var det metabola syndromets komponenter kopplade till vänsterkammarhypertrofi 20 år senare, men tvärsnittsmässigt starkare kopplade till en ökad relativ väggtjocklek i vänster kammare. Ultraljudsmässig och EKG-mässig vänsterkammarhypertrofi medförde ökad dödlighet, oberoende av varandra och det metabola syndromets komponenter. 52 References 1. Marcus R, Krause L, Weder AB, Dominguez-Meja A, Schork NJ, Julius S. Sexspecific determinants of increased left ventricular mass in the Tecumseh Blood Pressure Study. Circulation 1994;90:928-936. 2. Andren B, Lind L, Hedenstierna G, Lithell H. Left ventricular hypertrophy and geometry in a population sample of elderly males. Eur Heart J 1996;17:1800-1807. 3. Levy D, Anderson KM, Savage DD, Kannel WB, Christiansen JC, Castelli WP. Echocardiographically detected left ventricular hypertrophy: prevalence and risk factors. The Framingham Heart Study. Ann Intern Med 1988;108:7-13. 4. Hammond IW, Devereux RB, Alderman MH, Laragh JH. Relation of blood pressure and body build to left ventricular mass in normotensive and hypertensive employed adults. J Am Coll Cardiol 1988;12:996-1004. 5. Levy D, Garrison RJ, Savage DD, Kannel WB, Castelli WP. Left ventricular mass and incidence of coronary heart disease in an elderly cohort. The Framingham Heart Study. Ann Intern Med 1989;110:101-107. 6. Levy D, Garrison RJ, Savage DD, Kannel WB, Castelli WP. Prognostic implications of echocardiographically determined left ventricular mass in the Framingham Heart Study. N Engl J Med 1990;322:1561-1566. 7. Koren MJ, Devereux RB, Casale PN, Savage DD, Laragh JH. Relation of left ventricular mass and geometry to morbidity and mortality in uncomplicated essential hypertension. Ann Intern Med 1991;114:345-352. 8. Ghali JK, Liao Y, Simmons B, Castaner A, Cao G, Cooper RS. The prognostic role of left ventricular hypertrophy in patients with or without coronary artery disease. Ann Intern Med 1992;117:831-836. 9. Bikkina M, Levy D, Evans JC, Larson MG, Benjamin EJ, Wolf PA, Castelli WP. Left ventricular mass and risk of stroke in an elderly cohort. The Framingham Heart Study. JAMA 1994;272:33-36. 10. Bolognese L, Dellavesa P, Rossi L, Sarasso G, Bongo AS, Scianaro MC. Prognostic value of left ventricular mass in uncomplicated acute myocardial infarction and onevessel coronary artery disease. Am J Cardiol 1994;73:1-5. 11. Krumholz HM, Larson M, Levy D. Prognosis of left ventricular geometric patterns in the Framingham Heart Study. J Am Coll Cardiol 1995;25:879-884. 12. Verdecchia P, Schillaci G, Borgioni C, Ciucci A, Battistelli M, Bartoccini C, Santucci A, Santucci C, Reboldi G, Porcellati C. Adverse prognostic significance of concentric remodeling of the left ventricle in hypertensive patients with normal left ventricular mass. J Am Coll Cardiol 1995;25:871-878. 13. Verdecchia P, Schillaci G, Borgioni C, Ciucci A, Gattobigio R, Zampi I, Santucci A, Santucci C, Reboldi G, Porcellati C. Prognostic value of left ventricular mass and geometry in systemic hypertension with left ventricular hypertrophy. Am J Cardiol 1996;78:197-202. 53 14. Haider AW, Larson MG, Benjamin EJ, Levy D. Increased left ventricular mass and hypertrophy are associated with increased risk for sudden death. J Am Coll Cardiol 1998;32:1454-1459. 15. Schillaci G, Verdecchia P, Porcellati C, Cuccurullo O, Cosco C, Perticone F. Continuous relation between left ventricular mass and cardiovascular risk in essential hypertension. Hypertension 2000;35:580-586. 16. Quinones MA, Greenberg BH, Kopelen HA, Koilpillai C, Limacher MC, Shindler DM, Shelton BJ, Weiner DH. Echocardiographic predictors of clinical outcome in patients with left ventricular dysfunction enrolled in the SOLVD registry and trials: significance of left ventricular hypertrophy. Studies of Left Ventricular Dysfunction. J Am Coll Cardiol 2000;35:1237-1244. 17. Kannel WB, Gordon T, Castelli WP, Margolis JR. Electrocardiographic left ventricular hypertrophy and risk of coronary heart disease. The Framingham study. Ann Intern Med 1970;72:813-822. 18. Kannel WB. Left ventricular hypertrophy as a risk factor: the Framingham experience. J Hypertens Suppl 1991;9:S3-8. 19. Levy D, Salomon M, RB DA, Belanger AJ, Kannel WB. Prognostic implications of baseline electrocardiographic features and their serial changes in subjects with left ventricular hypertrophy. Circulation 1994;90:1786-1793. 20. Siegrist J, Peter R, Motz W, Strauer BE. The role of hypertension, left ventricular hypertrophy and psychosocial risks in cardiovascular disease: prospective evidence from blue-collar men. Eur Heart J 1992;13 Suppl D:89-95. 21. Verdecchia P, Schillaci G, Borgioni C, Ciucci A, Gattobigio R, Zampi I, Porcellati C. Prognostic value of a new electrocardiographic method for diagnosis of left ventricular hypertrophy in essential hypertension. J Am Coll Cardiol 1998;31:383-390. 22. Ganau A, Devereux RB, Roman MJ, de Simone G, Pickering TG, Saba PS, Vargiu P, Simongini I, Laragh JH. Patterns of left ventricular hypertrophy and geometric remodeling in essential hypertension. J Am Coll Cardiol 1992;19:1550-1558. 23. Devereux RB, Pini R, Aurigemma GP, Roman MJ. Measurement of left ventricular mass: methodology and expertise. J Hypertens 1997;15:801-809. 24. Levy D, Savage DD, Garrison RJ, Anderson KM, Kannel WB, Castelli WP. Echocardiographic criteria for left ventricular hypertrophy: the Framingham Heart Study. Am J Cardiol 1987;59:956-960. 25. Lorell BH, Carabello BA. Left ventricular hypertrophy: pathogenesis, detection, and prognosis. Circulation 2000;102:470-479. 