Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Personalized medicine wikipedia , lookup
Biosynthesis wikipedia , lookup
Butyric acid wikipedia , lookup
Citric acid cycle wikipedia , lookup
Biochemistry wikipedia , lookup
Plant nutrition wikipedia , lookup
Pharmacogenomics wikipedia , lookup
Glyceroneogenesis wikipedia , lookup
Basal metabolic rate wikipedia , lookup
Pharmacometabolomics wikipedia , lookup
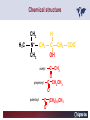
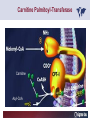
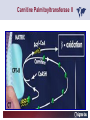
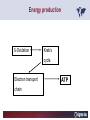
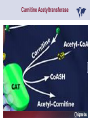
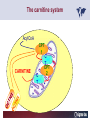
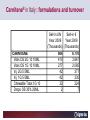
Carnitor Training Session India ST International Department Carnitine movie The essential role of Carnitine • Fatty acid transport • Detoxification of potentially toxic metabolites • Regulation of the mitochondrial acyl-CoA/CoA ratio • Stabilization of cell membranes Fatty acid transport • Transport of long-chain fatty acids across the inner mitochondrial membrane • Carnitine is the only transporter • Beta Oxidation – Krebs Cycle – Resp. Chain – ATP • Carnitine dependent transport of long fatty acids performed by the enzymes: Carnitine palmitoyltransferase I Carnitine–acylcarnitine translocase Carnitine palmitoyltransferase II Detoxification of potentially toxic metabolites • Carnitine is key in the transport of acyl-CoA compounds accumulated within the mitochondria • Metabolic disorders: amino acids, short & medium chain acyl CoA accumulates • Carnitine mediates transport of partially oxidized fatty acids and amino acid fragments out of the organelles in the form of acylcarnitines • Carnitine prevents the acculmulation of acyl-CoA, a surfactant, which destabilizes mitochondrial membranes • Drugs are conjugated with pivalic or valproic acid and metabolized to acyl-CoA esters – cytotoxic • Increased elimination of acylcarnitines: increasd consumption of Carnitine Regulation of the mitochondrial Acyl-CoA/ CoA ratio • Free CoA is an esssential element in the cell’s metabolic pathsways, cell membranes are impermeable to CoA • Carnitine controlls intracelluluar concentrations of acyl-CoA, acetyl-CoA and free CoA: reversible transforamtion of acylCoA and acetyl-CoA into acyl-Carnitine and acetyl-Carnitine, reactions which produde free CoA • Free CoA regulates pyruvate dehydrogenase and of the betaoxidation dehydrogenases, which control the supply of acetylCoA for the Krebs cycle Regulation of the mitochondrial Acyl-CoA/ CoA ratio Ketogenesis Results of high rates of fatty acid oxidation: high amounts of acetyl-CoA are generated, Krebs cycle is saturated, Formation of ketone bodies Results of a low or deficient carbohydrate metabolism: oxaloacetate is low leads to reduced ATP production, increased keton bodies formation – “The glucose sparing effect” Carnitine has a direct control over the rate of production of acetylCoA Stabilization of cell mebranes • Carnitine has a stabilizing effect on cell mebranes • Regulates the turnover of the fatty acids within the phospholipid membranes • The availability of free CoA is of paramount importance for the activation of free fatty acids as acyl-CoA. • CPT I controls the reacylation of phospholipids • Carnetine regulates the cellular acyl-CoA/free CoA ratio Carnitine Deficiencies • Primary deficiency: a genetic defect in the transport of Carnitine across the cell membrane: OCTN2 • Secondary deficiencies: – Inborm errors of Metabolism • Organic acidurias –amino acid oxidation errors • Fatty acid oxidation defects – Carnetine system enzyme defects – Beta oxidation defects – Mitochondrial respiratory chain enzyme defects – Drug-induced Carnetine deficiency – Physiopathologically-induced Carnitine deficiency • Total Parenteral Nutrition • Haemodyalisi • Myocardial Infarction Carnitine movie Mitochondrial carnitine pathway interplay between lipid and glucose metabolism Background 1905 Carnitine is isolated from muscle tissue 1952 Stimulation of fatty acid oxidation 1973 Congenital carnitine metabolism disorders are discovered 1995 Carnitine’s role in acyl-group transfer is recognised 2003 New indications Chemical structure CH3 H3C N+ H _ CH2 CH3 CH2 OH acetyl proprionyl palmitoyl C C CH3 O C CH2CH3 O C (CH2)14CH3 O COO Chemistry Chemical formula: C7H15O3N Molecular weight: 161 Appearance: White powder Solubility: Highly soluble in acetone In the organism The concentration in the tissues