Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
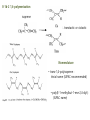
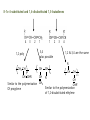
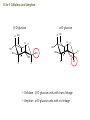
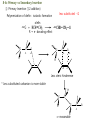
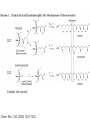
Chapter 8. Stereochemistry of Polymerization ※ Constitutional (Structural) isomerism i) From different monomers Nylon 6 Nylon 6,6 ii) From a single monomer • head-to-head & head-to-tail • 1,3-dienes : 1,2- and 1,4- polymers • monomers with alkene & carbonyl groups (5-7a) • linear or cyclized polymers (6.6b) From radicals From ions 8-1 Types of Stereoisomerism in Polymers → arises from different spatial arrangement (configuration) ↔ conformational isomer : interconvertible 8-1a Monosubstituted Ethylenes 8-1a-1 Site of Steric Isomerism H2C CH R ⇒ Possible chirality arises from the differences of the length Chiral property • Optical rotation, different properties in chiral environment • Same physical property → The carbons at the end of the polymer chain can have optical activity : Population is very negligible → Other carbons : not much optical activity ∴ The sum of optical activity is below the limits of detection ⇒ rather achirotopic (achiral) 8-1a-2 Tacticity Atactic : R groups on successive stereocenters randomly distributed Isotactic : the stereocenters has the same configuration Tactic Syndiotactic : the configuration of the stereo Stereospecific polymerization : polymerization that yields stereoregular polymers ※ Stereoregular : isotactic or syndiotactic 8-1b Disubstituted Ethylenes 8-1b-1 1,1-disubstituted ethylenes R H2C C R' R & R’ are the same : no stereo isomerism R & R’ are different : atactic, isotactic, syndiotactic 8-1b-2 1,2-disubstituted ethylenes -Diisotactic : each of the two stereocenters is isotactic - Disyndiotactic : each of the two stereocenters is syndiotactic - Erythro : R and R’ is anti, H and H is anti - Threo : R and H, : R’ and H is anti, then R and R’ is gauche ⇒ four possible isomeric structures; threodiisotactic, erithreodiisotactic, Threodisyndiotactic, erithreodisyndiotactic Except the End group they are identical, 8-1c Carbonyl and Ring-opening polymerizations • Polyacetaldehyde : Carbonyl O CH3CH H C O CH3 n Iso & Syndio 8-1c Carbonyl and Ring-opening polymerizations • Poly(propylene oxide) or Poly(oxy[1-methylethylene]) O OCHCH2 CH3 Iso or Syndio 8-1d 1,3-Butadiene and 2-substituted 1,3 butadienes 8-1d-1 1,2 and 3,4- polymerization butadiene 1,2 3,4 Same Product ⇒ iso, syndio, atactic ⇒ three different structures are possible Isoprene or chloroprene 4 1,2 poly ⇒ iso, syndio, atactic 3 2 1 3,4 poly ⇒ iso, syndio, atactic ⇒ six different structures are possible 8-1d-2 1,4-polymerization isoprene ∴ transtactic or cistactic Nomenclature • trans-1,4-polyisoprene : trivial name (IUPAC recommended) • poly(E-1-methylbut-1-ene-2,4-diyl): (IUPAC name) 8-1e 4-substituted and 1,4-disubstituted 1,3-butadienes 4 3 2 1 1 3,4 also possible 1,2 poly 2 3 4 1,2 & 3,4 are the same R CH2 CH CH n CH CHR CH n CH CH CH R CH CH 2 Similar to the polymerization CHR' Similar to the polymerization Of propylene of 1,2-disubstituted ethylene n * 1,4 polymerization of 4-substitutes cis chiral Still not optically active possibilities ① Trans Iso ② Cis Iso ③ Trans Syndio ④ Cis Syndio ① Trans atactic ① Cis atactic 8-1e-2 1,4-polymerization of 1,4-disubstituted 1,3-butadienes possibilities R&H anti Cis & Trans R & R’ anti Threo & Erythro disyndio & diiso See Fig. 8.2 + cis & trans See p 631 XIV : erythrodiisotactic R&R’ anti, R&R iso, R’&R’ iso Transerythrodiisotatic 1,4-poly(methyl sorbate) Diisotactic poly[erythro-3-(methoxycarbonyl)-4-E-methylbut-1-ene-1,4-diyl] 8-1f Other polymer Poly(cyclopetane-1,2-diyl) (IUPAC) or polycyclopetene Erythro: cis of chain bonds entering and leaving each ring Threo: trans of chain bonds entering and leaving each ring cis trans 8-2 Properties of stereoregular polymers 8-2a-1 Isotactic, Syndiotactic, and Atactic P.P • Atactic : Amorphous (noncrystalline), soft (tacky) material With no physical strength Application : asphalt blends, formulations sealant & adhesives • Isotactic & Syndiotactic : highly crystalline ( ∵ ordered structure ) high physical strength increased solvent & chemical resistance Application : plastic & fiber • Isotactic PP Tm ~176˚C → Easily synthesized from stereospecific polymerization • Syndiotactic PP Tm ~156˚C → not easily synthesized ! 