26. Weber KT, Brilla CG. Pathological hypertrophy and cardiac interstitium. Fibrosis and renin-angiotensin-aldosterone system. Circulation 1991;83:1849-1865. 27. Weber KT, Brilla CG, Cleland JG, Cohn JN, Hansson L, Heagerty AM, Laragh JH, Laurent S, Ollivier JP, Pauletto P, et al. Cardioreparation and the concept of modulating cardiovascular structure and function. Blood Press 1993;2:6-21. 54 28. Manolio TA, Levy D, Garrison RJ, Castelli WP, Kannel WB. Relation of alcohol intake to left ventricular mass: The Framingham Study. J Am Coll Cardiol 1991;17:717721. 29. Post WS, Larson MG, Myers RH, Galderisi M, Levy D. Heritability of left ventricular mass: the Framingham Heart Study. Hypertension 1997;30:1025-1028. 30. Garner C, Lecomte E, Visvikis S, Abergel E, Lathrop M, Soubrier F. Genetic and Environmental Influences on Left Ventricular Mass : A Family Study. Hypertension 2000;36:740-746. 31. Devereux RB, Drayer JI, Chien S, Pickering TG, Letcher RL, DeYoung JL, Sealey JE, Laragh JH. Whole blood viscosity as a determinant of cardiac hypertrophy in systemic hypertension. Am J Cardiol 1984;54:592-595. 32. Ganau A, Saba PS, Roman MJ, de Simone G, Realdi G, Devereux RB. Ageing induces left ventricular concentric remodelling in normotensive subjects. J Hypertens 1995;13:1818-1822. 33. Lauer MS, Anderson KM, Levy D. Influence of contemporary versus 30-year blood pressure levels on left ventricular mass and geometry: the Framingham Heart Study. J Am Coll Cardiol 1991;18:1287-1294. 34. Liebson PR. Clinical studies of drug reversal of hypertensive left ventricular hypertrophy. Am J Hypertens 1990;3:512-517. 35. Dahlof B, Pennert K, Hansson L. Reversal of left ventricular hypertrophy in hypertensive patients. A metaanalysis of 109 treatment studies. Am J Hypertens 1992;5:95-110. 36. Schmieder RE, Martus P, Klingbeil A. Reversal of left ventricular hypertrophy in essential hypertension. A meta-analysis of randomized double-blind studies. JAMA 1996;275:1507-1513. 37. Majahalme S, Turjanmaa V, Weder A, Lu H, Tuomisto M, Virjo A, Uusitalo A. Blood pressure levels and variability, smoking, and left ventricular structure in normotension and in borderline and mild hypertension. Am J Hypertens 1996;9:11101118. 38. Devereux RB, Pickering TG, Alderman MH, Chien S, Borer JS, Laragh JH. Left ventricular hypertrophy in hypertension. Prevalence and relationship to pathophysiologic variables. Hypertension 1987;9:II53-60. 39. Lauer MS, Anderson KM, Kannel WB, Levy D. The impact of obesity on left ventricular mass and geometry. The Framingham Heart Study. JAMA 1991;266:231236. 40. de Simone G, Devereux RB, Daniels SR, Meyer RA. Gender differences in left ventricular growth. Hypertension 1995;26:979-983. 41. Karason K, Wallentin I, Larsson B, Sjostrom L. Effects of obesity and weight loss on left ventricular mass and relative wall thickness: survey and intervention study. BMJ 1997;315:912-916. 55 42. MacMahon SW, Wilcken DE, Macdonald GJ. The effect of weight reduction on left ventricular mass. A randomized controlled trial in young, overweight hypertensive patients. N Engl J Med 1986;314:334-339. 43. Liebson PR, Grandits GA, Dianzumba S, Prineas RJ, Grimm RH, Jr., Neaton JD, Stamler J. Comparison of five antihypertensive monotherapies and placebo for change in left ventricular mass in patients receiving nutritional-hygienic therapy in the Treatment of Mild Hypertension Study (TOMHS). Circulation 1995;91:698-706. 44. Nikkari T. Serum fatty acids and coronary heart disease in Finnish populations. Prog Lipid Res 1986;25:437-450. 45. Zock PL, Mensink RP, Harryvan J, de Vries JH, Katan MB. Fatty acids in serum cholesteryl esters as quantitative biomarkers of dietary intake in humans. Am J Epidemiol 1997;145:1114-1122. 46. Ohrvall M, Berglund L, Salminen I, Lithell H, Aro A, Vessby B. The serum cholesterol ester fatty acid composition but not the serum concentration of alpha tocopherol predicts the development of myocardial infarction in 50-year-old men: 19 years follow-up. Atherosclerosis 1996;127:65-71. 47. Kylin E. Studien über das Hypertonie-Hyperglykämie-Hyperurikämiesyndrom. Zentralblatt für innere Medizin 1923;44:105-127. 48. DeFronzo RA, Tobin JD, Andres R. Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am J Physiol 1979;237:E214-223. 49. Haffner SM, Valdez RA, Hazuda HP, Mitchell BD, Morales PA, Stern MP. Prospective analysis of the insulin-resistance syndrome (syndrome X). Diabetes 1992;41:715-722. 50. Reaven GM. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes 1988;37:1595-1607. 51. Reaven GM, Lithell H, Landsberg L. Hypertension and associated metabolic abnormalities--the role of insulin resistance and the sympathoadrenal system. N Engl J Med 1996;334:374-381. 52. Modan M, Halkin H, Fuchs Z, Lusky A, Chetrit A, Segal P, Eshkol A, Almog S, Shefi M. Hyperinsulinemia--a link between glucose intolerance, obesity, hypertension, dyslipoproteinemia, elevated serum uric acid and internal cation imbalance. Diabete Metab 1987;13:375-380. 53. Lind L, Jakobsson S, Lithell H, Wengle B, Ljunghall S. Relation of serum calcium concentration to metabolic risk factors for cardiovascular disease. BMJ 1988;297:960963. 54. Beck-Nielsen H. General characteristics of the insulin resistance syndrome: prevalence and heritability. European Group for the study of Insulin Resistance (EGIR). Drugs 1999;58 Suppl 1:7-10; discussion 75-82. 55. WHO. Definition, diagnosis and classification of diabetes mellitus and its complications: Report of a WHO consultation. Part 1: Diagnosis and classification of diabetes mellitus. Geneva: WHO, 1999: 31-33. 