Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Ring-closing metathesis wikipedia , lookup
Metal carbonyl wikipedia , lookup
Evolution of metal ions in biological systems wikipedia , lookup
Metalloprotein wikipedia , lookup
Hydroformylation wikipedia , lookup
Jahn–Teller effect wikipedia , lookup
Stille reaction wikipedia , lookup
Spin crossover wikipedia , lookup
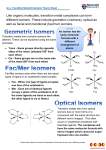
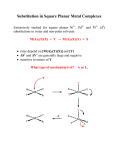
Inorganic reaction mechanisms Substitution reactions Thermodynamic factors Kinetic factors Energy profile Activated Complex Ea, ΔH#, ΔS# Kinetics Y + M―X Reactants Thermodynamics Products M―Y + X Thermodynamics Both kinetics and thermodynamics are important in the assesment of a reaction. This is because some thermodynamically feasible reactions may be kinetically hindered Ligand substitution reactions will be used to illustrate these concepts: Consider the reaction: Y + M ― X → M ― Y + X This is a typical substitution reaction Y = entering group X = leaving group The simplest illustration is a complex formation reaction: e.g. [Co(H2O)6]2+ + Cl- → [CoCl(H2O)5]+ + H2O Aquo complexes Fundamentally metal ions dissolved in water are complexed ÎAquo complexes Î [M(H2O)x]n+ Formation of coordination compounds from an aqueous medium Î requires displacement of aquo ligands by other ligands. Î Several geometries are possible: Typical examples of aquo compounds Î e.g. Co(II) [Co(H2O)6]2+ Octahedral [Co(H2O)4]2+ Terahedral In reality the value of x varies Î due to extensive hydrogen bonding around the coordination sphere of M Spectra and magnetic properties Î similar to hydrated salts of noncoordinating anions Î Co(ClO4)2.6H2O; Co(NO3)2.6H2O Aquo complexes Predominantly [Co(H2O)6]2+ Îoctahedral Also general for the 1st transition series Î [M(H2O)x]n+; n=+2 or +3 Note: Cr(II); Mn(III) and Cu(II) show distortions in the octahedral geometry due to Jahn-Teller effects 2nd and 3rd TS are much less certain Î octahedral probably Î higher coordination also possible Most M(H2O)6 exchange rapidly Î can easily be demonstrated by isotopic labeling with 18O enriched H2O. Aqua ions are more or less acidic Î dissociate as follows: [M(H2O)x]n+ → [M(H2O)x-1(OH)]n-1 + H+ e.g. [Co(NH3)5(H2O)]3+ → [M(NH3)5(OH)]2+ + H+ Formation constants Formation constant: the strength of a ligand relative to the strength of the solvent molecules (usually H2O) as a ligand. [Fe(OH2)6]3+(aq) + SCN-(aq) In dilute solutions [H2O] Î constant; [Fe(SCN)(OH2)5]2+(aq) + H2O(l) Kf [Fe(SCN)(OH2)52+] [Fe(OH2)6 3+][SCN-] Step-wise formation constant: formation constant of each solvent replacement stage. Kf1 ; Kf2 ........Kfn Overall formation constant: product of the step-wise formation constants. βn = Kf1Kf2........Kfn Successive formation constants [ML] Kf1 M + L ML [M][L] ML + L ML2 MLn-1 + L M + nL MLn MLn Kf2 Kfn βn ML [ML][L] [MLn] [MLn-1][L] [MLn] [M][L]n The inverse of each Kf is the dissociation constant Kd Î: M + L [ML2 ] Kd1 [M][L] 1 [ML] Kf1 Trends in successive formation constants The general trend Î Kf1 > Kf2 Formation constants of Ni(II) …… Kfn-1 > Kfn. 2+ ammines, [Ni(NH ) (H O) ] 3 n 2 6-n This trend is a result of the n Kf Kn/Kn-1 Kn/Kn-1 sequential decrease in number of Exp Stat H2O to be replaced. Situations do arise where Kfn > Kfn-1 1 525 Generally there are 2 reasons to 2 148 0.28 0.42 account for anomalies in the 3 45.7 0.31 0.53 trends of successive Kf values: 1 Î due to a major change in 4 13.2 0.29 0.56 electronic structure of the 4.7 0.35 0.53 complex, e.g. moving from a high 5 spin (due to weak field H2O) to a 6 1.1 0.20 0.42 low spin complex. 2 Î due to a major structural change e.g. from an octahedral to a tetrahedral or square planar geometry Î characteristic of some halo complexes Q. Consider the formation constants of the following Fe(II) complexes: Justify the observed trend? [Fe(bipy)(H2O)4]2+: [Fe(bipy)2(H2O)2]2+: [Fe(bipy)3]2+: Kf1: 4.2 Kf2: 3.7 Kf3: 9.3 Kf3: [Fe(bipy)3]2+ >> Kf2: [Fe(bipy)2(H2O)2]2+: The reason is due to a major electronic shift Î from a high spin (due to weak field H2O) t2g4eg2 (LFSE Δo = 0.4) config Î to a low spin t2g6 config (LFSE Δo = 2.4). Large increase Δo. Therefore [Fe(bipy)3]2+ is more stable than [Fe(bipy)2(H2O)2]2+ Q. Justify the following observation in the successive formation constants for complexes of cadmium with Br-: [Cd(Br)(H2O)5]+: Kf1: 36.3 [Cd(Br)2(H2O)4]: Kf2: 3.47 [Cd(Br)3(H2O)3]-: Kf3: 1.15 [Cd(Br)4]2-: Kf4: 2.34 