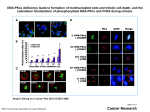
Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Mitochondrial Disease RITE Review Andrew Sas Neurology Resident 12/28/2015 Objectives • Discuss different types of mitochondrial disease • Recognize common features • Review options for treatment if they exist • Focus on RITE diseases/topics • RITE type questions Mitochondrial Diseases • Summary • • • • • • • • • • • Encephalopathy Myopathy Neuropathy Cardiomyopathy Retinopathy Exercise intolerance Short stature Hearing loss Elevated lactate Ragged red fibers Inheritance • Maternal, recessive, or sporadic • Treatment: • Creatine, Co-Q10, ketogenic diet (other than beta oxidation defects with high carb diet), carnitine, avoidance of fasting, supportive Mitochondrial Disorders or “Cytopathies” • Result from mutations in mitochondrial or nuclear genes resulting in failures of oxidative phosphorylation characterized by: • Genetic and clinical heterogeneity • Painful ACRONYMS and Eponyms (or both!) • Lactic acidosis • Some treatments exist, but no cures • Could try creatine or CoQ10 Types of disease • • • • • • • • • • • • • • • • • • • • • • Alpers Disease Barth syndrome Beta-oxidation Defects Carnitine-Acyl-Carnitine Deficiency Carnitine Deficiency Creatine Deficiency Syndromes Co-Enzyme Q10 Deficiency Complex I Deficiency Complex II Deficiency Complex III Deficiency Complex IV Deficiency Complex V Deficiency COX Deficiency CPEO CPT I Deficiency CPT II Deficiency Glutaric Aciduria Type II KSS Lactic Acidosis LCAD LCHAD Leigh Disease or Syndrome LHON LIC (Lethal Infantile Cardiomyopathy) Luft Disease MAD MCAD MELAS MERRF MIRAS Mitochondrial Cytopathy Mitochondrial DNA Depletion Mitochondrial Encephalopathy Mitochondrial Myopathy MNGIE NARP Pearson Syndrome Pyruvate Carboxylase Deficiency Pyruvate Dehydrogenase Deficiency POLG Mutations Respiratory Chain SCAD SCHAD VLCAD www.umdf.org – The United Mitochondrial Disease Foundation *found in RITE database Types of disease LHON MAD MELAS MERRF • Beta-oxidation Defects • Creatine Deficiency Syndromes • Co-Enzyme Q10 Deficiency • CPEO Mitochondrial DNA Depletion Mitochondrial Encephalopathy Mitochondrial Myopathy NARP Pyruvate Dehydrogenase Deficiency Respiratory Chain • Glutaric Aciduria Type II • KSS • Lactic Acidosis • Leigh Disease or Syndrome www.umdf.org – The United Mitochondrial Disease Foundation Mitochondrial Disorders: Inheritance ? Mitochondrial Disorders: Inheritance • About ½ of mitochondrial diseases are inherited from mitochondrial DNA in a maternal fashion • The other ½ are due to nuclear mutations that affect mitochondrial function • Many are sporadic mutations Mitochondrial Disorders: Inheritance Maternal Inheritance Autosomal recessive * Barth syndrome is X-linked recessive Respiratory Chain • Complex • • • • • I II III IV V Mitochondrial Myopathy - Pathology Normal SDH Succinyl Dehydrogenase) SDH + Ragged Red Fibers Paracrystalline inclusions (MELAS) COX – Cytochrome oxidase Answer This substance is found in high concentrations in serum and CSF of patients with mitochondrial cytopathies. Question What is lactic acid? Lactate and glucose levels in various disease: Mitochondrial cytopathy – low glucose, high lactate 14 questions in 14 years Answer This anesthetic infusion used for sedation inhibits mitochondrial fatty acid oxidation, oxidative phosphorylation, carnitine palmitoyl transferase, and secondarily inhibits Complex II. Question What is propofol? • Propofol infusion syndrome • More common in children than adults • metabolic acidosis, heart failure, renal failure, hyperkalemia and rhabdo • Prevent by giving carbohydrate load/infusion (children have limited carbohydrate reserves) • Treat by hemofiltration •3 times in 14 years Case 1 • 16 year-old boy • CC: Ataxia and droopy eyelids. • HPI: • His ptosis started at age 5 and his parents note that he turns his head more then usual when trying to look around. He also has noted that his balance is off and he occasionally drops objects. • PMH: • short stature • diabetes • complete heart