depends on the type of metabolism (lipid or glucose). The highest concentration is found in the skeletal muscle and heart. The following highest concentrations are found in adipose tissue, the liver and adrenal cortex. In the organism The total amount of carnitine contained in the body is between 15 and 20 g. More than 95% of which is located in the skeletal muscle and heart. Distribution within the body Body liquid/tissues L-carnitine content L-carnitine total amount nmol / g mg % 1,100 – 3,900 19000 96 600 – 1,200 ~ 60 0.30 Liver 600 – 1,000 ~ 300 1.50 Kidney 300 – 600 ~ 150 0.75 Brain 500 – 1,000 ~ 250 1.25 60 ~ 50 0.25 8,000 – 12,000 ~ 20 0.10 Muscle Heart Blood Epididymes 40 – Pharmacokinetics In metabolically healthy adults, 25 to 35% of normal carnitine requirements are satisfied by endogenous synthesis, the remainder by foods of animal origin. The primary reabsorption of L-carnitine in the small intestine is caused partly by an active and saturable mechanism and partly by a passive, concentrationdependent process. Sources Endogenous synthesis Diet 15 - 20 mg/day 2 - 100 mg/day L-carnitine content of foods (mg/100 g) Mutton 203.0 Where? Beef 61.0 In the liver and kidney Pork 27.0 Sheep’s milk 14.0 Cow’s milk 2.6 Eggs 0.8 Rice 1.8 Bread 0.5 Potatoes 0.05 Carrots 0.0 Cabbage 0.0 From what? The amino acids lysine and methionine Cofactors? Vitamins B6, Niacin, Vitamin C, and iron Pharmacokinetics The primary oral bioavailability for therapeutic doses is 10 to 18%. The free carnitine normal values are: – Serum/Plasma: 32-48 µmol/l (free + acyl-carnitine: 39-68 µmol/l); – Muscle: 24 µmol/g of non-collagen protein; – Myocardium: 6.5-10 µmol/g of non-collagen protein; – Urine: 80-200 µmol/24 h.; – Erythrocytes: 0.1 µmol/g. Excretion The excretion of free and acyl-carnitine is primarily renal. Long-chain acylcarnitine is excreted far more rapidly than free carnitine. Excretion Free carnitine clearance is dose-dependant. For poor intakes (e.g. in vegetarians) it is lower due to higher reabsorption. Conversely, in healthy adults, it increases with the therapeutic dose. Mitochondrial carnitine pathway interplay between lipid and glucose metabolism Acyl-CoA-Synthetase Acyl-CoA-Synthetase Long-chain fatty acids (>12 C) are extracted from triglycerides by an intravascular or intracellular lipase which is then activated by Acyl-CoA Synthetase, located in the outer mitochondrial membrane (and in the microsomes). Carnitine Palmitoyl-Transferase Carnitine Acyl-CoA CPT There are 2 sub-groups : • CPT I is found on the outside face of the mitocondrial membrane. • CPT II is located on the matrix side. CPT and I CPT II have a vast specificity for the medium- to long-chain acyl groups CPT CPT I and II catalyse the reversible transfer of activated fatty acids between CoA and L-carnitine. Acyl CoA + L-carnitine CPT I CPT II CoA + Acyl-L-carnitine Acylcarnitine Acylcarnitine acts as an energy reservoir for storing and transporting the temporarily excessive acyl groups caused by a reduction in fat deposits. Carnitine Translocase Carnitine Translocase The transportation of carnitine or acylcarnitine through the mitochondrial membrane is catalysed by an exchange system, carnitine translocase (CT). Carnitine Palmitoyltransferase II Energy production ß-Oxidation Kreb‘s cycle Electron transport chain ATP Carnitine Acetyltransferase Carnitine Acetyltransferase Carnitine acetyltransferase re-binds the acetyl group of acetyl-CoA to carnitine, to produce acetyl-carnitine and free CoA, which can once more integrate the Kreb’s cycle. Carnitine Translocase The carnitine system AcylCoA CARNITINE CPT 1 C T CPT 2 C T Mitochondrial carnitine pathway interplay between lipid and glucose metabolism Topics • Carnitine: overview on its metabolic role in health and disease • Carnitine biosynthesis, metabolism and functions • Carnitine deficiency: primary and secondary • Dialysis • Drug induced deficiency • Sigma-tau carnitines Primary Carnitine Deficiency (PCD): pathophysiology •PCD is caused by a deficiency in the plasma membrane OCTN2 carnitine transporter expressed in muscle, heart, kidney, lymphoblasts and fibroblasts, with restricted tissue uptake and urinary carnitine wasting causing systemic carnitine depletion. •Intracellular carnitine deficiency impairs the entry of long-chain fatty acids into the mitochondrial matrix. •Consequently, long-chain fatty acids are not available for beta-oxidation and energy production, and the production