20˚C lower than that of isotactic, more soluble in HC solvent 8-2a-2 cis- and trans- 1,4-poly-1,3-dienes see P633 Table 8-1 Tm, Tg of trans is higher than those of cis ∵ The trans isomer crystallize to a greater extent as a result of higher molecular symmetry. Natural rubber : 1,4-polyisoprene Mostly cis 8-2a-3 Cellulose and Amylose β-D-glucose α-D-glucose H OH H OH H H O O HO HO OH HO H H H HO H OH H OH H • Cellulose : β-D-glucose units with trans linkage • Amylose : α-D-glucose units with cis linkage OH 8-2a-3 Cellulose and Amylose • Cellulose : fully extended chain hydrogen bonded into sheet ∴ crystalline, physical strength, decreased solubility, stable to hydrolysis • Amylose : random-coil 8-2b Analysis of Stereoregularity → IR & NMR How :the difference of sequence distribution ① Dyad tacticity R R CH CH2 CH R CH2 CH CH2 CH R meso ≡ m racemic ≡ r CH2 8-2b Analysis of Stereoregularity ② Triad tacticity mm rr mr Example) 6 meso dyads , 2 racemic dyads ∴ ∴ High resolution NMR Tetrads, Pentads are also possible • Tetrads • Pentads 8-3 Forces of stereoregulation of alkene polymerizations depends, • rates of addition (Temp) • monomer • initiator • active end ⇒ case by case 8-3a Radical polymerization ⇒ freely propagating species : chain end can freely rotate ∴ don’t have specific configuration ⇒ configuration is determined after the monomer is added. Kr & Km exist (racemic or meso) ΔΔH+ ∼ = -4 ~ -8 kJ/mol + ΔΔS + ∼ = 0 ~ -4 J/mol∙K + ∴ Syndiotactic is favored over isotactic because of the enthalpy difference. Ex) bulkier + ΔΔH+ + ΔΔS + MMA -4.5 kJ/mol -4.2 J/mol∙K VC -1.3 kJ/mol -2.5 J/mol∙K * Syndiotactic is favored because of steric and/or electrostatic repulsions between the substituent ∙ T ↑ , Kr/Km → 1 Ex) PVC , Since radical polymerization is performed above 60˚C, → atactic with syndio rich. 8-3b Ionic and Coordination Polymerization 8-3b-1 Effect of Coordination • In good solvent : free ion or solvated ion pair → Similar to radical polymerization Syndio favored ! • In poor solvent (coordination is possible) → Stereospecific polymerization can occurs Usually isotactic , sometimes syndiotactic including Z-N Catalyst ? To form isotactic polymer, Key point : coordinates in one direction 8-4 Traditional Ziegler-Natta polymerization of nonpolar alkene monomers 8-4a History The significance of Ziegler-Natta initiators → can polymerize α-olefin Cannot be polymerized by radical & ionic mechanism ⇒ TiCl3 with Al(C2H5)2Cl or TiCl4 with Al(C2H5)3 in HC solvent with olefin ⇒ then polymers are obtained in situ as a precipitate ⇒ Many trial and errors ⇒ -TiCl3 (brown color) was found to be effective for the polymerization (prepared by mixing TiCl4 with H2, aluminium, or alkylaluminiums Still isotacticity 20 – 40 % with Al(C2H5)2Cl -, -, -crystals of TiCl3 were found to increase the isoctacticity Finally used initiator preparation method ① Mixing (by ball milling)of magnesium chloride (or alkoxide) as a solid support and TiCl4 ② Addition of Al(C2H5)3 with an organic Lewis base (external base; water, ester, phenol) ⇒ high activity (50- 200 kg of polymer per gram of initiator system ex) initiator 2-4% of Ti, so 1500-6000 kg polymer per gram of initiator So very small amount of initiator in PE or PP + 90~98% isotactic 8-4b Chemical nature of propagating species ⇒ Radical, ionic, or coordination is possible according to the conditions. ex) ① The use of alkyl lithium as group I-III metal component (early days) Not AlR3 ⇒ anionic polymerization ex) MMA ② Monomers with e--rich double bond cationic by TiCl4 ex) vinyl ether) ③ Radical can be generated Ionic character Possibly anioic Poly rate: Ethylene>propene>1-butene The reverse of cation stability ④ Stereospecific polymerization → generally coordination mech. Evidences for the anionic character • Polymerization rate: Ethylene>propene>1-butene • Carbanion attacks at the carbon to form less substituted (more stable carbanion) • Size -substituent ↑, reactivity ↓ • Termination with CH3O3H radioactive, while that with 14CH3OH is not radioactive 8-4c Primary vs Secondary Insertion ① Primary Insertion (1,2 addition) less substituted - G Polymerization of olefin : isotactic formation olefin R → e- donating effect C C -δ G C +δ R C -δ G +δ R +δ -δ Less steric hinderence * Less substituted carbanion is more stable H C R C C C -δ -δ R ⇒ reasonable G 8-4c Primary vs Secondary Insertion ② Secondary Insertion (2,1-addition) → substituted chain end is attached to G Attack to less substituted part like radcial -δ +δ When R is very bulky, then secondary insertion is possible -δ +δ C G R due to the size If R is very bulky, then secondary insertion is possible, like radical polymerization. So syndioselective polymerization proceeds through secondary insertion 8-4d Propagation at Carbon-Transition Metal Bond Ziegler-Natta catalyst Transition metal (Ti, Zr, V ···) + Group I-III metals (Li, Al ···) which is G Conclusion ⇒ Transition metal • No polymerization of olefins with Group I-III metals • With transition metal alone, olefins can be polymerized → less stereospecific ⇒ Transition metal + Group I-III metal • increased reactivity ! • stereospecific reaction ! 8-4e Mechanism of isoselective propagation Active sites are formed from tatanium chloride and alkyaluminiums TiCl4 with AlR3 or TiCl3 with AlR2Cl ① Surface structure of TiCl3 crystal I. Four Cl’s are shared with another Ti nearby. II. R : on the surface of TiCl3 could be polymer chain III. Empty site ⇒ monomer can approach Mixing with MgCl2 Mg-Cl···Ti linkage is helpful. Mg and Ti are interchangeable in The metal lattice The actual possible structure ② Other possible structure Since Group I-III metals are helpful Possibility R Al R Cl Cl Ti Cl Cl R Same as monometallic R & Cl is shared with Al. Propagating site is shared ! Polymerization mechanism for isoselective polymerization The whole polymer chain does not migrate, only the active end of the Polymer chain migrates. Ti is always bonded less substituted carbon The migratory insertion only the atoms of the first few repeat units move. migration different position CH3 isotactic ∵ migration regenerate the original conformation. ⇒ same position addition Ti CH2 C CH3 syndiotactic (∵ steric effect) alternating position addition If propagation continued with the vacant site contineously, the syndioselective propagation can occur. Then the Position changes! 8-4f Syndioselective propagation Syndiotactic rich polymerization is mostly possible from soluble initiator, vanadium comppounds, still the selectivity is not very high ex)rr=0.9 8-4g Direction of Double-bond opening for 1,2 disubstituted monomers Isotactic polymerization ⇒ monomer approaches the propagating center with the same face, then rotate to form threodiisotactic, trans monomer produces diisotatic incoming monomer is aligned so as to minimize steric repulsions between the R group of the chain end and the R groups on the monomer (R groups a re opposed to each other) Syn addition: It is possible the transition metal directs the stereochemistry. 8-4a-6 Mechanism of isotactic control If they coordinate in the same manner ⇒ isotactic If not ⇒ atactic In alternative way → active site changes ⇒ syndiotactic Ti • always same position , same direction ⇒ isotactic polymer R 8-4h Effect of component of Ziegler-Natta initiator 8-4h-1 Transition-metal TiCl3 → isotactic VCl3 → syndiotactic Order of stereospecificity α, γ, δ – TiCl3 > TiCl2 > TiCl4 ~ = β-TiCl3 layered structure Composed of bundle of linear TiCl3 chains β-TiCl3 : on the surface Half of Ti ⇒ one vacant site Half of Ti ⇒ two vacant site ∴ Two sites ⇒ stereospecific polymer is not possible Actual situation Use TiCl4 rather than TiCl3 ∵ TiCl4 : liquid → handling is easy → by adding AlR3 or AlR2Cl, TiCl4 is reduced to TiCl3 Actual initiator The extent of isotacticity for propene polymerization or α-TiCl3 > CrCl3 > VCl3 > FeCl3 TiCl4 ~ = VCl4 ~ = ZrCl4 ⇒ reasons are not very clear but there is a trend. stereoselectivity and activity are dependent on the crystal structure, steric size, ligands, and the electronegativity of the transition metal. • Ti3+ (trivalant oxidation state): more stereospecific and active • Ti4+ (tetravalant oxidation state): less stereospecific and active not much dependent on the ligand Decreasing the electronegativity of the transition metal (more easily polarized metal growth bond), increasing the stereoselectivity Increasing the size of the ligands decreases stereoselctivity probably because of a decreased coordinating ability. 