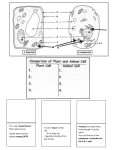
Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Chem Biol Drug Des 2011; 78: 199–210 ª 2011 John Wiley & Sons A/S doi: 10.1111/j.1747-0285.2011.01135.x Research Article Chemogenetic Analysis of Human Protein Methyltransferases Victoria M. Richon1,*, Danielle Johnston1, Christopher J. Sneeringer 1, Lei Jin1, Christina R. Majer1, Keith Elliston2, L. Fred Jerva2, Margaret Porter Scott1 and Robert A. Copeland1,* 1 Epizyme, Inc., 840 Memorial Drive, Cambridge, MA 02139, USA Genstruct Inc., One Alewife Center, Cambridge, MA 02140, USA *Corresponding authors: Victoria M. Richon, [email protected] or Robert A. Copeland, [email protected] 2 A survey of the human genome was performed to understand the constituency of protein methyltransferases (both protein arginine and lysine methyltransferases) and the relatedness of their catalytic domains. We identified 51 protein lysine methyltransferase proteins based on similarity to the canonical Drosophila Su(var)3-9, enhancer of zeste (E(z)), and trithorax (trx) domain. Disruptor of telomeric silencing-1-like, a known protein lysine methyltransferase, did not fit within the protein lysine methyltransferase family, but did group with the protein arginine methyltransferases, along with 44 other proteins, including the METTL and NOP2 ⁄ Sun domain family proteins. We show that a representative METTL, METTL11A, demonstrates catalytic activity as a histone methyltransferase. We also solved the co-crystal structures of disruptor of telomeric silencing-1-like with S-adenosylmethionine and S-adenosylhomocysteine bound in its active site. The conformation of both ligands is virtually identical to that found in known protein arginine methyltransferases, METTL and NOP2 ⁄ Sun domain family proteins and is distinct from that seen in the Drosophila Su(var)3-9, enhancer of zeste (E(z)), and trithorax (trx) domain protein lysine methyltransferases. We have developed biochemical assays for 11 members of the protein methyltransferase target class and have profiled the affinity of three ligands for these enzymes: the common methyl-donating substrate S-adenosylmethionine; the common reaction product S-adenosylhomocysteine; and the natural product sinefungin. The affinity of each of these ligands is mapped onto the family trees of the protein lysine methyltransferases and protein arginine methyltransferases to reveal patterns of ligand recognition by these enzymes. Key words: chemical biology, cheminformatics, drug discovery, enzyme structure, molecular recognition Abbreviations: DOT1L, disruptor of telomeric silencing-1-like; HGNC, HUGO Gene Nomenclature Committee; IPTG, isopropyl b-D-1thiogalactopyranoside; NSUN, NOP2 ⁄ Sun domain family; PKMT, protein lysine methyltransferase; PMT, protein methyltransferase; PRDM, (PRDIBF1 and RIZ homology) domain; PRMT, protein arginine methyltransferase; PSI-BLAST, position-specific iterated basic local alignment search tool; SAH, S-adenosylhomocysteine; SAM, S-adenosylmethionine; SETdomain, Drosophila Su(var)3-9, enhancer of zeste (E(z)), and trithorax (trx) domain. Received 16 March 2011, revised 23 April 2011 and accepted for publication 27 April 2011 Posttranslational modifications of proteins provide powerful mechanisms for selective and stringent temporal control of many cellular processes (1). For example, the timing and extent of protein maturation and degradation are often controlled by proteolytic processing of precursor proteins and by a combination of covalent ubiquitin modification and proteolysis. Also, cellular signaling pathways, which communicate binding events at cell surface receptors to nuclear-localized transcription events, are tightly controlled by specific protein phosphorylation and dephosphorylation; these processes are catalyzed respectively by protein kinases and protein phosphatases (1). In a like manner, other posttranslational modifications of proteins, catalyzed by specific enzymes, confer remarkable levels of specificity to critical biological systems. Not surprisingly, dysregulation of the enzymes that perform these critical functions often leads to pathologies such as cancer, inflammation, and other serious human diseases. Regulation of gene transcription is yet another example of a critical biological process that is controlled in large part by enzyme-catalyzed, covalent modification of proteins and nucleic acids (2). For example, DNA methyltransferase-mediated modification of the 5position of cytosine, within the CpG dinucleotide sites of genes, plays a direct role in silencing of gene transcription (3). The DNA of chromosomes is found in complex with a number of proteins, most notably the histone proteins; together, the DNA–histone complex constitutes chromatin, the main component of chromosomes (4). Conformational remodeling of chromatin regulates gene transcription and its silencing. This conformational remodeling has been shown to result from specific posttranslational modifications of the histone proteins of the chromatin complex (5). The types of posttranslational modification of histones known to contribute to the regulation of gene transcription include acetylation, methylation, phosphorylation, and ubiquitinylation (6). Recently, our group has reviewed the enzymes that methylate histones and other cellular 199 Richon et al. proteins, the protein methyltransferases (PMTs) (6), and has made the case that the enzymes of this class represent particularly attractive targets for drug discovery efforts, because of the disease association and druggability of these proteins. The enzymes of the PMT class can be subdivided into two major families: the protein lysine methyltransferases (PKMTs) and the protein arginine methyltransferases (PRMTs). These two families are distinguished from one another not only on the basis of the methyl-accepting amino acid side chain recognized but also on the basis of the primary sequence of their catalytic domains and their three-dimensional structures (6). All of the lysine methyltransferases, except one, contain a ca. 130amino acid domain – referred to as the Drosophila Su(var)3-9, enhancer of zeste (E(z)), and trithorax (trx) domain (SET-domain) – that constitutes the active site domain of these enzymes (7). The one exception to this structural generality is the enzyme disruptor of telomeric silencing-1-like (DOT1L), a PKMT that does not contain a SET-domain (8). To exploit most effectively the PMT target class for drug discovery and chemical biological studies, it is critical to understand the constituency of the class within an organism of interest (in the case of drug discovery, this would usually mean humans) and the structural relatedness of these proteins to one another. In terms of drug discovery and chemical biology, it is the structural relatedness of the active site domains in particular, rather than the overall structures of the