of ketone bodies (which are used by the brain) is also impaired. http://emedicine.medscape.com/article/942233-overview Rebouche CJ, 2005. Encyclopedia of Dietary Supplements Primary Carnitine Deficiency (PCD): pathophysiology • Plasma membrane OCTN2 carnitine transporter defect is caused by a rare autosomal recessive error of the gene SLC22A5 (located on chromosome 5q31) • The genetic deficiency of the transporter activity represents the only known form of PCD. PCD: epidemiology •In a Japanese study, primary systemic carnitine deficiency was estimated to occur in 1 per 40,000 births. •No incidence studies in the United States. However it may be similar to the incidence in Japan from the cases already reported •In Australia, the incidence has been estimated to be between 1:37,0001:100,000 newborns. •The frequency of this condition in adults is not known. However, in the United Kingdom, a previous report identified 4 affected mothers in 62,004 infants screened, with a frequency of 1:15,500. •Of particular interest is the population of the Faroe Islands, in which the condition is found in about 1 in every 500 people. http://emedicine.medscape.com/article/942233-overview http://www.cphpost.dk/news/135-science/48995-faeroes-susceptible-to-deadly-illness.html PCD: different forms Depending on the time of onset there are three distinct clinical entities: •the adult •the infantile •the perinatale Differences in severity/onset of the disease are due to different mutations in SLC22A5 Flanagan et al., Nutrition and Metabolism 2010, 7:30 PCD: different forms Depending on the system involved there are two forms: • The systemic form, caused by malabsorption of carnitine in the gastrointestinal tract, shows low carnitine in the liver and/or plasma. • The myopathic form, restricted to skeletal muscle, is caused by difficulties in carnitine‘s penetration of muscle cells. Lipid storage myopathy occurs with low muscle carnitine but normal liver and serum carnitine. Both forms cause severe lipid metabolism alterations with lipid storage myopathies and problems in cardiac and skeletal muscle contraction http://www.ncbi.nlm.nih.gov/omim/212160 PCD clinical findings Three tissues/organs are affected in PCD: • Cardiac muscle → progressive cardiomyopathy. Onset may occur with rapidly progressive heart failure or murmur. Cardiomegaly may be found associated with the presence of a heart murmur. A gallop rhythm can be found, associated with a dilated cardiomyopathy. Pericardial effusion. • Central nervous system → encephalopathy caused by hypoketotic hypoglycaemia. The patient may present limp, unresponsive, and comatose after a prolonged fast. Pyramidal movements or minimal athetoid movements can persist. Modest hepatomegaly also can be appreciated, elevated liver transaminases, and hyperammonemia. • Skeletal muscle → Myopathy. In the myopathic presentation, patients may have mild motor delays, hypotonia, or progressive proximal weakness. Other manifestations: • • • GI dysmotility, with recurrent episodes of abdominal pain and diarrhea Hypochromic anemia and recurrent infections Mild developmental delay can be the only manifestation in rare cases Flanagan et al., Nutrition and Metabolism 2010, 7:30. http://emedicine.medscape.com/article/942233-overview PCD clinical findings http://ods.od.nih.gov/news/carnitine_conference_summary.aspx Newborn screen PCD can be identified in infants by expanded newborn screening using tandem mass spectrometry by detection of low levels of free carnitine (C0)*. – Pediatrician needs to contact the family to inform them of the newborn screening result and ascertain clinical status and whether the newborn presents with poor feeding, lethargy or tachypnea. – Consultation with a pediatric metabolic specialist has to be immediately activated and the newborn should be evaluated for tachycardia, hepatomegaly, or reduced muscle tone. After obtaining confirmatory testing with total and free plasma carnitine levels in the newborn and mother, carnitine supplementation. *In addition, newborns’ low carnitine levels may result from PCD in their affected mothers or, on the contrary, can be within the reference range if obtained too early, due to the transfer of carnitine through the placenta to the fetus. Newborn screen – Confirmatory and diagnostic testing can be performed with carnitine uptake assay in cultured fibroblasts and OCTN2 gene sequencing. – Clinical availability of OCTN2 gene sequencing may preclude the need of a skin biopsy and carnitine uptake assay on cultured fibroblasts. – The family has to be educated about signs, symptoms and need for urgent treatment if infant becomes ill Laboratory Studies If the patient is suspected of having PCD and is presenting with a metabolic emergency, the following studies are indicated: 1. Immediate assessment: blood glucose and urine ketones if a child presents to the emergency room with lethargy, seizures, apnea, or any episode of decreased consciousness. 