8-4h-2 Group I-III Metal component • not an absolute necessity • but need to obtain high stereospecificity and high activity * Group I : Li , Na, K → not very attractive ∵ low solubility in hydrocarbon solvent Lithium → soluble but aggregation * Group II : Be , Mg , Zn , Cd Be : not used ( ∵ toxic ) Zn, Mg : widely used * Group III Al : most widely used ( ∵ active ) Ga : active but expensive 8-4h-3 Third component : Lewis base → oxygen, water, inorganic halide (KCl, NaF), organic halide, phenols, ethers, amines, phosphines, aromatics ∙∙∙ ⇒ case by case ( ∵ ) Internal base & External base The preparation of initiators ① Internal base is ball milled with support material and the transition-metal. (TiCl4 + MgCl2) ② Then group I-III compound + external base are added Ester : internal base , also external base Phenyl triethoxysilane : external base ⇒ change the stereospecificity 8-4i Kinetics → very complicated (∵ heterogeneous system !) ball milling after sometime large crystal aggregates Polymer chain cleaves the aggregates. ⇒ Ball milling can reduce the induction period. Different active sites are existing TiCl4+AlR3 MgCl2 milling TiCl2 + MgCl2 milling TiCl3 Low activity TiCl3 High activity 8-4i-2 Termination → probably no termination * Active site have a character of living polymer, not the propagating chain ① Spontaneous intramolecular β-hydride transfer still active Chain transfer to monomer active ② Chain transfer to the group I-III metal alkyl ③ Chain transfer to H2 or other compound ④ Many other complicated unknown reactions 8-4i-3 Rate expression Rp=kp[C*][M] Conc. Of active site → If homogeneous, then true * Langmuir-Hinshelwood model: Reaction occurs only after monomer is absorbed from the solution onto the transition metal active sites. • Group I-III metal compounds are also soluble in the solution & competes with monomers for the same sites, they are normally excess. Therefore the polymerization of monomer competes with the adsorption of I-III Metals. [A] and [M] are the group I-III metals and monomers and KA anf KM are equilibrium constant for the adsorption Then, Rp=kp[C*][M] becomes Xn → same as radical polymerization and 8-4i-4 Values of kinetics parameters [C*] : conc. of active site ⇒ determined by quenching with CH3OH, ∵ Ti 14CO, 14CO 2 ∙∙ coordinated with O the unpaired e-’s then reaction stops ⇒ measures the consumed MeOH, CO2, CO Problem : obtained [C*] could be higher than the real value ∵ The products from chain transfer to the group I-III metal alkyl Ti + AlR3 Ti R + Al both react with O ∙∙ [C*] is hundredthes of a percent (0.01%) to tenths of a percent (0.1%) of the metal concentration 0.65 M propene in heptane, 0.0001M Ti (TiCl4/Al Et3 on MgCl2) Internal base : ethyl benzoate a : atactic External base : methyl p-toluate -specific i : isotactic -aspecific With both bases, specificity increases ! * Summary of Ziegler-Natta initiator Possible monomers ① Ethylene & α-olefins propylene, 1-butene, 4-methyl-1-pentene vinyl cyclohexane, styrene ② 1,1-disubstituted ethylene isobutylene, α-methyl styrene by non-coordination cationic mechanism ③ 1,2- disubstituted No polymerization using Ziegler-Natta initiators because of steric hinderence except) I. 1,2-disubstituted ethylene → isomerization → polymerization II. cycloalkene (Prior to the polymerization) ④ Polar monomers vinyl acetate, acrylate, vinyl chloride → by non-coordinated radical or cationic mechanism How about stereospecific polymerization ? ⇒ Not easy ∙∙ ∵ coordination of N an oxygen on the transition metal ! 