full-length proteins, that is most germane. While several groups have reported alignments of known members of the PKMT and PRMT families, no systematic analysis of the constituency of PMTs encoded by the human genome has yet been reported (7,9–13). In this study, we report the results of such a systematic survey of the human genome for PKMT- and PRMT-related enzymes on the basis of amino acid sequence alignments of the active site domains of these enzymes. We find that both families are composed of >40 putative enzymes and that each family can be further subdivided into discrete clusters of related proteins. Interestingly, the non-SET-domain PKMT, DOT1L, could not be rationally included within the PKMT family, but could be reasonably associated with the PRMT family. This seemingly anomalous association of DOT1L with the PRMTs, based on amino acid sequence, is consistent with crystallographic data comparing the conformation of the substrate and product ligands (S-adenosylmethionine, SAM, and S-adenosylhomocysteine, SAH) bound to SETdomain PKMTs, PRMTs, and DOT1L. The implications of the data generated here for drug discovery and chemical biology studies are also discussed. Methods Sequence alignment and family tree construction Amino acid sequence alignments and family tree construction were performed by Genstruct, Inc. (Cambridge, MA, USA). Amino acid sequences were obtained from annotated protein sequence files obtained from the NCBI reference proteins database, RefSeq (http://www.ncbi.nlm.nih.gov/RefSeq/) or from Swiss-Prot (http:// www.expasy.ch/sprot/) (14). Rigorous identification of family member sequences was accomplished using position-specific iterated 200 basic local alignment search tool (PSI-BLAST) (http://blast.ncbi.nlm.nih.gov). Multiple sequence alignments for each family group were generated using the CLUSTALW2 software (http://www.clustal.org). Visualization and manual editing of alignments were performed using GeneDoc (http://www.nrbsc.org/gfx/genedoc/). Interfamily sequence alignment was accomplished using a 'sparse alignment' approach [S. Smith (2009), personal communication] that aligns representative members of each family using CLUSTALW2 to define appropriate anchor points for the interfamily alignments. The final alignments were generated by manually adding additional proteins to the alignment, ensuring alignment of key anchor points identified in the sparse alignments. The final multiple sequence alignments used for the phylogenetic analysis are illustrated in the supplemental materials as Figures S1 and S2 for the PKMTs and the PRMTs (plus DOT1L), respectively. The phylogenetic analysis of the aligned sequences was accomplished using the PHYLIP package developed at the University of Washington (http://evolution.genetics.washington.edu/phylip.html). To facilitate bootstrap analysis of the data, the SEQBOOT program was used to create 250 randomized data representations of the input alignment for analysis. The PROTDIST program was used to calculate pairwise distance measures between the sequences within the alignments using the BLOSUM45 matrix, and the program NEIGHBOR was used to construct phylogenetic trees from the distance data using the neighbor joining method. Consensus trees were built using the CONSENSE utility and uprooted trees were drawn using the DRAWTREE program. Determination of PMT enzymatic activity Recombinant protein production and activity assays were performed for the 11 PMTs as described elsewhere (15; S. R. Daigle, E. J. Olhava, C. A. Therkelsen, C. R. Majer, C. J. Sneeringer, J. Song, D. Johnston, M. Porter Scott, J. J. Smith, Y. Xiao, L. Jin, K. W. Kuntz, R. Chesworth, M. P. Moyer, K. M. Bernt, S. A. Armstrong, R. A. Copeland, V. M. Richon, R. M. Pollock, unpublished data). Determination of inhibitor IC50 values IC50 values for enzymes in the protein methyltransferase panel were determined under balanced assay conditions with both SAM and protein ⁄ peptide substrate present at concentrations equal to their respective KM values. Where a peptide was used as methyl-accepting substrate, the peptide is referred to here by the histone and amino acid residue numbers that it represents. For example, peptide H3:16–30 refers to a peptide representing histone H3 amino acid residues 16 through 30. Flag and his-tagged CARM1 (2–585) purified from 293 cells was assayed at a final concentration of 0.25 nM against a biotinylated peptide corresponding to histone H3:16–30 (R26-Me1). E. coli-expressed EHMT2 (913–1193) was assayed at a final concentration of 0.1 nM against a biotinylated peptide corresponding to H3:1–15. Full-length EZH1 and EZH2 were expressed as four-component complexes in insect cells and purified as described elsewhere (15). EZH1 (4 nM) and EZH2 (4 nM) four-component complexes were assayed against a biotinylated peptide corresponding to histone H3:21–44. Insect expressed full-length PRMT1 was assayed at a final concentration of 0.75 nM against biotinylated Chem Biol Drug Des 2011; 78: 199–210 Chemogenetic Analysis of Human Protein Methyltransferases peptide corresponding to H4:36–50. Flag-tagged full-length PRMT5 purified from 293 cells was assayed at a final concentration of 1.5 nM against a biotinylated peptide corresponding to H4:1–15. E. coli-expressed full-length PRMT8 was assayed at a final concentration of 1.5 nM against a biotinylated peptide corresponding to H4:31–45. Full-length SETD7 purified from E. coli was assayed at a final concentration of 1 nM against a biotinylated peptide corresponding to H3:1–15. Flag and his-tagged full-length WHSC1 was purified from 293 cells and assayed at a final concentration of 2.5 nM against avian oligonucleosomes. Determination of METTL11A enzymatic activity Histone H3 and Histone H4 (NEB, Ipswich, MA, USA) were diluted separately in assay buffer (20 mM Tris, pH 8.0, 0.002% Tween20, 0.005% bovine skin gelatin, and 0.5 mM DTT) to a final concentration of 200 nM in a 96-well plate (25 lL each well). To monitor the reaction, 300 nM S-[methyl-3H]-adenosyl-L-methionine (80 Ci ⁄ mmol, from ARC) together with 200 nM cold SAM (total SAM concentration = 500 nM) was used in each reaction. For initial determination of activity, METTL11A (Sino Biologics, Beijing, China) was added to initiate the reaction (25 lL each well for final concentration of 50 nM). The reaction was incubated at room temperature and quenched with 10 lL per well of 300 lM unlabeled S-adenosyl-Lmethionine (Sigma, St. Louis, MO, USA) at various time-points. For detection, 50 lL of reaction was transferred to a filter plate [Millipore (Billerica, MA, USA) Multiscreen HTS FB 1.0 ⁄ 0.65 lm], washed three times with 10% trichloroacetic acid, washed once with 95% ethanol, and read on the Top Count (Perkin Elmer, Waltham, MA, USA) after the addition of 30 lL scintillant. Having established histone H4 as the preferred substrate, the enzyme concentration was titrated from 5 