2. Ammonia level, liver enzymes, chemistry panel, uric acid, creatine kinase (CK), lactic acid, and coagulation tests *LCHAD=long-chain 3-hydroxyacyl-CoA dehydrogenase http://emedicine.medscape.com/article/942233-overview Laboratory Studies 3. Plasma carnitine level: In PCD, the carnitine level in plasma is usually less than 5% of normal, with acylcarnitines proportionately reduced. The ratio between acylcarnitine and free carnitine is normal. 4. Urine carnitine level: the transporter in kidney cells has decreased capacity for reabsorption, causing increased carnitine excretion. 5. Fasting test: blood samples at regular intervals to measure glucose, ketone bodies, free fatty acids and acylcarnitine profile. Fasting may be continued in children for up to 24 hours, unless blood glucose drops to less than 3 mmol/L. An inadequate production of ketones with a high free fatty acid–to–ketone bodies ratio suggests a defect in long-chain fatty acid oxidation. Laboratory Studies 6. Fatty acid oxidation study: This is used if a fatty acid oxidation defect is suspected. The most appropriate first line of investigation in these patients is to study the entire fatty acid oxidation pathway. 7. Enzyme assay: This criterion standard for demonstrating an enzyme defect measures the activity in cultured fibroblasts or in some other tissue, such as muscle or liver. 8. Carnitine transport assay in cultured fibroblasts (skin biopsy) specifically demonstrates the absence of active carnitine transport in cultured fibroblasts (specific for PCD). Laboratory Studies 9. Molecular diagnosis provides information on the gene for the carnitine transporter defective in PCD, which has been cloned (OCTN2) and can be screened for mutations. 10. Mutation analysis: the genes for most of the enzymes of fatty acid oxidation that are defective in fatty acid oxidation disorders and may cause secondary carnitine deficiency have been identified, and mutation analysis is available for numerous genes (eg, CPT I, CPT II, VLCAD, MCAD). For example in the adult form of CPT-II deficiency, a C439T mutation accounts for 60% of mutations in patients with adult onset. VLCAD= very long-chain acyl-CoA dehydrogenase MCAD=medium-chain 3-hydroxyacyl-CoA dehydrogenase CPT I and II= carnitine palmitoyltransferase I and II Imaging Studies and other tests Imaging studies: • X-rays reveal cardiac enlargement • The echocardiogram may reveal cardiac enlargement and increased thickness of the left ventricular wall. • Brain imaging studies (eg, cranial ultrasound, brain MRI) may show cystic lesions in glutaric aciduria type II or basal ganglia involvement in mitochondrial disorders that may be associated with secondary carnitine deficiency. Other tests: ECG: The ECG reveals left ventricular hypertrophy and peaked T waves in primary carnitine deficiency. Cardiac arrhythmias can be observed in translocase deficiency and in the lethal neonatal form of carnitine palmitoyltransferase II (CPT-II) deficiency. Therapy • In infants with carnitine deficiency oral L-carnitine supplementation is a lifesaving treatment. • Use of L-carnitine in PCD restores plasma carnitine levels to nearly normal. • Cardiomyopathy often responds well to carnitine supplementation. • Carnitine supplementation in fatty acid oxidation disorders and other organic acidurias is to correct carnitine deficiency and to allow removal of toxic intermediates. • The other goal of therapy is to restore CoA levels. • Carnitine supplementation in total parenteral nutrition (TPN) prevents secondary carnitine deficiency in preterm newborns. http://emedicine.medscape.com/article/942233-overview PCD Therapy: L-carnitine dosing Pediatric • 50 mg/kg/d PO initially; may gradually increase to 100-400 mg/kg/d PO divided bid/tid; not to exceed 3 g/d Adult • 1 g PO/IV tid; not to exceed 3 g/d L-carnitine therapy will need to be continued for life http://emedicine.medscape.com/article/942233-overview Thomson Pharma Drug Report, 