block • Exam: • Bilateral ptosis and restricted bilateral horizontal eye movements. His fundoscopic exam reveals pigmentary retinopathy. His has 4/5 strength in his proximal arms and legs and is unable to tandem walk. • Workup: • Serum lactate is slightly elevated. CSF shows elevated protein and lactate. Muscle biopsy shows ragged red fibers. • Diagnosis? Case 1 – Kearns-Sayre Syndrome 16 year-old boy CC: Ataxia and droopy eyelids. HPI: PMH: short stature diabetes complete heart block Exam: His ptosis started at age 5 and his parents note that he turns his head more then usual when trying to look around. He also has noted that his balance is off and he occasionally drops objects. Bilateral ptosis and restricted bilateral horizontal eye movements. His fundoscopic exam reveals pigmentary retinopathy. His has 4/5 strength in his proximal arms and legs and is unable to tandem walk. Workup: Serum lactate is slightly elevated. CSF shows elevated protein and lactate. Muscle biopsy shows ragged red fibers. Genetics: mtDNA deletions, usually sporadic 5 question in 14 years Kearns-Sayre Syndrome • Insidiously progressive disease with CPEO before the age of 20. • Symptoms/Clinical Features: • Mitochondrial Myopathy (proximal weakness • CPEO • retinal degeneration (pigmentary retinopathy) • cardiac conduction defects (heart block) • Ataxia/cerebellar syndrome • Other symptoms include small stature, deafness, dementia, delayed puberty, and endocrine dysfunction • Laboratory: Increased CSF protein and lactate • MRI- bilateral subcortical white matter T2 hyperintensities involving basal ganglia, thalamus, and brainstem • Pathology: ragged red fibers • Note: Pearson’s syndrome- sideroblastic anemia, pancreatic insufficiency, and low birth weight precede KSS symptoms. Chronic Progressive External Ophthalmoplegia Ptosis Symmetric ophthalmoplegia with relative sparing of downgaze Facial weakness, frontalis Dysarthria Sparing of ciliary and iris muscles Common presentation of any mitochondrial myopathy Lid surgery is usually not performed due to risk for exposure keratopathy Can be isolated or part of KSS Case 2 14 year old girl CC: New onset seizures HPI: Normal up until 2 months prior to presentation when she started having falls. She then had several seizures, when her parents brought her to the hospital. She became comatose and was intubated. After extubation and waking, she had difficulty using the right side of her body PMH: Normal childhood and development, previously healthy Exam: Cogwheel rigidity in the right arm and wrist, cervical dystonia, bradykinesia and apraxia on the right. Diffuse hyperreflexia, worse right than left, and right Babinksi sign. Workup: MRI shows changes in the basal ganglia, thalamus Spectroscopy shows increased lactate peak Diagnosis? Case 2 – Leigh’s Disease 14 year old girl CC: New onset seizures HPI: Normal up until 2 months prior to presentation when she started having falls. She then had several seizures, when her parents brought her to the hospital. She became comatose and was intubated. After extubation and waking, she had difficulty using the right side of her body PMH: Normal childhood and development, previously healthy Exam: Cogwheel rigidity in the right arm and wrist, cervical dystonia, bradykinesia and apraxia on the right. Diffuse hyperreflexia, worse right than left, and right Babinksi sign. Workup: MRI shows changes in the basal ganglia, thalamus Spectroscopy shows increased lactate peak Diagnosis? Case 2 – Leigh’s Disease Subacute Necrotizing Encephalomyelopathy (SNEM) Clinical Features Developmental delay or psychomotor regression Brainstem dysfunction Respiratory disorders (episodic hyperventilation) Ophthalmoplegia Ataxia Dystonia Seizures, lactic acidosis, vomiting, weakness Peripheral neuropathy, reduced nerve conduction velocity, demyelination Genetics Mutations in either mtDNA or nuclear DNA Pyruvate dehydrogenase (PDHC) most common Imaging/Pathology Bilateral, symmetric necrotizing lesions with spongy changes, microcysts in the basal