to 40 nM, and the reaction progress over time was determined at each enzyme concentration as described previously. Structure determination of DOT1L with SAH or SAM bound DOT1L-1-416 was produced in E. coli as either a GST or hexa-histidine N-terminal fusion protein. The GST-DOT1L-1-416 was cloned into the pGEX-KG vector and expressed in BL21 (DE3) cells (Novagen, Madison, WI, USA) with coexpression of chaperone plasmid pGTf2 (Takara Bio Inc., Shiga, Japan) with 0.2 mM isopropyl b-D-1thiogalactopyranoside (IPTG), at 18 C for 16 h. Cell pellets were suspended in buffer containing 50 mM Na2HPO4 ⁄ NaH2PO4, pH 7.5, 200 mM NaCl, 5% glycerol, and 5 mM b-mercaptoethanol. Cells were lysed by sonication. The supernatant was loaded onto a column of glutathione-sepharose Fast Flow resin (GE Healthcare, Pittsburg, PA, USA). Protein was eluted with buffer containing 10 mM reduced glutathione. The GST-tag was cleaved from DOT1L-1-416 by thrombin and removed by a second run on a glutathione-sepharose column. The tag-free protein was purified further by SP Sepharose Fast Flow (GE Healthcare) chromatography and eluted with 25 mM Tris–HCl, pH 8.0, 675 mM NaCl, 5% glycerol, and 5 mM bmercaptoethanol. The protein was further purified by Superdex75 (GE Healthcare) chromatography in buffer containing 20 mM Tris– HCl, pH 8.0, 200 mM NaCl, 1 mM EDTA, and 1 mM dithiothreitol. Fractions of pure protein were pooled and concentrated to Chem Biol Drug Des 2011; 78: 199–210 40 mg ⁄ mL for crystallization work. The His-DOT1L-1-416 was cloned into pET30a vector with N-terminal His tag and expressed in BL21Gold (DE3) (Stratagene, Cedar Creek, TX, USA). The culture was incubated at 37 C until OD600 reached 0.3 and continued at 25 C until OD600 reached 0.7. Expression was induced by adding 0.2 mM IPTG, and cells were harvested after 4 h. Cell pellet was suspended in buffer A (20 mM Tris–HCl, 500 mM NaCl, 10 mM imidazole, 10% glycerol, 5 mM b-mercaptoethanol, pH 7.8) with 1 mg ⁄ mL lysozyme and incubated on ice for 30 min. The cells were sonicated and centrifuged. The supernatant was loaded onto Ni-NTA column (Qiagen, Valencia, CA, USA) that preequilibrated with buffer A and washed with the same buffer. Protein was eluted with 200 mM imidazole in the buffer A and then dialyzed against 20 mM HEPES, 50 mM NaCl, 1 mM EDTA, 1 mM DTT, pH 7.5. The sample was loaded on a SP Sepharose Fast Flow column (GE Healthcare) and eluted with a linear gradient of 0–1 M NaCl. The target protein was concentrated and further purified by Superdex200 column (GE Healthcare) with 20 mM HEPES, 200 mM NaCl, 1 mM EDTA, 1 mM DTT, pH 7.8. Fractions of pure protein were pooled. SAH or SAM (Sigma-Aldrich) was dissolved in the final DOT1L protein buffer to 100 mM. The DOT1L–SAH or DOT1L–SAM binary complex was prepared at a final concentration of DOT1L of 20 mg ⁄ mL (0.4 mM) and SAH or SAM of 2 mM. Crystals were obtained in 1.94 M ammonium sulfate, 0.1 M sodium acetate, pH 5.3, and 2 mM TCEP by the hanging-drop method at 18 C. The crystals were cryoprotected in 30% glucose and flash-frozen in liquid nitrogen. The diffraction data set for DOT1L–SAH was collected at beamline 17 U at Shanghai Synchrotron Radiation Facility and the data set for DOT1L– SAM was collected at beamline 21-ID-F at Advanced Photon Source in Argonne National Laboratory. All data were processed by HKL2000 (16). The SAH-bound crystal diffracted to 2.3 and the SAM-bound crystal diffracted to 2.1 , in the space group of P65 with one protein molecule in the asymmetric unit. The structures were solved by molecular replacement (Molrep) (17) using the published DOT1L– SAM structure (PDB ID: 1NW3) as a search model (18). Refinement was carried out by Refmac5 (19), and the model building was carried out by COOT (20). Detailed information on the diffraction data, refinement, and structure statistics is provided in Table S1. The enzyme thus purified was tested for enzymatic activity in a radiometric assay of 3H-CH3 transfer from labeled SAM (Perkin-Elmer) to purified nucleosomes from chicken erythrocytes according to the method of Fang et al. (21). The DOT1L used for crystallographic studies was found to have reproducible activity as a histone methyltransferase and displayed the following steady state kinetic parameters: SAH = 0.67 lM, KNuc kcat = 0.3 per minute, KSAM M = 8.6 nM, Ki M = 0.26 lM. A more complete description of the enzymatic mechanism of DOT1L will be reported separately (A. Basavapathruni, C. R. Majer, R. A. Copeland and M. Porter Scott, manuscript in preparation). Results Construction of PKMT and PRMT family tree diagrams To facilitate the chemical biological and pharmacological exploration of the human PMTs, it would be useful to know the relationships 201 Richon et al. among members of this enzyme class and to illustrate these relationships in a convenient format. This is commonly done by the construction of family tree diagrams; such a diagram for the human protein kinases (the human kinome) has proved an invaluable tool for scientists exploring the chemical biology of these enzymes (22). Toward this end, we have systematically explored the protein sequences of human PKMTs and PRMTs with the goal of developing a complete map of the relatedness among the active sites of enzymes in each of these protein families. Our initial intent was to construct a single map that combined both the PKMT and PRMT families. However, we could find no clear rationale with which to define a point of intersection between the two families. For this reason, we present here separate maps for each family within the overall PMT class. Our interrogation of the human genome using PSI-BLAST and representative PMT sequences revealed a total of 51 proteins with clear relatedness to the canonical SET-domain of enzymes with demonstrated biochemical activity as PKMTs. The relationship of these proteins to one another was determined and is illustrated by the dendogram and alignments as shown in Figures S1 and S2. These putative PKMTs cluster into four major groups, illustrated as distinct branches on the family tree shown in Figure 1. Each of these clusters includes at least one example of a protein for which biochemical evidence of direct PKMT enzymatic activity has been reported in the literature. One large branch of the putative PKMT tree is composed of 16 PRDM proteins. The active site domain of these proteins is the most divergent from that of the canonical SET-domain, being instead referred to as a PR (PRDI-BF1 and RIZ homology) domain. Despite the significant structural divergence of these proteins from the other family members, there are literature reports that demonstrate the PKMT activity of PRDM2 (23), PRDM8 (24), and PRDM9 (Meisetz) (25). The tree diagram, dendogram, and alignments for the PRMT family are illustrated in Figure 2 and Figures S3 and S4, respectively. Prior to this report, 11 human proteins have been annotated as PRMTs in the literature, and most of these have been reported to have enzymatic activity as protein arginine methyltransferases (10). Early iterations of our search and alignment process using PSI-BLAST resulted in clustering of most, but not all, of the enzymatically active PRMTs. However, PRMT5 and PRMT9 were not represented within the Figure 1: Protein lysine methyltransferase (PKMT) family tree diagram. The relationship among the PKMT family members is shown in this unrooted tree, where the branch lengths are proportional to the distance calculated between the various PKMT family members. Sequence names are the HUGO Gene Nomenclature Committee standard gene names. 