56 56. Byberg L, McKeigue PM, Zethelius B, Lithell HO. Birth weight and the insulin resistance syndrome: association of low birth weight with truncal obesity and raised plasminogen activator inhibitor-1 but not with abdominal obesity or plasma lipid disturbances. Diabetologia 2000;43:54-60. 57. Lindahl B. Lifestyle and health. The role of the insulin resistance syndrome in cardiovascular disease and diabetes. The effects of lifestyle intervention: Umeå University, 1998. 58. Kannel WB. Risk stratification in hypertension: new insights from the Framingham Study. Am J Hypertens 2000;13:3S-10S. 59. Ohya Y, Abe I, Fujii K, Ohmori S, Onaka U, Kobayashi K, Fujishima M. Hyperinsulinemia and left ventricular geometry in a work-site population in Japan. Hypertension 1996;27:729-734. 60. Tomiyama H, Doba N, Kushiro T, Yamashita M, Kanmatsuse K, Kajiwara N, Yoshida H, Hinohara S. The relationship of hyperinsulinemic state to left ventricular hypertrophy, microalbuminuria, and physical fitness in borderline and mild hypertension. Am J Hypertens 1997;10:587-591. 61. Verdecchia P, Reboldi G, Schillaci G, Borgioni C, Ciucci A, Telera MP, Santeusanio F, Porcellati C, Brunetti P. Circulating insulin and insulin growth factor1 are independent determinants of left ventricular mass and geometry in essential hypertension. Circulation 1999;100:1802-1807. 62. Paolisso G, Galderisi M, Tagliamonte MR, de Divitis M, Galzerano D, Petrocelli A, Gualdiero P, de Divitis O, Varricchio M. Myocardial wall thickness and left ventricular geometry in hypertensives. Relationship with insulin. Am J Hypertens 1997;10:1250-1256. 63. Lind L, Andersson PE, Andren B, Hanni A, Lithell HO. Left ventricular hypertrophy in hypertension is associated with the insulin resistance metabolic syndrome. J Hypertens 1995;13:433-438. 64. Phillips RA, Krakoff LR, Dunaif A, Finegood DT, Gorlin R, Shimabukuro S. Relation among left ventricular mass, insulin resistance, and blood pressure in nonobese subjects. J Clin Endocrinol Metab 1998;83:4284-4288. 65. Nuutila P, Maki M, Laine H, Knuuti MJ, Ruotsalainen U, Luotolahti M, Haaparanta M, Solin O, Jula A, Koivisto VA, et al. Insulin action on heart and skeletal muscle glucose uptake in essential hypertension. J Clin Invest 1995;96:1003-1009. 66. Paternostro G, Pagano D, Gnecchi-Ruscone T, Bonser RS, Camici PG. Insulin resistance in patients with cardiac hypertrophy. Cardiovasc Res 1999;42:246-253. 67. Nuutila P, Knuuti MJ, Heinonen OJ, Ruotsalainen U, Teras M, Bergman J, Solin O, Yki Jarvinen H, Voipio Pulkki LM, Wegelius U, et al. Different alterations in the insulin-stimulated glucose uptake in the athlete's heart and skeletal muscle. J Clin Invest 1994;93:2267-2274. 68. Uusitupa M, Siitonen O, Pyorala K, Mustonen J, Voutilainen E, Hersio K, Penttila I. Relationship of blood pressure and left ventricular mass to serum insulin levels in 57 newly diagnosed non-insulin-dependent (type 2) diabetic patients and in nondiabetic subjects. Diabetes Res 1987;4:19-25. 69. Sharp SD, Williams RR. Fasting insulin and left ventricular mass in hypertensives and normotensive controls. Cardiology 1992;81:207-212. 70. Sasson Z, Rasooly Y, Bhesania T, Rasooly I. Insulin resistance is an important determinant of left ventricular mass in the obese. Circulation 1993;88:1431-1436. 71. Kupari M, Koskinen P, Virolainen J. Correlates of left ventricular mass in a population sample aged 36 to 37 years. Focus on lifestyle and salt intake. Circulation 1994;89:1041-1050. 72. Paolisso G, Galzerano D, Gambardella A, Varricchio G, Saccomanno F, d'Amore A, Varricchio M, d'Onofrio F. Left ventricular hypertrophy is associated with a stronger impairment of non-oxidative glucose metabolism in hypertensive patients. Eur J Clin Invest 1995;25:529-533. 73. Paolisso G, Galzerano D, Gambardella A, Gentile S, Lama D, Varricchio M. Low fasting and insulin-mediated intracellular magnesium accumulation in hypertensive patients with left ventricular hypertrophy: role of insulin resistance. J Hum Hypertens 1995;9:199-203. 74. Souza ML, Berardi EO, Gontijo JA, Leme Junior CA, Cavicchio JR, Saad MJ. Insulin resistance and myocardial hypertrophy in the attenuated reduction in mean arterial pressure after a glucose load in hypertensive patients. Braz J Med Biol Res 1995;28:967-972. 75. Costa CH, Batista MC, Moises VA, Kohlmann NB, Ribeiro AB, Zanella MT. Serum insulin levels, 24-hour blood pressure profile, and left ventricular mass in nonobese hypertensive patients. Hypertension 1995;26:1085-1088. 76. Kamide K, Nagano M, Nakano N, Yo Y, Kobayashi R, Rakugi H, Higaki J, Ogihara T. Insulin resistance and cardiovascular complications in patients with essential hypertension. Am J Hypertens 1996;9:1165-1171. 77. Rabkin SW, Dawson KG, Bhaumick B, E OB, Kendler DL. Serum insulin, IGF-I, IGF-II and growth hormone, and left ventricular mass in noninsulin-dependent mellitus. Can J Cardiol 1996;12:264-270. 78. Avignon A, du Cailar G, Ribstein J, Monnier L, Mimran A. [Determinants of the left ventricular mass in obese patients. Influence of lean body mass]. Arch Mal Coeur Vaiss 1997;90:1043-1046. 79. Vetta F, Cicconetti P, Ronzoni S, Rizzo V, Palleschi L, Canarile G, Lupattelli MR, Migliori M, Morelli S, Marigliano V. Hyperinsulinaemia, regional adipose tissue distribution and left ventricular mass in normotensive, elderly, obese subjects. Eur Heart J 1998;19:326-331. 80. Jelenc M, Zemva A, Marn-Pernat A, Zemva Z. Are insulin metabolism and nighttime blood pressure related to left ventricular hypertrophy? Int J Cardiol 1998;63:261265. 