ganglia, thalamus, brainstem, and spinal cord Prognosis Poor, survival of months from disease onset 5 questions in 14 years Case 2 – Leigh’s Disease Case 2 – Leigh’s Disease Case 3 20 year old woman CC: Right-sided weakness HPI: New symptom of right-sided face, arm, and leg weakness, started yesterday along with a headache Nausea, vomiting Roommate noticed she was sleepy today and not her normal self PMH: Headaches Sensorineural hearing loss Diabetes Exam: Short stature Right homonymous hemianopsia, right hemiparesis, language problems Workup: Serum lactic acidosis MRI Diagnosis? Mitochondrial Encephalopathy Lactic Acidosis and Stroke-Like Episodes (MELAS) • Clinical features• • • • Myopathy Encephalopathy with headaches and vomiting Stroke like symptoms: hemiparesis, hemianopsia most common Short stature, hearing loss, lactic acidosis and diabetes • Mutation- mtDNA point mutations • Laboratory: Lactic acidosis with exercise • MRI: T1-weighted hyperintense cortical signal that are compatible with cortical laminar necrosis & cytotoxic edema. Lesions do not conform to single vascular territories. Also see bilateral basal ganglia calcifications, cerebellar and cerebral atrophy. • Pathology: ragged red fibers • Treatment: Carnitine, Co-Enzyme Q10, Vitamin K, Vitamin C, L-Arginine • 8 questions in 14 years Case 4 22 year old man CC: Vision loss HPI: Blind spot in central visual field in right eye, onset over 5-6 days, painless Diagnosed originally with optic neuritis Presents for second time 2 months later with similar subacute vision painless central vision loss in left eye PMH: None – healthy otherwise Exam: Central visual field loss bilaterally, color desaturation, relatively preserved peripheral vision Workup: MRI brain - normal Visual field testing LP – negative for OCB, IgG index normal Case 4 Diagnosis? Hint Case 4 – Leber’s Hereditary Optic Neuropathy • Bilateral subacute optic neuropathy caused by mtDNA mutations. • Clinical Features: • Painless, severe and permanent subacute central visual loss. • Can be initially unilateral- with second eye often affected in following 1-2 months. • Usually starts in Late teens/early adulthood. • Can occasionally be associated with MS like symptoms/lesions • Fundoscopic exam: Telangiectatic microangiopathy, disc pseudoedema, microangiopathy, or vascular tortuosity. • Pathology: • mtDNA point mutations • No ragged red fibers seen on pathology- only mitochondrial disease with this. • 2 questions in 14 years Case 5 17 year old girl CC: Numbness and clumsiness HPI: Has always been clumsy, but recently she started falling and tripping over objects She says that she has a hard time feeling the ground, trips easily over cracks in the sidewalk or on carpet Feels a little weak in the legs, has some difficulty standing from a low-seated chair PMH: Poor vision, has glasses Learning disorder, has an IEP Exam: MS WNL. CNs show decreased visual acuity, ptosis, fundus exam shows… Proximal LE weakness and decreased sensation distally in lower extremities. Brisk patellar reflexes FNF and HKS shows significant dysmetria. Gait ataxia Workup: MRI brain - normal EMG shows peripheral sensory polyneuropathy, worse LE than UE Case 5 - Neuropathy, Ataxia, Retinitis Pigmentosa (NARP) Clinical features • • • • • Sensory neuropathy Cerebellar ataxia Retinitis pigmentosa Proximal weakness myopathy Ptosis • Mutation • mtDNA point mutation that is the same as seen in KSS and Leigh’s • EMG • Sensorimotor polyneuropathy, myopathic features on needle exam • Pathology • Ragged red fibers • Treatment? • Gene therapy and skin-derived pluripotent stem cell grafts are being studied • For retinitis pigmentosa • Vitamin A, lutein, docosahexaenoic acid, acetazolamide, calcium channel blockers, and valproic acid • 1 question in 14 years – does not have opsoclonus Case 6 22 year old man CC: Seizures and limb jerking, also has weakness and clumsiness HPI: In the last year, has developed seizures of staring, followed by rhythmic jerking of the upper extremities. Occasionally, he has jerking muscle twitches