202 Chem Biol Drug Des 2011; 78: 199–210 Chemogenetic Analysis of Human Protein Methyltransferases Figure 2: Protein arginine methyltransferase (PRMT) family tree diagram. The relationship among the PRMT family members is shown in this unrooted tree, where the branch lengths are proportional to the distance calculated between the various PRMT family members. Sequence names are the HUGO Gene Nomenclature Committee standard gene names. clusters. To ensure inclusion of these latter PRMTs within the family tree, additional search and alignment iterations were performed. Through this method, the full complement of enzymatically active PRMTs was incorporated into the family. This process also resulted in the inclusion of multiple members of the METTL and NOP2 ⁄ Sun domain family (NSUN) protein groups, both of which have previously been described to be RNA methyltransferases (26,27). Determination of methyltransferase activity of METTL11A The structural relatedness of the METTL and NSUN protein groups to the bona fide PRMTs suggests that efforts to identify selective ligands for PRMTs should consider carefully these other enzyme groups for a full understanding of ligand selectivity. Beyond this, the inclusion of these protein groups within the PRMT tree raises the question of whether the members of these protein groups may have heretofore unrecognized enzymatic activity as PMTs. To address this question, we obtained a representative member of the METTL group of proteins, METTL11A. The recombinant METTL11A was initially tested for PMT activity using recombinant histones H3 Chem Biol Drug Des 2011; 78: 199–210 and H4 as substrates. Both H3 and H4 were found to be methylated by the recombinant METTL11A, but H4 appeared to be utilized more efficiently by this enzyme (data not shown). We then studied the methyltransferase activity of METTL11A in more detail using H4 as a methyl-accepting substrate. As illustrated in Figure 3, linear progress curves of methyl group incorporation into H4 were observed over a range of enzyme concentrations, suggesting that the reactions at all tested enzyme concentration conformed to steady state conditions. A replot of reaction velocity as a function of enzyme concentration was linear over a range of 5–40 nM enzyme, again suggesting well-behaved steady state kinetic behavior (28,29). These data confirm the annotation of at least one METTL group member as a PMT and more specifically as a histone methyltransferase. Conformation of SAM and SAH in METTL, NSUN, and PRMT proteins To further understand the chemical biology relatedness of the METTL and NSUN proteins to PRMTs, we compared the conformation of SAM or SAH in the crystal structures of representative 203 Richon et al. 600 4.0 A Velocity (CPM/min) 500 400 CPM B 3.5 300 200 100 3.0 2.5 2.0 1.5 1.0 0.5 0 0.0 0 20 40 60 80 100 120 0 5 10 Time (min) 15 20 25 30 35 40 [E], nM Figure 3: METTL11A is an active protein methyltransferase. (A) Reaction progress curves for the incorporation of 3H-CH3 from S-adenosylmethionine (SAM) into recombinant human histone H4 catalyzed by varying concentrations of METTL11A. Symbols: open circles, [E] = 5 nM; closed circles, [E] = 10 nM; open squares, [E] = 20 nM; closed squares, [E] = 40 nM. (B) Replot of the reaction velocity (measured as the slope of the linear least-squares best fit for each progress curve in panel A) as a function of METTL11A nominal enzyme concentration. PRMTs, METTLs, and NSUNs that have been deposited at RCSB PDB (http://www.pdb.org). Figure 4A illustrates the remarkable overlap of SAM or SAH conformation in the active sites of NSUN, A B METTL, and PRMT enzymes although the overall structure for these enzymes is quite different (Figure 4C,D). Only part of the SAM ⁄ SAH binding site is structurally conserved (the region immediately above D C Figure 4: The superimposition of S-adenosylmethionine (SAM) ⁄ S-adenosylhomocysteine (SAH) in the crystal structures of NSUN5, METTL11A, and protein arginine methyltransferases (PRMTs). (A) Conformational similarity of SAM or SAH in NSUN5, METTL11A, and PRMTs. The SAM in NSUN5 (PDB ID: 2B9E) is presented as a ball-and-stick representation in yellow. The SAH in METTL11A (PDB ID: 2EX4) is presented as a ball-and-stick representation in green. The SAH in PRMT1 (PDB ID: 1OR8), PRMT3 (PDB ID: 2FYT), and CARM1 (PDB ID: 3B3F) is presented as a stick representation with carbon atom in gray, oxygen atom in red, nitrogen atom in blue, and sulfur atom in yellow. (B) The crystal structure of NSUN5 with SAM bound (PDB ID: 2B9E) in ribbon representation. The N- and C-termini are labeled. The b-sheets are colored in blue, a-helices in red, and loops in gray. The bound SAM molecule is in the same representation and color as in A. (C) The crystal structure of METTL11A with SAH bound (PDB ID: 2EX4). (D) The crystal structure of CARM1 with SAH bound (PDB ID: 3B3F). The general conformations of PRMT1 and PRMT3 (not shown) are similar to that of CARM1. 204 Chem Biol Drug Des 2011; 78: 199–210 Chemogenetic Analysis of Human Protein Methyltransferases the SAM ⁄ SAH ligand in the presented orientation). The structures for the rest of the proteins vary significantly. Inclusion of DOT1L in the PRMT family tree Despite its known activity as a PKMT, all attempts to include the non-SET-domain enzyme DOT1L in the PKMT family were unsuccessful. In contrast, DOT1L could be readily incorporated into the PRMT family tree (Figure 2). Again, we questioned whether the amino acid alignment of the DOT1L active domain with that of the PRMTs translated into a structural relatedness that would be important from the perspective of ligand binding affinity and specificity. The crystal structure of DOT1L with the substrate SAM bound in the active site has been reported in the literature (PDB ID: 1NW3) (18). The crystal structures of several PRMTs have been reported with the product ligand, SAH, bound in the active site (PDB ID: 1ORH, 2FYT, 3B3F and 2V74). Preliminary comparisons of the ligand conformation of the DOT1L–SAM complex with that of PRMT–SAH complex suggested high similarity. However, some differences in specific torsion angles could be realized. Hence, we have solved the co-crystal structure of the DOT1L–SAH complex to 