Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
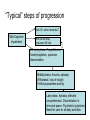
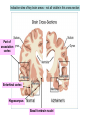
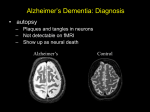
Alzheimer’s Disease A progressive neurodegenerative disease first characterised by Alios Alzheimer, Germany 1906 Alois Alzheimer 1906 wrote…. • “A woman (Frau Auguste D) exhibited ideas of jealousy against her husband, rapidly progressing loss of memory, inability to find her way around the home, dragged objects back and forth, hid them; at times she believed that someone wanted to kill her and began to shout loudly”. • Frau Auguste D died aged 51 in a local mental asylum following a number of years of progressive dementia. • Alzheimer carried out microscopic examination of her brain tissue. He observed: – structures previously described in the brains of elderly people – neuritic or senile plaques – structures contained within brain cells that had not been previously observed – neurofibrillary tangles. • Both of these microscopic changes are used to characterise the form of dementia known as Alzheimer’s Disease (AD) AD – Brief history of diagnostic criteria • In the early part of the 20th Century a positive diagnosis of AD was: “a person with dementia who was less than 65 years old”. • In individuals “senile dementia” was assumed to be vascular disease associated with old age. • During 1962 – 1970 studies showed that neuronal cellular inclusions, plaques and tangles were present in older individuals in the absence of vascular degeneration. • Recognised that AD could not be restricted to under 65s. The term Alzheimer’s Disease now describes dementia of the Alzheimer type irrespective of the age of onset. • A definite diagnosis requires demonstration of the presence of plaques and tangles – post mortem microscopic analysis • Prior to death, the diagnosis is “probably AD” if the patient has a history of progressive dementia-but new scans enable more certain diagnosis. Alzheimer’s Disease-Background • Alzheimer's Disease (AD) is most commonly diagnosed in older age groups (65+) • Currently estimated that there are 400,000+ AD sufferers in the UK • Death often occurs 5-10 years after diagnosis. • Sufferers include Ronald Regan, Charlton Heston Maggie Thatcher, Terry Pratchett Iris Murdoch. Alzheimer’s Disease-Background Broad clinical characteristics of AD • Symptoms – – – – Memory lapses of increasing frequency and extent Problems with word finding Disturbed/repetitive behaviour Effects on social function • Confusion, disorientation • Lack of memory affects organisational skills • Mood swings-depression, anger, paranoia • Loss of confidence, withdrawal • Loss of life skills and independence “Typical” steps of progression Not AD –other dementia? Mild Cognitive Impairment Not AD but may represent AD risk AD early phase. Forgetfulness, Anxiety/agitation, paranoia, disorientation. Middle phase. Anomia, aphasia, Withdrawal, loss of insight Fretful purposeless activity. Late phase. Aphasia, affected comprehension. Disorientation in time and space. Psychiatric symptoms Need for care for all daily activities Alzheimer’s Disease - clinical • AD is the most common dementia (accounts for 6080% of dementia in elderly) • 5-10% >65yr affected (up to 45% >85yr) • Slow, progressive – 5-10yrs • Complex presentation and progression • Becoming a major health burden as population ages CNS damage correlates with symptoms • Changes most prominent in: • • • • Hippocampus Entorhinal cortex Association cortex Basal forebrain • Accounts for early signs – memory loss + disturbance of higher cortical functions – preservation of primary sensory/motor functions until later Indicative sites of key brain areas – not all visible in this cross-section Part of association cortex Entorhinal cortex Hippocampus Basal forebrain nuclei PET Scan Normal Positron emission tomography Alzheimer’s Radiolabelled 2-deoxyglucose Development of AD pathology • Gradual onset • Accumulation of neuronal pathology – Protein build up – Neuronal loss • Compensation ability of CNS overwhelmed • Clinical symptoms of AD • Continued pathology and symptom development Main pathology Generation of inclusion bodies Molecules Central to Pathology • Proteins/peptides • Amyloid b-peptide • Presenilins • Apolipoprotein E • tau: microtubule-associated Protein accumulation pathology Alzheimer’s disease - Pathology • Extracellular plaques (Amyloid plaques) • Cerebral cortex • Cerebral/meningeal blood vessel wall • Plaques • Dense amyloid core • Dystrophic neurites + reactive glia surround • Intraneuronal neurofibrillary tangles (Tau protein fibrils) • • • • Neuronal + synaptic loss Reactive astrocytosis Microglia proliferation Later inflammation • Pathology correlation with dementia • Plaque no. – some correlation • Tangles no. – more correlation Examples of Cellular Inclusions B. Tau Protein in Alzheimer's Disease C. a-Synuclein in Parkinson’s Disease Diffuse plaque • Amyloid b protein deposit (A b1-42) • Non-aggregated (fibrillar) form • Intact neurones within plaque boundaries Neuritic plaques • Develop from diffuse/immature plaques • Spherical areas, 10-150nm diameter • Foci of enlarged axons, synaptic terminals, dendrites – Associated with extracellular b-amyloid – Dystrophic neurites within plaque – tau accumulation – Proliferating astrocytes/microglia surround AD - Neurofibrillary tangle Neurofibrillary tangles • Major pathological feature • Brain area affected correlates with severity of dementia • Flame shaped or globoid intracellular inclusions • Persists as