58 81. Chen CH, Ting CT, Lin SJ, Hsu TL, Ho SJ, Chou P, Chang MS, O'Connor F, Spurgeon H, Lakatta E, Yin FC. Which arterial and cardiac parameters best predict left ventricular mass? Circulation 1998;98:422-428. 82. Watanabe K, Sekiya M, Tsuruoka T, Funada J, Kameoka H. Effect of insulin resistance on left ventricular hypertrophy and dysfunction in essential hypertension. J Hypertens 1999;17:1153-1160. 83. Rheeder P, Stolk RP, Mosterd A, Pols HA, Hofman A, Grobbee DE. Insulin resistance syndrome and left ventricular mass in an elderly population (The Rotterdam Study). Am J Cardiol 1999;84:233-236, A239. 84. Galvan AQ, Galetta F, Natali A, Muscelli E, Sironi AM, Cini G, Camastra S, Ferrannini E. Insulin resistance and hyperinsulinemia : No independent relation to left ventricular mass in humans. Circulation 2000;102:2233-2238. 85. Ferrara N, Furgi G, Longobardi G, Nicolino A, Acanfora D, Leosco D, Rengo F. Relation between age, left ventricular mass and ventricular arrhythmias in patients with hypertension. J Hum Hypertens 1995;9:581-587. 86. Maron BJ, Wolfson JK, Ciro E, Spirito P. Relation of electrocardiographic abnormalities and patterns of left ventricular hypertrophy identified by 2dimensional echocardiography in patients with hypertrophic cardiomyopathy. Am J Cardiol 1983;51:189-194. 87. Devereux RB, de Simone G, Roman MJ. Relations of left ventricular geometry and function to prognosis in hypertension. Adv Exp Med Biol 1997;432:1-12. 88. Hedstrand H. A study of middle-aged men with particular reference to risk factors for cardiovascular disease. Ups J Med Sci Suppl 1975;19:1-61. 89. WHO. Diabetes mellitus: Report of a WHO study group. Technical Report Series 727. Geneva: WHO, 1985. 90. Becker W, Enghardt H, Robertson A-K. Kostundersökningar i Sverige 1950-1990. Uppsala: Livsmedelsverkets förlag, 1994. 91. Devereux RB, Alonso DR, Lutas EM, Gottlieb GJ, Campo E, Sachs I, Reichek N. Echocardiographic assessment of left ventricular hypertrophy: comparison to necropsy findings. Am J Cardiol 1986;57:450-458. 92. Blackburn H, Keys A, Simonson E, Rautaharju P, Punsar S. The electrocardiogram in population studies. A classification system. Circulation 1960;21:1160-1170. 93. Bazette HC. Analysis of time relations of electrocardiograms. Heart 1920;7:353360. 94. Dahlof B, Devereux RB, Julius S, Kjeldsen SE, Beevers G, de Faire U, Fyhrquist F, Hedner T, Ibsen H, Kristianson K, Lederballe-Pedersen O, Lindholm LH, Nieminen MS, Omvik P, Oparil S, Wedel H. Characteristics of 9194 patients with left ventricular hypertrophy: the LIFE study. Losartan Intervention For Endpoint Reduction in Hypertension. Hypertension 1998;32:989-997. 95. Pollare T, Vessby B, Lithell H. Lipoprotein lipase activity in skeletal muscle is related to insulin sensitivity. Arterioscler Thromb 1991;11:1192-1203. 59 96. Sobey WJ, Beer SF, Carrington CA, Clark PM, Frank BH, Gray IP, Luzio SD, Owens DR, Schneider AE, Siddle K, Temple RC, Hales CN. Sensitive and specific two-site immunoradiometric assays for human insulin, proinsulin, 65-66 split and 3233 split proinsulins. Biochem J 1989;260:535-541. 97. Patlak CS, Blasberg RG. Graphical evaluation of blood-to-brain transfer constants from multiple- time uptake data. Generalizations. J Cereb Blood Flow Metab 1985;5:584590. 98. Ng CK, Soufer R, McNulty PH. Effect of hyperinsulinemia on myocardial fluorine-18-FDG uptake. J Nucl Med 1998;39:379-383. 99. Vessby B, Uusitupa M, Hermansen K, Riccardi G, Rivellese A, Tapsell L, Nalsen C, Berglund L, Louheranta B, Rasmussen B, Calvert G, Maffetone A, Pedersen E, Gustafsson I-B, Storlien L. Substituting dietary saturated for monounsaturated fat impairs insulin sensitivity in healthy men and women - the KANWU study. Diabetologia 2001;in press. 100. Storz P, Doppler H, Wernig A, Pfizenmaier K, Muller G. Cross-talk mechanisms in the development of insulin resistance of skeletal muscle cells. Palmitate rather than tumour necrosis factor inhibits insulin-dependent protein kinase B (PKB)/Akt stimulation and glucose uptake. Eur J Biochem 1999;266:17-25. 101. Desvergne B, Wahli W. Peroxisome Proliferator-Activated Receptors: Nuclear Control of Metabolism. Endocr Rev 1999;20:649-688. 102. Kersten S, Desvergne B, Wahli W. Roles of PPARs in health and disease. Nature 2000;405:421-424. 103. Vessby B, Tengblad S, Lithell H. Insulin sensitivity is related to the fatty acid composition of serum lipids and skeletal muscle phospholipids in 70-year-old men. Diabetologia 1994;37:1044-1050. 104. Glatz JF, Soffers AE, Katan MB. Fatty acid composition of serum cholesteryl esters and erythrocyte membranes as indicators of linoleic acid intake in man. Am J Clin Nutr 1989;49:269-276. 105. Mori TA, Bao DQ, Burke V, Puddey IB, Beilin LJ. Docosahexaenoic acid but not eicosapentaenoic acid lowers ambulatory blood pressure and heart rate in humans. Hypertension 1999;34:253-260. 106. Stanley WC, Lopaschuk GD, Hall JL, McCormack JG. Regulation of myocardial carbohydrate metabolism under normal and ischaemic conditions. Potential for pharmacological interventions. Cardiovasc Res 1997;33:243-257. 107. Nuutila P, Koivisto VA, Knuuti J, Ruotsalainen U, Teras M, Haaparanta M, Bergman J, Solin O, Voipio-Pulkki LM, Wegelius U, et al. Glucose-free fatty acid cycle operates in human heart and skeletal muscle in vivo. J Clin Invest 1992;89:17671774. 108. van der Lee KA, Vork MM, De Vries JE, Willemsen PH, Glatz JF, Reneman RS, Van der Vusse GJ, Van Bilsen M. Long-chain fatty acid-induced changes in gene expression in neonatal cardiac myocytes. J Lipid Res 2000;41:41-47. 