2.3 resolution (PDB ID 3QOX). The conformation of SAH within the DOT1L active site is compared with that in PRMT–SAH complexes in Figure 5A. Here too we see remarkable overlap of the ligand conformation among DOT1L and PRMT enzymes, but clearly distinct from that of PKMTs (Figure 5B). To understand and confirm the torsional angle differences seen between SAM and SAH bound to DOT1L, we have also obtained a higher-resolution structure (2.1 ) of the DOT1L–SAM complex (PDB ID 3QOW; Figure 5C). To our surprise, the SAM conformation in this higher-resolution structure is identical to that of SAH in the enzyme–product complex. The difference in SAM conformation seen here and in the previously published structure may relate to the resolution difference. Ligand affinity mapping One important application of family tree diagrams, such as those developed here for the PKMTs and PRMTs, is the assessment of ligand affinity and selectivity from a target class perspective. To illustrate the power of this approach, we have evaluated three ligands across a representative sampling of 11 PMTs comprising both PKMTs and PRMTs. We chose to profile SAM and SAH as the universal methyl donor substrate and reaction product of PMT catalysis, respectively, and the natural product analog sinefungin (Figure 6). The PMTs all carry out an SN2 group transfer reaction involving a methyl donor (SAM) and methyl acceptor [e.g., histone; (6)]. To date every human PMT that has been studied has been found to conform to a ternary complex mechanism of catalysis, involving formation of a SAM–enzyme–acceptor complex and direct transfer of the methyl group from SAM to the acceptor protein (6). This commonality notwithstanding, bisubstrate-utilizing enzymes that conform to a ternary complex mechanism can vary in the preferred order (or lack thereof) of substrate binding and the degree to which individual substrate binding affinity is influenced by the state of enzyme saturation with respect to the other substrate (28). Hence, the conditions of subChem Biol Drug Des 2011; 78: 199–210 A B C Figure 5: The conformations for S-adenosylmethionine (SAM) and S-adenosylhomocysteine (SAH) in disruptor of telomeric silencing-1-like (DOT1L). (A) The SAH conformation in DOT1L (ball-andstick, in green) is similar to that in PRMT1, PRMT3, and CARM1 (stick, colored as carbon atom in gray, oxygen atom in red, nitrogen atom in blue, and sulfur atom in yellow). (B) The SAH conformation in DOT1L (ball-and-stick, in green) is distinct from those in protein lysine methyltransferase (PKMTs) [stick, colored as protein arginine methyltransferases (PRMTs) in A]. The PKMTs include SETD7 (PDB ID: 1O9S), SETD8 (1ZKK), EHMT2 (2O8J), EHMT1 (2RFI), SUV39H2 (2R3A), SETMAR (3BO5), SETD2 (3H6L), and SMYD3 (3MEK). (C) The SAM and SAH conformations in DOT1L. SAH is presented in stick representation, and SAM in our 2.1 DOT1L–SAM structure is presented in ball-and-stick representation with the same color scheme as the PRMTs in A. The conformations for SAM and SAH in our structures are identical and superimposed upon each other. SAM in the published structure (PDB ID: 1NW3) is presented in ball-andstick representation in green, with the torsion angle differences between the previously published structure and the current structure evident. strate saturation level for each substrate can have a profound effect on the apparent affinity of other ligands, such as inhibitors and activators. This is a critical issue for chemical biology studies aimed at identification of ligands through high-throughput screening and for 205 Richon et al. NH2 N N A SAM O CH3 N Table 1: Ligand affinity for S-adenosylmethionine (SAM), S-adenosylhomocysteine (SAH), and sinefungin for various protein methyltransferases O NH2 H H H OH OH H NH2 N B SAH EZH2 EZH1 WHSC1 SETD7 EHMT2 SMYD2b DOT1L PRMT1 CARM1 PRMT5 PRMT8 12 25 3.7 0.4 18 £0.01 4.5 16 1.6 1.5 5.7 N (10–14)a (10–30) (2.0–5.5) (0.2–0.6) (16–20) (3.1–5.9) (14–18) (1.3–1.9) (1.0–2.0) (3.1–8.3) SAH Ki (M · 107) Sinefungin Ki (M · 107) 75 83 35 230 10 3.6 2.7 4.3 1.2 1.6 2.5 >250 >250 150 1.7 93 0.9 37 2.0 1.0 5.1 3.1 (58–91) (60–100) (26–45) (180–270) (8–13) (2.3–4.8) (2.0–3.3) (3.6–4.9) (1.0–1.3) (1.0–2.1) (2.0–3.0) (100–200) (1.1–2.3) (80–110) (0.6–1.2) (25–48) (1.8–2.2) (0.7–1.1) (3.3–7.1) (2.8–3.4) a O N Values in table represent the best fit value of the parameter. Values in parentheses represent the 95% confidence interval for the parameter value. b For SMYD2, an accurate SAM KM value could not be determined. Hence, the KM and Ki values reported here should be viewed as apparent values. N S HO O NH2 H H H OH H OH an apparent value; likewise, the SAH and sinefungin Ki values should also be considered apparent values. NH2 C Sinefungin N NH2 O N HO N N O NH2 H H H OH H OH Figure 6: Chemical structures of (A) S-adenosylmethionine (SAM), (B) S-adenosylhomocysteine (SAH), and (C) sinefungin. the characterization of ligand selectivity among potential target proteins. This issue has been quantitatively addressed in general by Copeland (28–30) and specifically for the case of bisubstrate enzymes by Yang et al. (31). The results of these studies consistently demonstrate that quantitative comparisons of apparent ligand affinity across a spectrum of targets and ⁄ or ligands are best performed with assays under balanced conditions, in which all substrate concentrations are poised at their apparent KM values (31). In keeping with this widely accepted and common assay design tenet, we performed dual titrations of SAM and methyl-accepting substrate for all of the enzymes studied here to determine the apparent KM values for each substrate. With this information in hand, we have adjusted the substrate concentrations to match their respective apparent KM values (31) and thus achieved balanced conditions for all subsequent assays in which ligands were evaluated. For SMYD2, the KM value for SAM was below the detection limits of our assay methods. Hence, the KM value for this enzyme should be viewed as 206 SAM KM (M · 107) N S HO Enzyme For the methyl-donating substrate SAM, the Michaelis constant, KM, was determined as a relative measure of ligand affinity, despite the fact that the KM term is composed of kinetic rate constants representing steps in addition to initial substrate binding (28). Nevertheless, we felt that apparent KM values across various members of the PMT class would serve well as a measure of relative substrate recognition and utilization. Based on their close structural relatedness to SAM, we have assumed that both SAH and sinefungin are SAM-competitive inhibitors of PMTs, an assumption that is consistent with a significant body of crystallographic data. To support this assumption further, we have determined the modality of SAH inhibition for the 11 PMTs studied here by mutual titration of substrate and inhibitor. In all cases, SAH was found to be competitive with SAM (data not shown). A similar systematic analysis of sinefungin modality was precluded by the modest potency of this compound and its relatively limited solubility. Based on this preponderance of cumulative evidence, we felt confident in assuming that both SAH and sinefungin were competitive inhibitors with respect to SAM and have therefore used the Cheng–Prusoff equation for competitive inhibition (28,29,32) to calculate the apparent Ki values of SAH and sinefungin from the IC50 values obtained for each ligand under balanced assay conditions (vide supra). The results of this profiling are summarized in Table 1 and in Figure 7. For illustrative purposes, we have converted the Ki and KM values to pKi and pKM ()log (X), where X is either Ki or KM) so that the parameter value increases with increasing ligand affinity and have represented these values as spheres of varying diameter for the enzymes studied in Figure 7; the diameter of the individual spheres relates directly to the magnitude of the pKM ⁄ pKi value. Chem Biol Drug Des 2011; 78: 199–210 Chemogenetic Analysis of Human Protein Methyltransferases A SMYD2 EZH1 EZH2 SETD7 PRMT5 PRMT1 DOT1L CARM1 PRMT8 EHMT2 LEGEND: WHSC1 Km, [M] Sized by pKm 10–9 B SMYD2 EZH1 10–8 10–7 10–6 10–5 >2.5 × 10–5 EZH2 SETD7 PRMT5 PRMT1 DOT1L CARM1 PRMT8 EHMT2 LEGEND: WHSC1 Ki, [M] Sized by pKi 10–9 C SMYD2 EZH1 10–8 10–7 Chem Biol Drug Des 2011; 78: 199–210 10–5 >2.5 × 10–5 EZH2 SETD7 Figure 7: Ligand affinity maps for S-adenosylmethionine (SAM) (A), S-adenosylhomocysteine (SAH) (B), and sinefungin (C) binding to representative enzymes of the protein lysine methyltransferase (PKMT) family (left) and the protein arginine methyltransferase (PRMT) family (right). The diameter of the spheres for each enzyme is directly related to the magnitude of the pKM (for SAM) or pKi (for SAH and sinefungin) as described in the text. 10–6 PRMT5 PRMT1 DOT1L CARM1 PRMT8 EHMT2 LEGEND: WHSC1 Ki, [M] Sized by pKi 10–9 10–8 10–7 10–6 10–5 >2.5 × 10–5 207 Richon et al. Discussion Together, the human PRMTs and PKMTs (the PMTs) constitute an important class of enzymes that play critical roles in the methylation of a number of cellular proteins. Most notably, from the perspective of epigenetic regulation of gene transcription, a number of these enzymes have been shown to methylate arginine and lysine residues of histones and to thus effect chromatin remodeling (5,6). The dysregulation of PMT enzyme activity – through gene amplification, gene rearrangement, point mutations, and other genetic changes – has been directly associated with various cancer types as well as with other human diseases [see Copeland et al. for review, (6)]. For example, overexpression of the SET-domain PKMT EZH2 has been associated with prostate, breast, bladder, colon, skin, liver, endometrial, lung, gastric, lymphoid, and myeloid cancers (33,34). More recently, a subset of patients with non-Hodgkin's lymphoma have been found to be heterozygous for point mutations within the catalytic active site of EZH2 (35). These mutations were demonstrated to result in a change of function that, in concert with the wild-type enzyme, results in elevated levels of the tumorigenic product histone H3K27me3 (15). Likewise, expression levels of a number of other PMTs have been linked to tumorigenesis and ⁄ or tumor invasiveness in human cancers (6). Based on this disease association and the presumed druggability of the SAM binding pockets of these enzymes, we have made the case that the PMTs constitute an important, novel drug target class that should be exploitable for small-molecule drug therapies against a number of serious human diseases (6). As stated in the introduction of this article, our aim here was to define the number of PMT-related proteins that exist in humans and to understand their relatedness with respect to catalytic active domain structure. Toward this goal, we have performed a systematic survey of the human genome for proteins that display structural alignment with SET-domain PKMTs, and likewise with PRMTs, for which biochemical evidence of enzymatic activity has been demonstrated. Through multiple, iterative database searches and multiple sequence alignments, we have arrived at the family trees for human PKMTs and PRMTs that are displayed in Figures 1 and 2, respectively. A number of features of these family trees deserve comment. For the SET-domain PKMTs, our analysis indicates that there are 51 related proteins with sequences that suggest putative SAM binding and MT activity. These cluster into four major branches or groupings of putative enzymes. One such branch is the PRDM cluster. The proteins within this branch display the greatest divergence from the canonical SET-domain structure of the rest of the PKMTs. Whether these proteins are indeed bona fide PKMTs is unclear based on the current literature. Three PRDM family members (PRDM2, PRDM8, and PRDM9) have been reported to demonstrate methylation of a protein or peptide substrate (23–25). This highlights an important area for biochemical follow-up to the data reported here. For the PRMTs, the majority of enzymes with well-documented biochemical activity as arginine methyltransferases cluster together to form a single branch of the family tree. However, the enzymes PRMT5 (36) and PRMT9 (37) have been clearly demonstrated to 208 have PRMT enzymatic activity and do not fall within the branch defined for the other enzymes. Inclusion of these two enzymes in the alignments resulted in the additional incorporation of PRMT11, a known PRMT, and two groups of proteins for which protein methylation has not been previously suggested, the METTL and NSUN. Both of these latter groups of proteins have been experimentally demonstrated to be RNA methyltransferases, and the crystal structures of representative members of both groups display a welldefined SAH binding pocket for each. We do not wish to imply that the METTL and NSUN group members are necessarily PRMTs per se. Rather, we include them within the PRMT tree on the basis of their catalytic active site relatedness, which bears directly of chemical biology studies aimed at understanding small-molecule ligand affinity and cross-reactivity (i.e., selectivity) among structurally related proteins. This caveat notwithstanding, the data presented here suggest that at least one member of the METTL group, METTL11A, may be biochemically defined as a PMT and more specifically as a histone methyltransferase (Figure 3). The results illustrated in Figure 3 clearly demonstrate that METTL11A has enzymatic activity as a PMT, but the current data do not define the substrate specificity of the enzyme with respect to methyl-accepting group. While this report was being prepared, Clarke's group (38) also reported PMT activity for METTL11A and specifically demonstrated that the protein acted as a peptide N-terminal methyltransferase. As illustrated in Figure 