2010. Thomson Reuters L-carnitine therapy: left ventricular dimensions http://ods.od.nih.gov/news/carnitine_conference_summary.aspx SCD: patophisiology • SCD can be caused mainly by : inherited metabolic disorders [eg organic acidurias, FA oxidation defects, (carnitine system enzyme defects, β-oxidation defects, respiratory chain enzime defects)] acquired medical conditions (eg haemodialysis, peritoneal dialysis) iatrogenic states (eg drug induced carnitine deficiency) or other factors like poor diet or malabsorption of carnitine or increased renal tubular loss of free carnitine (Fanconi syndrome), SCD: patophisiology • SCD is characterized by increased carnitine excretion in urine in the form of acyl-carnitine due to an accumulation of organic acids • At least 15 syndromes in which carnitine deficiency seems to be secondary to genetic defects of intermediary metabolism or to other conditions • SCD is less severe than PCD with respect to its short-term clinical impact and is much more common SCD: clinical findings • Breastfed infants may experience a catabolic state shortly after birth, when the production of milk is not adequate to meet nutritional requirements. • Acute metabolic decompensation with hypoketotic or nonketotic hypoglycemia usually occurs in infancy, whereas cardiac and skeletal muscle disease manifest later. Patients may have a history of developmental delay. • Patients with organic acidemias causing secondary carnitine deficiency may present with crises consisting of hypoglycemia, ketoacidosis, and hyperammonemia. • Carnitine deficiency has been observed in children with urea cycle defects, and it may exacerbate episodes of hyperammonemia. • Signs and symptoms related to carnitine deficiency are not completely defined in the newborn. Apnea, cardiac death, and sudden death have been found in infants with carnitine depletion. SCD: clinical findings • Episodes of metabolic decompensation triggered by infection or fasting may present with lethargy that may be accompanied by seizures or apnea. • This encephalopathy may also present with hypotonia and hepatomegaly. • Signs of cardiac hypertrophy may be evident, with gallop or heart murmur on the cardiac examination. • Less frequently, these patients may have other findings, such as pigmentary retinopathy, peripheral neuropathy, cardiac arrhythmias, or myoglobinuria. • Carnitine deficiency can develop in children with renal Fanconi tubulopathy; it may be idiopathic and present with renal tubular acidosis or secondary to acquired or inherited conditions. Therapy Administration of carnitine improves clinical evolution and reduces the frequency of metabolic attacks. SCD Therapy: Pediatric dosing • Initial: -50 mg/kg IV bolus (over 2 to 3 min) or infusion; repeat 50 mg/kg IV daily if clinically indicated; max dose 300 mg/kg -50 mg/kg orally in divided doses daily, titrate slowly to therapeutic response; max dose 3 g daily • Severe metabolic crisis, initial, 50 mg/kg IV over 2 to 3 min bolus injection or infusion, repeat 50 mg/kg IV in divided dose over 24 hr (every 3 to 4 hr and not less than every 6 hr); repeat 50 mg/kg IV daily if clinically indicated; MAX dose 300 mg/kg Thomson Pharma Drug Report, 2010. Thomson Reuters SCD Therapy: adult dosing • Initial, 50 mg/kg IV bolus (over 2 to 3 min) or infusion; repeat 50 mg/kg IV daily if clinically indicated; MAX dose 300 mg/kg • Severe metabolic crisis, initial, 50 mg/kg IV over 2 to 3 min bolus injection or infusion, repeat 50 mg/kg IV in divided dose over 24 hr (every 3 to 4 hr and not less than every 6 hr); repeat 50 mg/kg IV daily if clinically indicated; MAX dose 300 mg/kg • Tablet, 1 g ORALLY 2 to 3 times daily • Oral solution, initial, 1 g orally daily, titrate slowly to therapeutic response; average dose 1 to 3 g orally daily for 50 kg adult Thomson Pharma Drug Report, 2010. Thomson Reuters Secondary genetic carnitine deficiency • CPT-I deficiency Carnitine palmitoyltransferase 1 • CPT-II deficiency Carnitine palmitoyltransferase 2 Secondary genetic carnitine deficiency CPT-I deficiency • Carnitine palmitoyltransferase I (CPTI) deficiency is thought to cause serious disorders of fatty acid metabolism. • The nucleotide sequences of cDNA and genomic DNA encoding human CPTI have been characterized • However, a relationship between disease and mutation of the human CPTI gene has not been reported Secondary genetic carnitine deficiency CPT II deficiency • The adult CPT II clinical phenotype is