60 109. Christe ME, Rodgers RL. Altered glucose and fatty acid oxidation in hearts of the spontaneously hypertensive rat. J Mol Cell Cardiol 1994;26:1371-1375. 110. Massie BM, Schaefer S, Garcia J, McKirnan MD, Schwartz GG, Wisneski JA, Weiner MW, White FC. Myocardial high-energy phosphate and substrate metabolism in swine with moderate left ventricular hypertrophy. Circulation 1995;91:1814-1823. 111. Allard MF, Henning SL, Wambolt RB, Granleese SR, English DR, Lopaschuk GD. Glycogen Metabolism in the Aerobic Hypertrophied Rat Heart. Circulation 1997;96:676-682. 112. Barger PM, Brandt JM, Leone TC, Weinheimer CJ, Kelly DP. Deactivation of peroxisome proliferator-activated receptor-alpha during cardiac hypertrophic growth. J Clin Invest 2000;105:1723-1730. 113. Watanabe K, Fujii H, Takahashi T, Kodama M, Aizawa Y, Ohta Y, Ono T, Hasegawa G, Naito M, Nakajima T, Kamijo Y, Gonzalez FJ, Aoyama T. Constitutive regulation of cardiac fatty acid metabolism through peroxisome proliferatoractivated receptor alpha associated with age-dependent cardiac toxicity. J Biol Chem 2000;275:22293-22299. 114. Murphy MG, Wright V, Ackman RG, Horackova M. Diets enriched in menhaden fish oil, seal oil, or shark liver oil have distinct effects on the lipid and fatty-acid composition of guinea pig heart. Mol Cell Biochem 1997;177:257-269. 115. Brandle M, Jacob R. Effects of a diet rich in Q-3 fatty acids on left ventricular geometry and dynamics in spontaneously hypertensive rats. Eur J Appl Physiol 1990;61:177-181. 116. Litwin SE, Raya TE, Gay RG, Bedotto JB, Bahl JJ, Anderson PG, Goldman S, Bressler R. Chronic inhibition of fatty acid oxidation: new model of diastolic dysfunction. Am J Physiol 1990;258:H51-56. 117. Singh RB, Niaz MA, Sharma JP, Kumar R, Rastogi V, Moshiri M. Randomized, double-blind, placebo-controlled trial of fish oil and mustard oil in patients with suspected acute myocardial infarction: the Indian experiment of infarct survival--4. Cardiovasc Drugs Ther 1997;11:485-491. 118. Dietary supplementation with n-3 polyunsaturated fatty acids and vitamin E after myocardial infarction: results of the GISSI-Prevenzione trial. Gruppo Italiano per lo Studio della Sopravvivenza nell'Infarto miocardico. Lancet 1999;354:447-455. 119. Hense HW, Gneiting B, Muscholl M, Broeckel U, Kuch B, Doering A, Riegger GA, Schunkert H. The associations of body size and body composition with left ventricular mass: impacts for indexation in adults. J Am Coll Cardiol 1998;32:451-457. 120. Andren B, Lind L, Larsson K, Hedenstierna G, Ljunghall S, Lithell H. The influence of body composition on left ventricular mass and other echocardiographic and Doppler measurements in 70-year-old males. Clin Physiol 1995;15:425-433. 121. Julius S. Effect of sympathetic overactivity on cardiovascular prognosis in hypertension. Eur Heart J 1998;19 Suppl F:F14-18. 61 122. Paternostro G, Camici PG, Lammerstma AA, Marinho N, Baliga RR, Kooner JS, Radda GK, Ferrannini E. Cardiac and skeletal muscle insulin resistance in patients with coronary heart disease. A study with positron emission tomography. J Clin Invest 1996;98:2094-2099. 123. Paternostro G, Clarke K, Heath J, Seymour AM, Radda GK. Decreased GLUT-4 mRNA content and insulin-sensitive deoxyglucose uptake show insulin resistance in the hypertensive rat heart. Cardiovasc Res 1995;30:205-211. 124. Holmang A, Yoshida N, Jennische E, Waldenstrom A, Bjorntorp P. The effects of hyperinsulinaemia on myocardial mass, blood pressure regulation and central haemodynamics in rats. Eur J Clin Invest 1996;26:973-978. 125. Kissling G, Rupp H, Malloy L, Jacob R. Alterations in cardiac oxygen consumption under chronic pressure overload. Significance of the isoenzyme pattern of myosin. Basic Res Cardiol 1982;77:255-269. 126. Mercadier JJ, Bouveret P, Gorza L, Schiaffino S, Clark WA, Zak R, Swynghedauw B, Schwartz K. Myosin isoenzymes in normal and hypertrophied human ventricular myocardium. Circ Res 1983;53:52-62. 127. Hill DJ, Milner RD. Insulin as a growth factor. Pediatr Res 1985;19:879-886. 128. Frystyk J, Orskov H. IGF-I, IGF-II, IGF-binding proteins and diabetes. In: Alberti KGMM, Zimmet P, DeFronzo RA, eds. International textbook of diabetes mellitus. 2 ed. Chichester: John Wiley & Sons Ltd, 1997: 417-436. 129. Ito H, Hiroe M, Hirata Y, Tsujino M, Adachi S, Shichiri M, Koike A, Nogami A, Marumo F. Insulin-like growth factor-I induces hypertrophy with enhanced expression of muscle specific genes in cultured rat cardiomyocytes. Circulation 1993;87:1715-1721. 130. Delaughter MC, Taffet GE, Fiorotto ML, Entman ML, Schwartz RJ. Local insulinlike growth factor I expression induces physiologic, then pathologic, cardiac hypertrophy in transgenic mice. Faseb J 1999;13:1923-1929. 131. Ebensperger R, Acevedo E, Melendez J, Corbalan R, Acevedo M, Sapag-Hagar M, Jalil JE, Lavandero S. Selective increase in cardiac IGF-1 in a rat model of ventricular hypertrophy. Biochem Biophys Res Commun 1998;243:20-24. 132. Donohue TJ, Dworkin LD, Lango MN, Fliegner K, Lango RP, Benstein JA, Slater WR, Catanese VM. Induction of myocardial insulin-like growth factor-I gene expression in left ventricular hypertrophy. Circulation 1994;89:799-809. 133. Serneri GG, Modesti PA, Boddi M, Cecioni I, Paniccia R, Coppo M, Galanti G, Simonetti I, Vanni S, Papa L, Bandinelli B, Migliorini A, Modesti A, Maccherini M, Sani G, Toscano M. Cardiac growth factors in human hypertrophy. Relations with myocardial contractility and wall stress. Circ Res 1999;85:57-67. 