in his arms without loss of consciousness Has fallen twice in the last 6 months at home walking down the stairs and feels a little unsteady while walking, feeling both weak in his legs and having difficulty feeling the ground with his feet Noted some problems with coursework, now getting failing grades in college – he previously graduated high school without difficulty PMH: Hearing loss Short stature Idiopathic cardiomyopathy diagnosed on echo, ordered for mild murmur on exam Exam: MS, CNs, reflexes are normal Proximal LE weakness and decreased sensation distally in lower extremities. FNF and HKS shows mild dysmetria. Gait ataxia Case 6 Workup: Biopsy: EMG: Small amplitude, short duration motor unit action potentials LP: Elevated lactate and pyruvate Diagnosis ? Case 6 - Myoclonic Epilepsy with Ragged Red Fibers (MERRF) • Clinical Features: • • • • • • • Starts in 20’s to 30’s Proximal muscle weakness Epilepsy with Myoclonus Dementia Ataxia Cardiomyopathy Also get pyramidal tract signs, neuropathy, optic nerve atrophy, and neurosensory hearing loss. • Mutation: mtDNA point mutations or deletions or POLG-1 mutation • Laboratory: Increased CSF lactate and pyruvate. • EMG: Myopathic features • Pathology: Neuronal loss in dentate, inferior olive, substantia nigra, gliosis in the cerebellar cortex, and degeneration of the posterior column. • Treatment: Treat Seizures with Keppra rather than VPA, as VPA can cause hepatic failure in these pts with increased frequency. • 3 question in 14 years Cases 7, 8, and 9 3 month old girl Recurrent episodes of hypoglycemia, hypotonia, and profound acidosis 35 year old man Elevated CK and recurrent rhabdomyolysis after vigorous exercise 60 year old woman Asymptomatic What type of mitochondrial disease could they all have? Beta Oxidation Defects • Autosomal recessive • • • • • • • Very Long-Chain Acyl-CoA Dehydrongenase Deficiency Long-Chain Acyl-CoA Dehydrongenase Deficiency Long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD) deficiency Multiple Acyl-CoA Dehydrogenase Deficiency/Glutaritic Aciduria Type II Medium-Chain Acyl-CoA Dehydrongenase Deficiency Short-Chain Acyl-CoA Dehydrogenase Deficiency. Short-chain-3-hydroxyacyl-CoA dehydrogenase (SCHAD) deficiency • Common features include liver disease, heart disease, encephalopathy • May include pigmentary retinopathy, neuropathy, myoglobinuria Treatment: High carbohydrate, low fat diet, administration of medium-chain triglyceride oil, and diet supplementation with carnitine and/or riboflavin. Avoidance of fasting. 5 questions in 14 years Cases 7, 8, and 9 3 month old girl Recurrent episodes of hypoglycemia, hypotonia, and profound acidosis More likely shorter chain defect 35 year old man Elevated CK and recurrent rhabdomyolysis after vigorous exercise More likely longer chain defect 60 year old woman Asymptomatic Any beta oxidation defect Case 10 14 year old girl CC: Dystonia and cognitive regression HPI: Met normal milestones until about 18 months and began toe walking Initially spoke in sentences, but regressed to single words only Significant cognitive impairment, but able to physically keep up with peers until last year when she began to have dystonic posturing of extremities Now is having agitation, outbursts, crying and screaming episodes Dystonia also now involves face, jaw, neck, trunk, and tongue PMH: Cognitive impairment Epilepsy Exam: MS – impaired language, follows only simple 1-word commands CNs – abnormal dystonic posturing of head with neck rotation Strength full but tone increased, reflexes brisk but equal, no obvious sensory changes, cannot do complicated coordination test, gait is stiff-appearing Case 10 Workup: MRI – no significant abnormalities Muscle biopsy – no ragged red fibers CK – not elevated, if anything ~ low Nerve conduction/EMG – no neuropathic or myopathic changes Skin biopsy - normal Copper level, ceruloplasmin – normal range Urine organic acids, plasma amino acids normal VLCFA (Beta oxidation diseases) normal Niemann-Pick A and B, Fragile X, Rett – negative