somewhat benign and requires additional external triggers such as high intensity exercise before the predominantly myopathic symptoms are elicited. • The perinatal and infantile forms involve multiple organ systems. • The perinatal disease is the most severe form and is invariably fatal. Secondary genetic carnitine deficiency CPT II deficiency • The most frequent symptom of muscle palmitoyltransferase CPT II deficiency is an exercise induced myalgia. • Myoglobinuria, is the traditional hallmark of this disease • Myalgia typically starts in childhood while myoglobinuria starts later in adolescence or early adulthood. Secondary genetic carnitine deficiency CPT II deficiency • One case study found a novel mutation in CPT II (del1737C), an autosomal recessive disease with a distinct phenotype. A two- day old boy died due to severe hepatocardiomuscular disease with an extreme early onset. His sister also died. Upon autopsy the brother showed massive pulmonary atelectasis with intra-alveolar hemorrhage, cardioand hepato-megaly. The sister died of sudden cardiopulmonary arrest due to the increase of long-chain (C16-18) acylcarnitines. Decreased CPT II activity was found in her liver, heart and kidney. The cause of death was neonatal CPT II deficiency. Topics • Carnitine: overview on its metabolic role in health and disease • Carnitine biosynthesis, metabolism and functions • Carnitine deficiency: primary and secondary • Dialysis • Drug induced deficiency • Sigma-tau carnitines DIALYSIS Dialysis Severe kidney failure is still a serious health problem. The greatest problems for patients are the kidney failure itself and the side effects of dialysis. Post-dialysis syndrome Symptoms • Chronic fatigue • Impaired performance • Low cardiac output • Poor state of nutrition • Hypotension during dialysis • Muscle pains and/or cramps during dialysis L-carnitine effects on skeletal muscle in hemodialysed patients Dialysis Haemodialysis extracts carnitine directly from the “fast equilibrating pool” (blood and liver). After a dialysis session, the plasma carnitine concentration normalises through the mobilisation of carnitine from the “slow equilibrating pool” towards the “fast equilibrating pool”. Dialysis The consequence of this process is a loss of carnitine from the “slow equilibrating pool“ and the onset of secondary carnitine deficiency. Post-dialysis carnitine supplementation compensates this loss in the “fast equilibrating pool“ and reduces the loss in the “slow equilibrating pool“. Dialysis The plasma levels of free carnitine at the end of a dialysis session must not be below the threshold of 40 µmol/l. To maintain this level, plasma concentrations prior to dialysis must be within the 120 - 150 µmol/l range. Dialysis and anaemia Anaemia represents one of the greatest problems for dialysis patients. L-carnitine alters red blood cell deformability. L-carnitine administration reduces erythropoietin requirements in about 45% of patients. Carnitine therapy Administration of carnitine : • increases plasma and muscle carnitine levels. • improves tolerance of exertion. • reduces intradialytic muscle cramps and hypotension. • improves cardiac function. Carnitine therapy • improves general clinical condition. • improves quality of life. • improves serum markers related with nutrition: it increases albumin and reduces urea and creatinine. • improves erythropoietin-resistant anaemia. Therapeutic applications In dialysed patients, L-carnitine helps to compensate the loss of carnitine and prevents the onset of postdialysis syndrome. L-carnitine is consequently indispensable for the treatment of these patients. L-Carnitine and renal anaemia Improvement in haematocrit Source: Trovato et al. Current Therapeutic Research1982 L-Carnitine and renal anaemia Reduced rh-EPO levels Source: Veséla et al. Nephron 2001 Reduction in Rh-EPO levels whilst taking L-Carnitine a) Kavadias et al. EDTA Congress 1995 b) Labonia et al. American Journal of Kidney Diseases, 1995 c) Boran et al. d) Patrikarea et al. Nephron, 1996 EDTA Congress 1996 Therapy-dialisys Pediatric and Adult SCD- Treatment and Prophylaxis - End stage renal disease - Hemodialysis • initial, 10 to 20 mg/kg (dry weight) over 2 to 3 min bolus injection after dialysis; subsequent dose titration determined by trough (predialysis) levocarnitine concentrations (Prod Info CARNITOR(R) injection, 2004) Thomson Pharma Drug Report, 2010. Thomson Reuters Topics • Carnitine: overview on