134. Modesti PA, Vanni S, Bertolozzi I, Cecioni I, Polidori G, Paniccia R, Bandinelli B, Perna A, Liguori P, Boddi M, Galanti G, Serneri GG. Early sequence of cardiac adaptations and growth factor formation in pressure- and volume-overload hypertrophy. Am J Physiol Heart Circ Physiol 2000;279:H976-985. 62 135. Fazio S, Sabatini D, Capaldo B, Vigorito C, Giordano A, Guida R, Pardo F, Biondi B, Sacca L. A preliminary study of growth hormone in the treatment of dilated cardiomyopathy. N Engl J Med 1996;334:809-814. 136. Devereux RB, Roman MJ, Paranicas M, O'Grady MJ, Lee ET, Welty TK, Fabsitz RR, Robbins D, Rhoades ER, Howard BV. Impact of diabetes on cardiac structure and function: the strong heart study. Circulation 2000;101:2271-2276. 137. Felicio JS, Ferreira SR, Plavnik FL, Moises V, Kohlmann O, Jr., Ribeiro AB, Zanella MT. Effect of blood glucose on left ventricular mass in patients with hypertension and type 2 diabetes mellitus. Am J Hypertens 2000;13:1149-1154. 138. Bahr M, Spelleken M, Bock M, von Holtey M, Kiehn R, Eckel J. Acute and chronic effects of troglitazone (CS-045) on isolated rat ventricular cardiomyocytes. Diabetologia 1996;39:766-774. 139. Ghazzi MN, Perez JE, Antonucci TK, Driscoll JH, Huang SM, Faja BW, Whitcomb RW. Cardiac and glycemic benefits of troglitazone treatment in NIDDM. The Troglitazone Study Group. Diabetes 1997;46:433-439. 140. Abel ED, Kaulbach HC, Tian R, Hopkins JC, Duffy J, Doetschman T, Minnemann T, Boers ME, Hadro E, Oberste-Berghaus C, Quist W, Lowell BB, Ingwall JS, Kahn BB. Cardiac hypertrophy with preserved contractile function after selective deletion of GLUT4 from the heart. J Clin Invest 1999;104:1703-1714. 141. Inoguchi T, Battan R, Handler E, Sportsman JR, Heath W, King GL. Preferential elevation of protein kinase C isoform beta II and diacylglycerol levels in the aorta and heart of diabetic rats: differential reversibility to glycemic control by islet cell transplantation. Proc Natl Acad Sci U S A 1992;89:11059-11063. 142. King GL, Wakasaki H. Theoretical mechanisms by which hyperglycemia and insulin resistance could cause cardiovascular diseases in diabetes. Diabetes Care 1999;22 Suppl 3:C31-37. 143. Wakasaki H, Koya D, Schoen FJ, Jirousek MR, Ways DK, Hoit BD, Walsh RA, King GL. Targeted overexpression of protein kinase C beta2 isoform in myocardium causes cardiomyopathy. Proc Natl Acad Sci U S A 1997;94:9320-9325. 144. DeFronzo RA, Cooke CR, Andres R, Faloona GR, Davis PJ. The effect of insulin on renal handling of sodium, potassium, calcium, and phosphate in man. J Clin Invest 1975;55:845-855. 145. Muscelli E, Natali A, Bianchi S, Bigazzi R, Galvan AQ, Sironi AM, Frascerra S, Ciociaro D, Ferrannini E. Effect of insulin on renal sodium and uric acid handling in essential hypertension. Am J Hypertens 1996;9:746-752. 146. McNulty PH, Louard RJ, Deckelbaum LI, Zaret BL, Young LH. Hyperinsulinemia inhibits myocardial protein degradation in patients with cardiovascular disease and insulin resistance. Circulation 1995;92:2151-2156. 147. Rowe JW, Young JB, Minaker KL, Stevens AL, Pallotta J, Landsberg L. Effect of insulin and glucose infusions on sympathetic nervous system activity in normal man. Diabetes 1981;30:219-225. 63 148. Berne C, Fagius J, Pollare T, Hjemdahl P. The sympathetic response to euglycaemic hyperinsulinaemia. Evidence from microelectrode nerve recordings in healthy subjects. Diabetologia 1992;35:873-879. 149. Watanabe K, Sekiya M, Tsuruoka T, Funada J, Kameoka H, Miyagawa M, Kohara K. Relationship between insulin resistance and cardiac sympathetic nervous function in essential hypertension. J Hypertens 1999;17:1161-1168. 150. Simpson P. Norepinephrine-stimulated hypertrophy of cultured rat myocardial cells is an alpha 1 adrenergic response. J Clin Invest 1983;72:732-738. 151. Grassi G, Seravalle G, Mancia G. Left ventricular hypertrophy and sympathetic activity. Adv Exp Med Biol 1997;432:173-179. 152. Julius S, Li Y, Brant D, Krause L, Buda AJ. Neurogenic pressor episodes fail to cause hypertension, but do induce cardiac hypertrophy. Hypertension 1989;13:422429. 153. Laks MM, Morady F, Swan HJ. Myocardial hypertrophy produced by chronic infusion of subhypertensive doses of norepinephrine in the dog. Chest 1973;64:75-78. 154. Kelm M, Schafer S, Mingers S, Heydthausen M, Vogt M, Motz W, Strauer BE. Left ventricular mass is linked to cardiac noradrenaline in normotensive and hypertensive patients. J Hypertens 1996;14:1357-1364. 155. Pikkujamsa SM, Huikuri HV, Airaksinen KE, Rantala AO, Kauma H, Lilja M, Savolainen MJ, Kesaniemi YA. Heart rate variability and baroreflex sensitivity in hypertensive subjects with and without metabolic features of insulin resistance syndrome. Am J Hypertens 1998;11:523-531. 156. Liao D, Sloan RP, Cascio WE, Folsom AR, Liese AD, Evans GW, Cai J, Sharrett AR. Multiple metabolic syndrome is associated with lower heart rate variability. The Atherosclerosis Risk in Communities Study. Diabetes Care 1998;21:2116-2122. 157. Mandawat MK, Wallbridge DR, Pringle SD, Riyami AA, Latif S, Macfarlane PW, Lorimer AR, Cobbe SM. Heart rate variability in left ventricular hypertrophy. Br Heart J 1995;73:139-144. 158. Petretta M, Marciano F, Bianchi V, Migaux ML, Valva G, De Luca N, Salemme L, Berardino S, Bonaduce D. Power spectral analysis of heart period variability in hypertensive patients with left ventricular hypertrophy. Am J Hypertens 1995;8:12061213. 159. Piatti PM, Monti LD, Conti M, Baruffaldi L, Galli L, Phan CV, Guazzini B, Pontiroli AE, Pozza G. Hypertriglyceridemia and hyperinsulinemia are potent inducers of endothelin-1 release in humans. Diabetes 1996;45:316-321. 