4A, the conformation of SAH within the binding pockets of METTL and NSUN proteins shows remarkable overlap with that seen in bona fide PRMTs. However, this extended conformation of product ligand is also similar to that seen in the unrelated DNA methyltransferases. Conspicuously absent from the PKMT family tree is the non-SETdomain enzyme DOT1L. DOT1L has been well demonstrated to have histone H3K79 methyltransferase activity when presented with nucleosomes as the methyl-accepting substrate. In addition to lacking a SET-domain, DOT1L is further differentiated from the other PKMTs by its strict substrate specificity for nucleosomes; in contrast, many of the SET-domain enzymes have been shown to utilize recombinant histones or peptides as methyl-accepting substrates. DOT1L is additionally distinct from the SET-domain enzymes in how it binds ligands. As previously reported for SAM (18), and reported here for SAH, DOT1L binds substrate and product ligands in an extended conformation that is significantly different from the 'Ushaped' conformation seen in SET-domain PKMTs and is in fact highly overlapping with the ligand conformation seen for the PRMTs (Figure 5A,B). All attempts to include DOT1L within the PKMT family tree using were unsuccessful. However, we were able to rationally include DOT1L within the PRMT family tree. Inclusion of DOT1L also resulted in the inclusion of additional METTL and NSUN group members, but these protein groups were already represented within the PRMT tree when PRMT5, PRMT9, and PRMT11 were brought in (vide supra). Hence, the incorporation of additional METTL and NSUN proteins here seems reasonable. The biochemical literature leaves no question as to the ability of DOT1L to act as a highly specific protein lysine methyltransferase (8). The literature is silent, however, on whether or not this enzyme may have additional methyltransferase activities, including arginine methylation. To our PSI-BLAST Chem Biol Drug Des 2011; 78: 199–210 Chemogenetic Analysis of Human Protein Methyltransferases knowledge, no systematic evaluation of DOT1L activity beyond H3K79 methylation has been made. Again, on the basis of the current data, we do not suggest that DOT1L is in fact an arginine methyltransferase. Nevertheless, the inclusion of DOT1L within the PRMT family tree raises the question of what the full spectrum of methyltransferase activity for DOT1L might be. Regardless of whether or not DOT1L and ⁄ or the NSUN and METTL proteins are bona fide arginine methyltransferases, the similarity of the active sites of these enzymes to those of the PRMTs is nevertheless striking. Hence, any attempt to identify inhibitors of the PRMTs will require selectivity assessment against these other enzymes as this may be a case where the structural determinants of pharmacologic selectivity and biological specificity are distinct. The data reported here provide a clear and simple mechanism for understanding the full complement of human PMTs and their relatedness to one another. The illustration of these relationships in the form of a family tree or dendogram has proved to be a very powerful tool for other enzyme families, most notably the protein kinases (22); it is from examination of this type of family tree that the kinome concept was born. We believe that the data presented here will be equally valuable as a research tool for investigators interested in studying the PMT enzymes. In particular, the information reported here will be of use in chemical biology and drug discovery efforts aimed at identifying selective ligands for these enzymes. The PKMT and PRMT trees provide a cogent basis for investigating ligand selectivity among these enzymes, as illustrated here for the ligands SAM, SAH, and sinefungin (Figure 7). Inspection of Figure 7 reveals some interesting patterns of ligand affinity among the PMTs, even with the limited ligand set considered here. For example, for all three ligands, one observes a significant range of affinity among the PKMTs, but a much narrower affinity range among the PRMTs. Also, for the PKMTs, one generally observes greater relative affinity for the substrate SAM compared with the product SAH, which is typical of enzyme reactions; weaker product affinity results in facile release of product, thus facilitating subsequent rounds of substrate binding and turnover. In contrast, the PRMTs display similar affinity for substrate and product. The more indiscriminate binding of substrate and inhibitors observed for the PRMTs may relate to the greater substrate promiscuity that is inherent to the biological function of these enzymes. Thus, while the substrate specificity among PKMTs is thought to be fairly restrictive, the PRMTs are known to methylate a broader range of protein substrates, including histones and cytosolic proteins. The data summarized in Figure 7 might lead one to infer that development of selective inhibitors for the PRMTs may prove to be a greater challenge than for the PKMTs. However, examples of selective inhibitors for enzymes in each of these families have already been reported [see (6) for a review]. Thus, as just described, the selectivity of ligands for PKMTs and PRMTs can be rationally evaluated by reference to the trees provided here. Drawing on the experience with the kinome, it will often be the case that enzymes in closest proximity to a desired target on a family tree will present the greatest challenge with respect to ligand selectivity. However, the kinome experience also teaches that there are important exceptions to the foregoing generChem Biol Drug Des 2011; 78: 199–210 ality. In several instances, inhibitory ligands have been found to 'leap frog' over closely related enzymes to inhibit enzymes more distal to the desired target within the kinome. The same phenomenon is observed for the PMTs with the ligands tested here. For example, EZH2 and EZH1 are very similar in active domain structure and are nearest neighbors on the PKMT tree. As expected from this proximity, the two enzymes display identical KM values for the substrate SAM and similar Ki values for the inhibitor sinefungin (Figure 7 and Table 1). In contrast, however, these two enzymes show more than an order of magnitude difference in product affinity. In this latter respect, the affinity of EZH2 for SAH is more similar to that for SMYD2 and the PRMTs than to its nearest neighbor EZH1. As additional inhibitors of PMTs are discovered, determining the pattern of selectivity among the PKMTs and PRMTs, as illustrated here, will be an important aspect of compound optimization, especially for pharmacologic utility. It is our hope that the information reported here will provide an important starting point for the further exploration of the human PMTs as an enzyme class. The family trees that have been developed in this report will be of value to the chemical biology and drug discovery communities, as described earlier. The results also point to specific experiments to explore further the full repertoire of PMT activities within humans. Whether or not DOT1L is a dual lysine ⁄ arginine methyltransferase and whether the NSUN proteins may methylate both nucleic acid and protein substrates are open questions that have been raised by the current analysis. The experimental resolution of these questions will be important tasks for the community to address in the continued efforts to understand protein methylation and its impact on biology and disease. References 1. Walsh C.T., Garneau-Tsodikova S., Gatto G.J. Jr (2005) Protein posttranslational modifications: the chemistry of proteome diversifications. Angew Chem Int Ed Engl;44:7342–7372. 