its metabolic role in health and disease • Carnitine biosynthesis, metabolism and functions • Carnitine deficiency: primary and secondary • Dialysis • Drug induced deficiency • Sigma-tau carnitines Valproic acid intoxication Valproic acid Most patients on valproate treatment have low plasma carnitine levels. An even greater reduction is observed in patients on valproate therapy who are simultaneously taking supplementary antiepileptic medication. Valproic acid Valproate (Depakine®) is an antiepileptic medication. This illness often requires long-term therapy and in addition to valproate, patients are often taking other substances. Patients on valproate medication experience more consistent reductions in acylcarnitine and a higher proportion of acylcarnitine to free carnitine. Carnitine deficiency may present in children being treated with valproic acid and may be associated with fulminant liver failure and presentation similar to that in Reye syndrome. It also may present with a myopathy and increased lipid storage in patients with AIDS who are being treated with zidovudine. Valproic acid Treatment with carnitine can block a tendency to hyperammonaemia and hypocarnitinaemia and result in an improvement in terms of carnitine and greater acylcarnitine clearance without reducing the effects. Carnitine supplements also eliminate toxic acyl CoA as acylcarnitine. Topics • Carnitine: overview on its metabolic role in health and disease • Carnitine biosynthesis, metabolism and functions • Carnitine deficiency: primary and secondary • Dialysis • Drug induced deficiency • Sigma-tau carnitines Sigma-tau Carnitines: know-how • In 1977 sigma-tau synthesizes the biologically active L-carnitine • In 1978 sigma-tau is the first inventor of L-Carnitine industrial production process (Italian Patent no.1156852) • First launched in 1982 in Italy • US FDA Orphan Drug designation in 1985* (primary deficit) followed by those in 1992 (secondary deficit) and in 1999 (dialysis deficit) • A wide library on carnitines present in sigma-tau *second Orphan Drug Designation awarded by US FDA Sigma-tau Carnitines Three active ingredients: • L-Carnitine (LC) • Acetyl L-Carnitine (ALC) • Propionyl L-Carnitine (PLC) Three product families Sigma-tau Carnitines Three presentations: • End Products • API • Nutraceuticals containing Carnitines Three forms requested by the MKT Sigma-tau Carnitines: End Products REGION North America Central/South America COUNTRY Product name USA Carnitor CANADA Carnitor COSTA RICA Cardispan GUATEMALA Cardispan HONDURAS Cardispan MEXICO Cardispan NICARAGUA Cardispan PANAMA Cardispan ARGENTINA Albicar CHILE Carnicor COLOMBIA Carnitene ECUADOR Carnitine VENEZUELA Carnitene ALBANIA Carnitene L-Carnitine Fresenius Carnitene L-Carnitin "Fresenius" Levocarnil AUSTRIA BELGIUM CZECH REPUBLIC FRANCE 37 country registrations 12 Brands GERMANY L-Carn GREECE Superamin Carnitene Sigma-Tau HOLLAND Europe ITALY Carnitene Nicetile Dromos MALTA Carnitene PORTUGAL Disocor SPAIN Carnicor Carnitene Sigma-Tau SWITZERLAND A wide diffusion Asia Middle East UNITED KINGDOM Carnitor HONG KONG Carnitor INDIA Carnitor KOREA L-Carn PEOPLE'S REP. OF CHINA Carnitene PHILIPPINES Carnicor RUSSIAN FED. Carnitene TAIWAN Carnitene AZERBAIJAN Carnitene ISRAEL Carnitine TURKEY Carnitene Sales: End Products in Units END PRODUCTS BASED ON: Units 2009 L-CARNITINE ACETYL L-CARNITINE PROPIONYL L-CARNITINE TOTAL Relevant revenues 4.944.415 1.277.693 268.981 6.491.089 Units 2008 4.735.636 1.224.602 280.358 6.240.596 +-% 4 4 -4 4 Total 2009 turnover for ST: 56 mio € Internal data € ST Carnitines Trend: End Products (Units) 7,0 6,0 6,0 6,2 6,5 5,5 5,0 4,7 4,0 Units (mio) 3,0 2,0 1,0 0,0 2005 2006 2007 2008 2009 A positive trend since several years Internal data Sales: API (Kg) API L-Carnitine Acetyl L-Carnitine TOTAL Relevant revenues Kg 2009 34.885 36.000 70.885 Kg 2008 29.850 32.550 62.400 +-% 17 11 14 Total 2009 turnover For ST: 8,9 mio € Internal data Nutraceuticals containing Carnitines PRODUCT NAME ITALY SWITZERLAND ITALY 2 AMEDIAL AZERBAIJAN BELGIUM BULGARIA HOLLAND ITALY PAKISTAN PERU POLAND 3 PROXEED_plus SAUDI ARABIA SERBIA SINGAPORE TUNISIA TURKEY UK UNITED ARAB EMIRATES USA ITALY 4 CARNIDYN plus AZERBAIJAN ITALY LIBIA 5 EZEREX PAKISTAN POLAND SERBIA AZERBAIJAN BELGIUM 6 BIO-RECORD PLUS HOLLAND ITALY AZERBAIJAN BELGIUM HOLLAND HUNGARY 7 PHOTOTROP ITALY POLAND SERBIA US ITALY 8 AVANT ITALY 9 RIABYLEX ITALY 10 RESTORFAST ITALY 11 CARNIFAST US 12 