160. Cardillo C, Nambi SS, Kilcoyne CM, Choucair WK, Katz A, Quon MJ, Panza JA. Insulin stimulates both endothelin and nitric oxide activity in the human forearm. Circulation 1999;100:820-825. 161. Ito H, Hirata Y, Adachi S, Tanaka M, Tsujino M, Koike A, Nogami A, Murumo F, Hiroe M. Endothelin-1 is an autocrine/paracrine factor in the mechanism of angiotensin II-induced hypertrophy in cultured rat cardiomyocytes. J Clin Invest 1993;92:398-403. 64 162. Gaboury CL, Simonson DC, Seely EW, Hollenberg NK, Williams GH. Relation of pressor responsiveness to angiotensin II and insulin resistance in hypertension. J Clin Invest 1994;94:2295-2300. 163. Iyer SN, Katovich MJ. Vascular reactivity to phenylephrine and angiotensin II in hypertensive rats associated with insulin resistance. Clin Exp Hypertens 1996;18:227242. 164. Kobayashi R, Nagano M, Nakamura F, Higaki J, Fujioka Y, Ikegami H, Mikami H, Kawaguchi N, Onishi S, Ogihara T. Role of angiotensin II in high fructose-induced left ventricular hypertrophy in rats. Hypertension 1993;21:1051-1055. 165. Mazzolai L, Pedrazzini T, Nicoud F, Gabbiani G, Brunner HR, Nussberger J. Increased cardiac angiotensin II levels induce right and left ventricular hypertrophy in normotensive mice. Hypertension 2000;35:985-991. 166. Hamawaki M, Coffman TM, Lashus A, Koide M, Zile MR, Oliverio MI, DeFreyte G, Cooper Gt, Carabello BA. Pressure-overload hypertrophy is unabated in mice devoid of AT1A receptors. Am J Physiol 1998;274:H868-873. 167. Harada K, Komuro I, Shiojima I, Hayashi D, Kudoh S, Mizuno T, Kijima K, Matsubara H, Sugaya T, Murakami K, Yazaki Y. Pressure overload induces cardiac hypertrophy in angiotensin II type 1A receptor knockout mice. Circulation 1998;97:1952-1959. 168. Koide M, Carabello BA, Conrad CC, Buckley JM, DeFreyte G, Barnes M, Tomanek RJ, Wei CC, Dell'Italia LJ, Cooper Gt, Zile MR. Hypertrophic response to hemodynamic overload: role of load vs. renin- angiotensin system activation. Am J Physiol 1999;276:H350-358. 169. Weinberg EO, Lee MA, Weigner M, Lindpaintner K, Bishop SP, Benedict CR, Ho KK, Douglas PS, Chafizadeh E, Lorell BH. Angiotensin AT1 receptor inhibition. Effects on hypertrophic remodeling and ACE expression in rats with pressureoverload hypertrophy due to ascending aortic stenosis. Circulation 1997;95:1592-1600. 170. Homcy CJ. Signaling hypertrophy: how many switches, how many wires. Circulation 1998;97:1890-1892. 171. Laakso M, Edelman SV, Brechtel G, Baron AD. Decreased effect of insulin to stimulate skeletal muscle blood flow in obese man. A novel mechanism for insulin resistance. J Clin Invest 1990;85:1844-1852. 172. Mather K, Laakso M, Edelman S, Hook G, Baron A. Evidence for physiological coupling of insulin-mediated glucose metabolism and limb blood flow. Am J Physiol Endocrinol Metab 2000;279:E1264-1270. 173. Lithell H, Lindgarde F, Hellsing K, Lundqvist G, Nygaard E, Vessby B, Saltin B. Body weight, skeletal muscle morphology, and enzyme activities in relation to fasting serum insulin concentration and glucose tolerance in 48-year-old men. Diabetes 1981;30:19-25. 174. Westerbacka J, Uosukainen A, Makimattila S, Schlenzka A, Yki-Jarvinen H. Insulin-induced decrease in large artery stiffness is impaired in uncomplicated type 1 diabetes mellitus. Hypertension 2000;35:1043-1048. 65 175. Andersson PE, Lind L, Andren B, Hanni A, Reneland R, Berne C, Lithell H. Regression of left ventricular wall thickness during ACE-inhibitor treatment of essential hypertension is associated with an increase in insulin mediated skeletal muscle blood flow. Blood Press 1998;7:118-126. 176. Di Bello V, Pedrinelli R, Giorgi D, Bertini A, Talarico L, Caputo MT, Massimiliano B, Dell'Omo G, Paterni M, Giusti C. Ultrasonic Videodensitometric Analysis of Two Different Models of Left Ventricular Hypertrophy : Athlete's Heart and Hypertension. Hypertension 1997;29:937-944. 177. Di Bello V, Giampietro O, Pedrinelli R, Matteucci E, Giorgi D, Bertini A, Bianchi M, Ferdeghini M, Boldrini E, Dell'Omo G, Paterni M, Giusti C. Can insulin action induce myocardial texture alterations in essential hypertension? Am J Hypertens 1999;12:283-290. 178. Mizushige K, Yao L, Noma T, Kiyomoto H, Yu Y, Hosomi N, Ohmori K, Matsuo H. Alteration in left ventricular diastolic filling and accumulation of myocardial collagen at insulin-resistant prediabetic stage of a type II diabetic rat model. Circulation 2000;101:899-907. 179. Haenni A, Reneland R, Lind L, Lithell H. Serum aldosterone changes during hyperinsulinemia are correlated to body mass index and insulin sensitivity in patients with essential hypertension. J Hypertens 2001;19:in press. 180. Brilla CG, Matsubara LS, Weber KT. Antifibrotic effects of spironolactone in preventing myocardial fibrosis in systemic arterial hypertension. Am J Cardiol 1993;71:12A-16A. 181. Lacka K, Piszczek I, Kosowicz J, Gembicki M. Echocardiographic abnormalities in acromegalic patients. Exp Clin Endocrinol 1988;91:212-216. 182. Santos AD, Miller RP, Mathew PK, Wallace WA, Cave WT, Jr., Hinojosa L. Echocardiographic characterization of the reversible cardiomyopathy of hypothyroidism. Am J Med 1980;68:675-682. 183. Gans RO, Bilo HJ, von Maarschalkerweerd WW, Heine RJ, Nauta JJ, Donker AJ. Exogenous insulin augments in healthy volunteers the cardiovascular reactivity to noradrenaline but not to angiotensin II. J Clin Invest 1991;88:512-518. 184. Lakatta EG. Similar myocardial effects of aging and hypertension. Eur Heart J 1990;11 Suppl G:29-38. 185. Post WS, Larson MG, Levy D. Impact of left ventricular structure on the incidence of hypertension. The Framingham Heart Study. Circulation 1994;90:179-185. 