The anomaly here is that Kf4: > Kf3: The reason may not be electronic since both H2O and Br- are considered weak field ligands Î The reason is due to a major structural shift from an octahedral [Cd(Br)3(H2O)3]- configuration Î to a tetrahedral [Cd(Br)4]2- geometry with the simultaneous expulsion of 3 molecules of water from a restricted geometry Most halo complexes of M2+ have tetrahedral or square geometry Irving-Williams series 10 log Kf The Irving-Williams series summarises the relative 8 stabilities of complexes formed by M2+ ions. The series reflect a combination of electrostatic 6 effects and LFSE. For the series Ba2+ < Sr2+ < Ca2+ < Mg2+ the 4 observed trend is purely electrostatic. For the TMs in addition to electrostatic effects , the 2 high values of Kf is due to additional stability from 0 LFSE. Ba Sr Ca Mg Mn Fe Co Ni Cu Zn In terms of ionic radii Î RECALL: Δo is Mn2+ > Fe2+ > Co2+ > Ni2+ > Cu2+ > Zn2+. highest at d3 and d8 But, Generally for strong field ligands the observed order is: Mn2+ < Fe2+ < Co2+ < Ni2+ < Cu2+ > Zn2+. The additional stability of d9 Cu(II) is due to the influence of JahnTeller distortion Î results in the strong binding of the 4 planar ligands in tetragonally distorted Cu(II) complexes Î higher Kf values. Other thermodynamic factors In addition to the foregone discussion the following thermodynamic factors are also important to the stability of TM complexes: The chelate effect: this is largely an entropy effect Î represents the greater stability of a complex containing a chelated polydentate ligand compared with the equivalent complex with an analogous monodentate ligands. H O H2O N N N N Ru N 2 OH2 Ru N H2O H2O OH2 Steric Effect: The size of ligands are important in determining the most stable configuration Î e.g. octahedral vs. tetrahedral Electron delocalisation: This is important in complexes containing chelated ring structures. The empty ring π* orbitals act as electron sinks that drain electrons from the full metal t2g non-bonding orbitals. Labile and non-labile complexes The rate at which one complex inter-converts into another is determined by the height of the activation barrier between the two: Labile complexes: A complex with a half life of the order of milliseconds; e.g. [Ni(H2O)6]2+ is said to be labile Non-labile or inert complex has half-life of the order of minutes; e.g [Co(HH3)5(H2O)]3+ Factors affecting lability of complexes: Complexes with no stabilising LFSE or chelate effects are very labile Complexes of small metal ions are less labile due to greater M-L bonds Complexes of M(III) ions are less labile than those of M(II) ions Complexes of d3 and low spin d6 config are non-labile Î high LFSE Chelate complexes of metal ions with high LFSE (e.g. [Fe(phen)3]2+) are very stable Complexes of d10 ions (zero LFSE: Zn2+, Cd2+, Hg2+) are generally labile Classification of mechanisms Associative (A); Dissociative (D); Interchange (Ia or Id) Classification of mechanisms Dissociative: 5-coordinate intermediate D Interchange Id or Ia MLnX + Y Associative: 7-coordinate intermediate A MLnY + X MLn+ X + Y MLnX + Y MLnXY MLnY+X MLnX + Y MLnY+X Energy profile for Associative Energy profile for Dissociative mechanism: Intermediate has lower mechanism: Intermediate has higher coordination number than reactant. coordination number than reactant. Classification of mechanisms No intermediate With intermediate Intermediate Î occurs at a local E minimum Î may be isolated Transition state occurs at an E maximum Î cannot be isolated Interchange I In most TM substitution reactions Î bond formation between M + Y happens concurrently with bond cleavage between M + X Î Interchange mechanism: MLnX + Y Y……MLn…….X MLnY + X For an interchange mechanism Î there is no intermediate formed, but various TS may be possible: Hence there are 2 types of I: Dissociative Id: bond breakage dominates over formation and Associative Ia: bond formation dominates over breakage It is often difficult to distinguish between: A and Ia D and Id Ia and Id Substitution reactions in square planar complexes Referring to complexes of d8 ion TM Under a large crystal field: Rh(I) Ir(I) Pt(II) Pd(II) Au(III) Steric Effects Remember 4-coord. Ni(II) can be tetrahedral or sq planar (why?) Nucleophilic subst. in sq. planar complexes Î usually A or Ia Î the rate of the reaction is dependent on the nature of the entering group Y But steric crowding at the reaction centre by bulky groupsÎ usually inhibits A or Ia reactions Î results in D or Id mechanism k values for cis-[PtClL(PEt3)2] + H2O → cis-[Pt(H2O)L(PEt3)2] L= k/s-1 pyridine 8x 10-2 2-Mepy 2x 10-4 2,6-diMepy 1x 10-6 The rate ↓ with ↑ in steric bulk around N→M bond Î The Me groups hinder the attack by H2O Me PEt3 M Cl PEt3 Stereochemistry Substitution reactions at square complexes are stereospecific Î the original geometry of the complex is preserved: cis reactants lead to cis products trans reactants lead to trans products most of these are 16 electron complexes Î useful in catalysis Î oxidative addition-reductive elimination (16 e↔18 e) chemistry Y trans reactant X 5 coord TS via A mech trans product The kinetic trans-effect and thermodynamic trans-influence Since the substitution reactions of sq. planar TM complexes are usually A or Ia, what factors determine the nature of the TS and Why a 5 coordinate (trigonal bipyramidal or square pyramidal) TS? Which ligand leaves is determined by the ligand trans to it Î trans influence Î trans effect σ-donor ligands Î Ligands capable of contributing more electron density to the shared orbital between itself and the trans ligand, thereby weakening the bond to the leaving group Î trans influence π-acceptor ligands Î drain away electrons in A or Ia substitution reactions Î TS characterised by high electron density at the metal center due to a 5-coordinate intermediate Î trans effect The kinetic trans-effect and thermodynamic trans-influence trans influence and trans-effect are related Î weakening the bond to the trans-ligand and increasing the rate of substitution, Note: there is NO close correlation between the relative magnitudes of the two phenomena Why a 5 coordinate (trigonal bipyramidal or square pyramidal) TS? L2 The leaving group (X), the entering group (Y) and the trans ligand L3 all lie in the same plane. The TS is stabilised if L3 is a good π acceptor L3 dxy ligand such as CO. The three are able to communicate electronically via L1 the π-bonding orbitals. good π acceptor ligand e.g. CO Remember the influence of π bonding on the crystal field splitting Δ X Y The kinetic trans-effect and thermodynamic trans-influence The trans effect The trans effect states that the bond holding a group trans to an electronegative group is weakened. This trans group is the first to be removed in a substitution reaction Kinetic Studies to determine trans-effect of ligands L k (s-1) C2H4, CO too fast P(OMe)3 10.3 PEt3 6.6 PPh3 3.1 Me2SO 0.0082 Et2S 0.0024 Me2S 0.0015 NH3 6.3x10-6 H2O 8.0x10-8 Trans-influence from x-ray crystallography Trans-influence from vibrational spectroscopy L ν Pt-Cl (cm-1) L ν Pt-Cl (cm-1) L ν Pd-Cl (cm-1) CO 322 py 336 py 342 SMe2 310 SMe2 336 Cl 330 C2H4 309 COD 327 NH3 327 SEt2 307 SEt2 324 EtSCH2CH2SEt 323 PPh3 279 NH3 321 H2NCH2CH2NH2 307 PEt3 271 PPh3 305 PCH3 297 PEt3 294 ❑ Ligands with high trans-influence have high trans-effect. ❑ Exceptions to this rule include CO, C2H4, and DMSO. ❑ Frequency order for Rh(III)-Cl: H- >PR3 >Me- >CO >I- >Br- >Cl-. ❑ For octahedral complexes, trans-influence parallels trans-effect. Trans effect vs. trans influence ❑ The trans effect is the influence of a ligand (L) on the rate of substitution of the ligand trans to it (X). It is a kinetic effect (ground and transition states) H- = CH3- = CN- = C2H4 = CO >> PR3 = SR2 > NO2 = SCN- = I-> Br-> Cl-> py > RNH2 = NH3 > OH- > H2O ❑ The trans influence is the extent to which a ligand (L) weakens the bond that is trans to itself. It is a thermodynamic effect (ground state). Apply primarily to the leaving group R3Si- = R- = H- > PEt3 > PMe2Ph > PPh3 > P(OPh)3 = CN- > SEt2> Et2NH > py > OSMe2 = C2H4 = CO > Cl❑ The trans effect is often the manifestation of the trans influence although there are some ligands (DMSO, CO, C2H4) which do not show significant trans-influences yet show strong effects Theory on trans influence ❑ Grinberg Polarization Theory (1935): M→ L and then L→ M induced dipole results in repulsion of electrons in X Î weakening of M-X bond. ❑ Chatt/Orgel theory of back-bonding: Stabilisation of a 5 coordinate (trigonal bipyramidal) activated complex. Y X Q. Does back-bonding generally indicate stronger trans effect? YES………. ……………and NO Q. Given Pt(NH3)4]2+, [PtCl4]2-, HCl and NH3 suggest synthetic routes to a) cis-[PtCl2(NH3)2] b) trans-[PtCl2(NH3)2] H3N NH3 HCl Pt NH3 H3N Cl Cl NH3 Cl NH3 Cl HCl Pt H3N Cl Pt Cl H3N NH3 Pt Cl Cl H3N Cl Pt Cl NH3 Cl NH3 NH3 Pt Cl NH3