2. Kouzarides T. (2007) Chromatin modifications and their function. Cell;128:693–705. 3. Feinberg A.P., Tycko B. (2004) The history of cancer epigenetics. Nat Rev Cancer;4:143–153. 4. Kornberg R.D., Lorch Y. (1999) Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell;98:285–294. 5. Strahl B.D., Allis C.D. (2000) The language of covalent histone modifications. Nature;403:41–45. 6. Copeland R.A., Solomon M.E., Richon V.M. (2009) Protein methyltransferases as a target class for drug discovery. Nat Rev Drug Discov;8:724–732. 7. Schneider R., Bannister A.J., Kouzarides T. (2002) Unsafe SETs: histone lysine methyltransferases and cancer. Trends Biochem Sci;27:396–402. 8. Okada Y., Feng Q., Lin Y., Jiang Q., Li Y., Coffield V.M., Su L., Xu G., Zhang Y. (2005) hDOT1L links histone methylation to leukemogenesis. Cell;121:167–178. 9. Dillon S.C., Zhang X., Trievel R.C., Cheng X. (2005) The SETdomain protein superfamily: protein lysine methyltransferases. Genome Biol;6:227. 209 Richon et al. 10. Cheng X., Collins R.E., Zhang X. (2005) Structural and sequence motifs of protein (histone) methylation enzymes. Annu Rev Biophys Biomol Struct;34:267–294. 11. Bachand F. (2007) Protein arginine methyltransferases: from unicellular eukaryotes to humans. Eukaryot Cell;6:889–898. 12. Krause C.D., Yang Z.H., Kim Y.S., Lee J.H., Cook J.R., Pestka S. (2007) Protein arginine methyltransferases: evolution and assessment of their pharmacological and therapeutic potential. Pharmacol Ther;113:50–87. 13. Wu H., Min J., Lunin V.V., Antoshenko T., Dombrovski L., Zeng H., Allali-Hassani A., Campagna-Slater V., Vedadi M., Arrowsmith C.H., Plotnikov A.N., Schapira M. (2010) Structural biology of human H3K9 methyltransferases. PLoS ONE;5:e8570. 14. Boeckmann B., Bairoch A., Apweiler R., Blatter M.C., Estreicher A., Gasteiger E., Martin M.J., Michoud K., O'Donovan C., Phan I., Pilbout S., Schneider M. (2003) The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res;31:365–370. 15. Sneeringer C.J., Scott M.P., Kuntz K.W., Knutson S.K., Pollock R.M., Richon V.M., Copeland R.A. (2010) Coordinated activities of wild-type plus mutant EZH2 drive tumor-associated hypertrimethylation of lysine 27 on histone H3 (H3K27) in human B-cell lymphomas. Proc Natl Acad Sci U S A;107:20980–20985. 16. Otwinowski Z., Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol;276:307– 326. 17. Vagin A., Teplyakov A. (1997) MOLREP: an automated program for molecular replacement. J Appl Cryst;30:1022–1025. 18. Min J., Feng Q., Li Z., Zhang Y., Xu R.M. (2003) Structure of the catalytic domain of human DOT1L, a non-SET domain nucleosomal histone methyltransferase. Cell;112:711–723. 19. Vagin A.A., Steiner R.A., Lebedev A.A., Potterton L., McNicholas S., Long F., Murshudov G.N. (2004) REFMAC5 dictionary: organization of prior chemical knowledge and guidelines for its use. Acta Crystallogr D Biol Crystallogr;60:2184–2195. 20. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr;60:2126– 2132. 21. Fang J., Wang H., Zhang Y. (2004) Purification of histone methyltransferases from HeLa cells. Methods Enzymol;377:213–226. 22. Manning G., Whyte D.B., Martinez R., Hunter T., Sudarsanam S. (2002) The protein kinase complement of the human genome. Science;298:1912–1934. 23. Kim K.C., Geng L., Huang S. (2003) Inactivation of a histone methyltransferase by mutations in human cancers. Cancer Res;63:7619–7623. 24. Eom G.H., Kim K., Kim S.M., Kee H.J., Kim J.Y., Jin H.M., Kim J.R., Kim J.H., Choe N., Kim K.B., Lee J., Kook H., Kim N., Seo S.B. (2009) Histone methyltransferase PRDM8 regulates mouse testis steroidogenesis. Biochem Biophys Res Commun;388:131–136. 25. Hayashi K., Yoshida K., Matsui Y. (2005) A histone H3 methyltransferase controls epigenetic events required for meiotic prophase. Nature;438:374–378. 26. Alexandrov A., Martzen M.R., Phizicky E.M. (2002) Two proteins that form a complex are required for 7-methylguanosine modification of yeast tRNA. RNA;8:1253–1266. 210 27. Sakita-Suto S., Kanda A., Suzuki F., Sato S., Takata T., Tatsuka M. (2007) Aurora-B regulates RNA methyltransferase NSUN2. Mol Biol Cell;18:1107–1117. 28. Copeland R.A. (2000) Enzymes: A Practical Introduction to Structure, Mechanism and Data Analysis, 2nd edn. Hoboken: Wiley. 29. Copeland R.A. (2005) Evaluation of enzyme inhibitors in drug discovery. A guide for medicinal chemists and pharmacologists. Methods Biochem Anal;46:1–265. 30. Copeland R.A. (2003) Mechanistic considerations in highthroughput screening. Anal Biochem;320:1–12. 31. Yang J., Copeland R.A., Lai Z. (2009) Defining balanced conditions for inhibitor screening assays that target bisubstrate enzymes. J Biomol Screen;14:111–120. 32. Cheng Y., Prusoff W.H. (1973) Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol;22:3099–3108. 33. Simon J.A., Lange C.A. (2008) Roles of the EZH2 histone methyltransferase in cancer epigenetics. Mutat Res;647:21–29. 34. Martinez-Garcia E., Licht J.D. (2010) Deregulation of H3K27 methylation in cancer. Nat Genet;42:100–101. 35. Morin R.D., Johnson N.A., Severson T.M., Mungall A.J., An J., Goya R., Paul J.E. et al. (2010) Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet;42:181–185. 36. Rho J., Choi S., Seong Y.R., Cho W.K., Kim S.H., Im D.S. (2001) Prmt5, which forms distinct homo-oligomers, is a member of the protein-arginine methyltransferase family. J Biol Chem;276: 11393–11401. 37. Cook J.R., Lee J.H., Yang Z.H., Krause C.D., Herth N., Hoffmann R., Pestka S. (2006) FBXO11 ⁄ PRMT9, a new protein arginine methyltransferase, symmetrically dimethylates arginine residues. Biochem Biophys Res Commun;342:472–481. 38. Webb K.J., Lipson R.S., Al-Hadid Q., Whitelegge J.P., Clarke S.G. (2010) Identification of protein N-terminal methyltransferases in yeast and humans. Biochemistry;49:5225–5235. Supporting Information Additional Supporting Information may be found in the online version of this article: Figure S1. PKMT dendogram. Figure S2. PKMT alignment. Figure S3. PRMT dendogram. Figure S4. PRMT alignment. Table S1. Data collection and refinement statistics. Please note: Wiley-Blackwell is not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article. Chem Biol Drug Des 2011; 78: 199–210