186. van Hooft IM, Grobbee DE, Waal-Manning HJ, Hofman A. Hemodynamic characteristics of the early phase of primary hypertension. The Dutch Hypertension and Offspring Study. Circulation 1993;87:1100-1106. 187. Julius S, Gudbrandsson T, Jamerson K, Andersson O. The interconnection between sympathetics, microcirculation, and insulin resistance in hypertension. Blood Press 1992;1:9-19. 188. Pfeffer MA, Braunwald E. Ventricular remodeling after myocardial infarction. Experimental observations and clinical implications. Circulation 1990;81:1161-1172. 66 189. Sandoe E, Sigurd B. Bundle-branch blocks. Arrhythmia. 1 ed. St. Gallen: Fachmed AG. Verlag fur Fachmedien., 1984: 271-298. 190. Sasson Z, Rakowski H, Wigle ED. Hypertrophic cardiomyopathy. Cardiol Clin 1988;6:233-288. 191. Sokolow M, McIlroy MB, Cheitlin MD. Hypertrophic cardiomyopathy. Clinical Cardiology. 5 ed. Connecticut: Appleton & Lange, 1990: 549-557. 192. Messerli FH, Ventura HO, Elizardi DJ, Dunn FG, Frohlich ED. Hypertension and sudden death. Increased ventricular ectopic activity in left ventricular hypertrophy. Am J Med 1984;77:18-22. 193. Gonzalez Fernandez RA, Rivera M, Rodriguez PJ, Fernandez Martinez J, Soltero LH, Diaz LM, Lugo JE. Prevalence of ectopic ventricular activity after left ventricular mass regression. Am J Hypertens 1993;6:308-313. 194. Carroll JD, Carroll EP. Diastolic function in coronary artery disease. Herz 1991;16:1-12. 195. Dawson JR, Gibson DG. Left ventricular filling and early diastolic function at rest and during angina in patients with coronary artery disease. Br Heart J 1989;61:248-257. 196. Appleton CP, Hatle LK. The natural history of left ventricular filling abnormalities: assessment by two-dimensional and Doppler echocardiography. Echocardiography 1992;4:437-457. 197. Merlo J, Lindblad U, Pessah-Rasmussen H, Hedblad B, Rastam J, Isacsson SO, Janzon L, Rastam L. Comparison of different procedures to identify probable cases of myocardial infarction and stroke in two Swedish prospective cohort studies using local and national routine registers. Eur J Epidemiol 2000;16:235-243. 198. Agabiti-Rosei E. Hypertension and left ventricular hypertrophy. In: Hansson L, ed. Hypertension and concomitant disorders. London: Science Press Ltd, 1997: 67-77. 199. Pollare T, Lithell H, Selinus I, Berne C. Application of prazosin is associated with an increase of insulin sensitivity in obese patients with hypertension. Diabetologia 1988;31:415-420. 200. Andersson PE, Johansson J, Berne C, Lithell H. Effects of selective alfa 1 and beta 1-adrenoreceptor blockade on lipoprotein and carbohydrate metabolism in hypertensive subjects, with special emphasis on insulin sensitivity. J Hum Hypertens 1994;8:219-226. 201. Andersson PE, Lithell H. Metabolic effects of doxazosin and enalapril in hypertriglyceridemic, hypertensive men. Relationship to changes in skeletal muscle blood flow. Am J Hypertens 1996;9:323-333. 202. Lind L, Berne C, Pollare T, Lithell H. Metabolic effects of isradipine as monotherapy or in combination with pindolol during long-term antihypertensive treatment. J Intern Med 1994;236:37-42. 67 203. Pollare T, Lithell H, Morlin C, Prantare H, Hvarfner A, Ljunghall S. Metabolic effects of diltiazem and atenolol: results from a randomized, double-blind study with parallel groups. J Hypertens 1989;7:551-559. 204. Pollare T, Lithell H, Berne C. A comparison of the effects of hydrochlorothiazide and captopril on glucose and lipid metabolism in patients with hypertension. N Engl J Med 1989;321:868-873. 205. Haenni A, Andersson PE, Lind L, Berne C, Lithell H. Electrolyte changes and metabolic effects of lisinopril/bendrofluazide treatment. Results from a randomized, double-blind study with parallel groups. Am J Hypertens 1994;7:615-622. 206. Reneland R, Andersson PE, Haenni A, Lithell H. Metabolic effects of long-term angiotensin-converting enzyme inhibition with fosinopril in patients with essential hypertension: relationship to angiotensin-converting enzyme inhibition. Eur J Clin Pharmacol 1994;46:431-436. 207. Reneland R, Alvarez E, Andersson PE, Haenni A, Byberg L, Lithell H. Induction of insulin resistance by beta-blockade but not ACE-inhibition: long-term treatment with atenolol or trandolapril. J Hum Hypertens 2000;14:175-180. 208. Pollare T, Lithell H, Selinus I, Berne C. Sensitivity to insulin during treatment with atenolol and metoprolol: a randomised, double blind study of effects on carbohydrate and lipoprotein metabolism in hypertensive patients. BMJ 1989;298:1152-1157. 209. Haenni A, Lithell H. Treatment with a beta-blocker with beta 2-agonism improves glucose and lipid metabolism in essential hypertension. Metabolism 1994;43:455-461. 210. Muscelli E, Camastra S, Catalano C, Galvan AQ, Ciociaro D, Baldi S, Ferrannini E. Metabolic and cardiovascular assessment in moderate obesity: effect of weight loss. J Clin Endocrinol Metab 1997;82:2937-2943. 211. Thurmann PA, Kenedi P, Schmidt A, Harder S, Rietbrock N. Influence of the angiotensin II antagonist valsartan on left ventricular hypertrophy in patients with essential hypertension. Circulation 1998;98:2037-2042. 212. Tedesco MA, Ratti G, Aquino D, Limongelli G, di Salvo G, Mennella S, Galzerano D, Iarussi D, Iacono A. Effects of losartan on hypertension and left ventricular mass: a long- term study. J Hum Hypertens 1998;12:505-510. 213. Kuperstein R, Sasson Z. Effects of antihypertensive therapy on glucose and insulin metabolism and on left ventricular mass : A randomized, double-blind, controlled study of 21 obese hypertensives. Circulation 2000;102:1802-1806. 68