Diagnosis? Creatine Deficiency Syndromes • Autosomal recessive • Guanidinoaceteate Methyltransferase Deficiency (GAMT Deficiency) • L-Arginine:Glycine Amidinotransferase Deficiency (AGAT Deficiency) • X-linked • SLC6A8-Related Creatine Transporter Deficiency (SLC6A8 Deficiency) • All have mental retardation, seizures, speech delay. • GAMT - behavioral disorder - including autistic behaviors; movement disorders • SLC6A8 • Growth retardation • Males: mild to severe mental retardation • Females: learning and behavior problems • In a review of 27 patients with GAMT deficiency, oral creatine supplementation improved behavior abnormalities in all patients, epilepsy in most patients and movement disorder in half of patients. Intellectual ability and speech did not respond. Treatment response appeared more favorable in younger patients.* • 1 question in 14 years * Mercimek-Mahmutoglu S, Stoeckler-Ipsiroglu S, Adami A, et al. GAMT deficiency: features, treatment, and outcome in an inborn error of creatine synthesis. Neurology. 2006 Aug 8;67(3):480-4. Answer Mitochondrial depletion, caused by this drug, can result in myalgias, weakness, and elevated CK in patients with HIV Question • What is AZT? • AZT – a thymidine analog that inhibits reverse transcriptase and mtDNA polymerase (POLG) causing mitochondrial depletion, weakness, myalgias, elevated CK, ragged red fibers\ 4 questions in 14 years Answer This anti-epileptic should not be used in patients with mitochondrial depletion syndromes because of the risk of hepatotoxicity Question • What is valproic acid? • Because of the risk of liver damage, valproic acid is contraindicated in patients with mitochondrial disease, as they are particularly susceptible Other conditions to consider CPT Deficiency • Transport of LCFA from cytosol into the mitochondrial matrix via β-oxidation • Autosomal recessive • CPT I • Hepatomegaly, mostly occurring in infants • Reye’s syndrome “attacks” with fasting or illness • Treatment can include maintaining high glucose intake and medium-chain triglyceride supplementation • CPT II • “Benign” adult form (more than 200 families reported) is characterized by episodes of rhabdomyolysis triggered by prolonged exercise. • Infantile-type CPT2 presents as severe attacks of hypoketotic hypoglycemia, occasionally associated with cardiac damage commonly responsible for sudden death before 1 year of age. In addition to these symptoms, features of brain and kidney dysorganogenesis are frequently seen in the neonatal-onset CPT2 deficiency, almost always lethal during the first month of life. • Treatment involves avoidance of fasting and/or exercise, a low fat diet enriched with medium chain triglycerides and carnitine. • 3 questions in 14 years contained carnitine deficiency, but none were the correct answer Carnitine/Acylcarnitine Translocase Deficiency • Deficiency of this transport protein results in impaired long-chain fatty acid oxidation and causes the accumulation of long-chain acylcarnitines outside the mitochondria and in plasma. • Begins soon after birth • • • • • • Seizures Arrhythmias Hypoketotic hypoglycemia Hyperammoniemia Hepatomegaly Cardiomyopathy • Many infants with CACT deficiency do not survive the newborn period. Some affected individuals have a less severe form of the condition and do not develop signs and symptoms until early childhood. These individuals are at risk for liver failure, nervous system damage, coma, and sudden death. Co-Enzyme Q10 Deficiency • Just like all other mitochondrial disease • • • • Ataxia Encephalopathy Myopathy Endocrine abnormalities Causes in myopathy in patients started on a statin. 3 question in 14 years Mitochondrial Diseases • Summary • • • • • • • • • • • Encephalopathy Myopathy Neuropathy Cardiomyopathy Retinopathy Exercise intolerance Short stature Hearing loss Elevated lactate Ragged red fibers Inheritance • Maternal, recessive, or sporadic • Treatment: • Creatine, Co-Q10, ketogenic diet (other than beta oxidation defects with high carb diet), carnitine, avoidance of fasting, supportive