ALCAR 1 CARNIDYN 19 country registrations 12 Brands A wide diffusion COUNTRY Sales: Nutraceuticals containing Carnitines API L-Carnitine (LC) Acetyl L-Carnitine Propionyl L-Carnitine (PLC) Both LC and PLC TOTAL Relevant revenues Un. 2009 420.815 797.976 115.889 112.193 1.446.873 Un. 2008 279.352 703.019 68.024 74.368 1.124.763 +-% 51 14 70 51 29 Total 2009 turnover For ST: 16,1 mio € Internal data Sigma-tau Carnitines: Total 2009 business • L-Carnitine (LC) • Acetyl L-Carnitine (ALC) • Propionyl L-Carnitine (PLC) 81 mio € A very important business Internal data Carnitene®: key facts Carnitene® Product name Active ingredient L-Carnitine ATC A16A other metabolic products - C1X all other cardiac preps Reference market A16A other metabolic products - C1X all other cardiac preps Indications Primary and secondary carnitine deficiencies Formulations (packs) 1g/5mL solution for injection-5 ampoules of 5 ml 2g/5mL solution for injection-5 ampoules of 5 ml 1g/10mL oral solution-10 vials of 10 mL 2g/10mL oral solution-10 vials of 10 mL 1g chewable tablets-10 tabs in blister 1,5g/5mL oral solution-bottle of 20 mL Daily dosage Oral: Primary carnitine deficiencies and deficiencies secondary to inborn errors: adults and over 12 years: 2-4 g (from 0 to12 years: depending on age and body weight) Secondary deficiency due to haemodialysis: 2-4 g Injection: Secondary deficiency due to haemodialysis: 2 g at the end of the dialytic session (slow i.v.infusion) Medical target Pediatrician, Nefrologist, Cardiologist Registered in (date of first reg.) Europe (1969), America, Asia Legal classification Inj.forms : Rx only; Oral forms: no subject to medical prescription Carnitene®: Top down Targeting KOL in Pediatrics Pediatricians, Nephrologists Cardiologists, Geriatricians Other specialists and GPs Carnitor® in UK: an example of sigma-tau L-carnitine Country registration C O UN T R Y D is t ribut o r P ro duc t na m e F o rm ula t io ns 1s t re g. O ra l s ingle do s e 1g T a ble t s 1g T a ble t s 3 3 0 m g 19 9 3 19 9 4 19 9 0 Indic a t e d in t he t re a t m e nt o f prim a ry a nd s e c o nda ry c a rnit ine de f ic ie nc y in a dult s a nd c hildre n o v e r 12 ye a rs o f a ge . 19 9 2 Indic a t e d in t he t re a t m e nt o f prim a ry a nd s e c o nda ry c a rnit ine de f ic ie nc y in a dult s a nd c hildre n up t o 12 ye a rs o f a ge , inf a nt a nd ne w- bo rns 19 9 4 Indic a t e d in t he t re a t m e nt o f prim a ry a nd s e c o nda ry c a rnit ine de f ic ie nc y in a dult s , c hildre n, inf a nt s a nd ne o na t e s . S e c o nda ry c a rnit ine de f ic ie nc y in ha e m o dia lys is pa t ie nt s . Seco ndary carnitine deficiency sho uld be suspected in lo ng-term haemo dialysis patients who have the fo llo wing co nditio ns: 1) Severe and persistent muscle cramps and/o r hypo tensive episo des during dialysis. 2) Lack o f energy causing a significant negative effect o n the quality o f life. 3) Skeletal muscle weakness and/o r myo pathy. 4) Cardio myo pathy. 5) A nemia o r uremia unrespo nsive to o r requiring large do ses o f erythro po ietin. 6) M uscle mass lo ss caused by malnutritio n. 3 0 % P a e dia t ric s o l. UN IT E D KIN G D O M S - T P ha rm a Lt d C a rnit o r Inje c t io n 1g Indic a t io ns Carnitene® in Italy : formulations and turnover Sell-in € Sell-in UN Year 2009 Year 2009 (Thousands) (Thousands) CARNITENE Vials OS 2G 10 10ML Vials OS 1G 10 10ML Inj. 2G 5 5ML Inj. 1G 5 5ML Chewable Tabs 1G 10 Drops OS 30% 20ML 804 410 277 42 42 32 2 6.770 3.847 2.085 377 230 224 7 Carnitene® in Italy : Current Target Pediatrician, Nefrologist, Cardiologist, Geriatrician Carnitene® in Italy : Current Positioning Pediatrician Carnitene® is a unique efficacious drug in primary carnitine deficiency Nephrologists Carnitene® is efficacious in secondary deficiencies due to haemodialysis. Carnitene® may reduce erythropoietin resistance in hemodialyzed patients (HD). Carnitene® is associated with decreased hospital utilization among HD. This results have important implications for healthcare cost containment. Cardiologist-Geriatrician Carnitene® is a unique efficacious drug in asthenia derived from secondary carnitine deficiency eg in the elderly, in multi drug treated patients, in malnourished or in cardiopathic patients GPs Carnitene® is a unique efficacious drug in asthenia derived from secondary carnitine deficiency eg in the elderly, in multi drug treated patients, in malnourished or in cardiopathic patients