extraneuronal structure – “ghost” • May represent cell damage by impaired intracellular transport systems Neurofibrillary tangles Tangles thought to comprise isoforms of Tau proteins • Paired helical fibrils (insoluble) and PHFs soluble straight filaments (SSfs) • Abnormally (hyper)phosphorylated microtubuleassociated protein tau = subunit of PHF • PHFs found also in dystrophic neurites. b – Amyloid • Cause or an effect of AD? (protein misfolding?) • Neuritic plaque major protein (70%) – Toxic to cultured neurons • Amyloid deposits aggregates of Ab containing 40 or 42 residues - insoluble • Ab40 normally produced in small amounts • Ab 42 overproduced due to genetic mutations (ADP transcripts found in AD brains) Presenilins • Presenilin 1 linked with 70% familial cases • 7 transmembrane protein, • Presenilin 2 linked with 20% familial cases • 67% similarity • Expressed in neurons – ER/golgi apparatus • Processing/transport role for proteins Tau Protein • Microtubule-associated protein • Abnormal concentration in neuron cell bodies • Normally synthesised in the body and transported to the axon in high concentrations Pathogenesis of AD [From Rang et al ‘Pharmacology’ 5th Ed] Genetics APP: chrom 21q21. >23 mutations PSEN1: chrom 14q24.3 PSEN2: 1q42.1 APOE4: epsilon 4 isoform + others = genetically complex disorder Potential protein manufacture control route Neurochemical changes in AD • Changes in many transmitter systems • Relatively selective loss of cholinergic neurons (ACh releasing) – Choline acetyltransferase activity in cortex/hippocampus reduced (30-70%) – Acetylcholinesterase activity reduced – Muscarinic R density unaffected – Nicotinic R reduced (cortex particularly) • Excess glutamate? Excitotoxicity Treatment • No current treatment can arrest AD • Some treatments can slow progress • Cholinesterase inhibitors (Tacrine, Donepezil, Rivastigmine, Galantamine) • Memantine • Other medicines can help control behavioural symptoms – sleeplessness, anxiety, depression • Novel targets – protein construction Drug therapy (1) • No real therapy until the mid 1970s when it was observed that the activity of the enzyme responsible for synthesising ACh was reduced in the cerebral cortex of AD patients. • Hypothesis that AD was a consequence of dysfunction of cholinergic neurones. • Attempts to increase cholinergic function by increasing dietary choline not appropriate results patients smelling of rotting fish! • Introduction of AChE inhibitors which work by prolonging the half-life of ACh in synaptic clefts – first drug used was tacrine Cholinesterase inhibitors • Mild to moderate AD • Delay or prevent worsening for limited time • Prevent breakdown of ACh • Cholinergic neurons damaged and lost linked to memory loss Presynaptic neuron Stag e1 Stage 2 Stage 3 Stage 5 ACh Stage 4 Stage 9 X astrocyte AChE ACh Postsynaptic neuron Drug therapy (2) Second generation AChE inhibitors with less side effects were developed – donepezil, rivastigmine and galantamine • AChE inhibitors modest effects- some patients showing no obvious benefit. Anticholinersterases treat mild to moderate AD Attempts to activate ACh receptors more directly using ACh agonists: – Nicotine shown to improve attention span in AD patients – Nicotinic agonist ABT-418 has cognitive-enhancing properties • Muscarinic antagonists such as atropine have been shown to induce memory loss in humans. • Potent muscarinic agonists (e.g.arecoline, oxotremorine and pilocarpine) have too many side effects to be useful but have been shown to improve memory in some patients & animal models. Drug therapy (3) • Muscarinic autoreceptors have been shown to control the release of ACh and attempts have been made to develop specific antagonists to these presynaptic cholinergic autoreceptors. • One such molecule, AF-DX-116 has been shown to improve cognitive performance in experimental animals while SCH-217443 has shown efficacy in increasing ACh release . • …but alvameline, a combined muscarinic agonist and antagonist potential treatment for AD phase II/III trials failed to demonstrate therapeutic efficacy. • Glutamate NMDA receptors also play a role in cognitive function and neurodegeneration and the NMDA antagonist memantine was launched in 2002 as the only treatment for moderate to severe AD ( NMDA Receptor Antagonist • • • • • N-methyl-D-aspartate receptor is normally activated by glutamate Excess glutamate can be detrimental and lead to cell death Memantine (Namenda) dampens glutamate effects via NMDA R Allows physiological NMDA R activities to continue Can be given with cholinesterase inhibitors Dissolving amyloids? HSP 104 Education – a risk factor? • BBC report 16 Feb 06 • High levels of education may help people to better resist AD initially. • However once accumulated damage reaches a critical level, decline inevitable and swift. • Whether truly related to schooling or associated factors (wealth/ occupation/lifestyle) yet to be determined. http://news.bbc.co.uk/1/hi/ health/4713570.stm Exercise 'cuts Alzheimer's risk' BBC report: January 2006 • Regular exercise reduces the risk of dementia and Alzheimer's disease by up to 40%. (US research). http://news.bbc.co.uk/1/hi/h ealth/4616502.stm Common agents and AD • Aspirin (NSAID) – Evidence that decreases risk and slows onset of AD • HRT – Unconfirmed observation that reduces incidence or slows onset in women • Nicotine – Controversial suggestion that it protects against AD (as for PD) Are we accurately presented with the Facts by the Media? How is this figure potentially misleading?