8-5 Metallocene Polymerization of nonpolar alkene monomers Metallocene : LL’MtX2 where LL’ are η5-cyclopentadienyl (Cp) or alkyl-substituted η5-cyclopentadienyls 1-indenyl (Ind) 4,5,6,7-tetrahydro-1-indenyl (H4Ind) Mt : group 4 transition metal ex) zirconium, titanium, nafnium group 3 transition metal ex) scandium, yttrium, lanthanum X : Cl or CH3 9-fluorenyl (Flu) Advantage of metallocene over Ziegler-Natta system I. soluble (homogeneous) → 100-fold more active (every site is active) II. stereoselectivity : by changing ligands & reaction conditions III. Narrower distributions of molecular weight ∵ same coordination environment of metals : single site initiation → Ziegler-Natta : multisite initiation MAO (methylaluminoxane), [Al(CH3)O,]n, Lewis acid for the initiation like AlR3 in Z-N system AlR3 does not work in mtallocene system Polymer site Monomer site Two functions alkylation, abstraction of second chloride 8-5a Metallocene symmetry MAO + Cp2TiCl2 ⇒ non stereoselective * unbridged metallocene : non stereoselective ⇒ free rotation of η5-cyclopentadienyl ligands to make achiral environment at the active sites Stereoselective polymerization is possible from chiral & stereorigid metallocene ex) η5-cyclopentadienyl ligands are linked by a bridging goups such as CH2, CH2CH2, (CH3)2C, Si(CH3)2 ⇒ called ansa metallocene See p 667 Table 8-5 next page CEC CSC Catalyst site control Chem. Rev., 100 (2000) 1223-1252. Chem. Rev., 100 (2000) 1223-1252. Bent metallocene η5-cyclopentadienyl ligands are not parallel : bite angle 60-75o : 115-125o : less than 90o * Preparation of ansa (bridged) metallocene initiator E: ex) Si(CH3)2 8-5b C2v-symmetric metallocenes Ex) (CH3)2SiFlu2ZrCl2 → unsubstituted biscyclopentadienyl → two coordination (active) sites are both achiral & homotopic ⇒ atactic polymer ! 8-5c C2-symmetric metallocenes Ex) rac-(dimethylsilyl)bis(1-indenyl)zirconium dichloride, rac-(CH3)2SiInd2ZrCl2 enantiomers and mesocompound are synthesized, then seperation (R, R) (S, S) or (R, S) Meso compound Cs-symmetry can be separated → different stereoselectivity 8-5c-1 Effect of initiator structure Angle + steric + bridge * Bite angle M small M large if bite angle is too large or stereorigidity is too low → stereoselectivity ↓ ∵ polymer chain & monomer loosely held in plane if bite angle too small or stereorigidity is too high → stereoselectivity ↓ ∵ Polymer chain and monomers have difficulty to coordinate ⇒ general rule but not always 1. Zr, Hf, Ti : metals Zr : most commonly used Hf : less active but higher molecular weight polymer produce Ti : less active & less stereoselective 2. Cp or Ind ligands: Substituents in the 3- and 4-positions have the greatest effect for increasing isoselectivity, activity, and polymer molecular weight. 3. bridge Isoselectivity and molecular weight ↑ in the order of CH2 < CH2CH2 < (CH3)C < Si(CH3)2 4. hetero atoms into ligands such as alkoxy or amine → not promising 8-5c-2 Effect of reaction variables - Temp ↑ , reaction rate ↑ , stereoselectivity ↓ High temp makes the initiator more flexible. So the effect for the very stereorigid initiators is less! ex) propene with rac-C2H4(Ind)2ZrCl2/MAO 20˚C , Mv : 56,000 , regio irregular 0.4% 70˚C , Mv : 19,600 , regio irregular 0.7% * If the initiator is more stereorigid, the effect of temperature is less. - Monomer conc. ↓ , stereoselectivity ↓ - Initiator (metallocene) & MAO ↑ , stereoselectivity ↓ same thing Low monomer concentration lower the propagation rate, then epimerization might occurs (scrambling the chirality of the last monomer unit inserted into the propagating chain end) 8-5d Cs-metallocene Two types Cs Table 8-5 XXX and XXXI Plane of symmetry: horizontal plane XXX, vertical plane XXXI XXX: atactic, XXXI: syndiotactic XXX type Cs -symmetric metallocene is called meso Cs XXXI type Cs -symmetric metallocene is called Cs ⇒ highly stereoselective , more than C2 8-5e C1-metallocene No element of symmetry Cs becomes C1 when on of the ligand is unsymetric Two coordinate sites are both chiral (chirotopic) ⇒ selectivity & molecular weight varies with the structures ! (as usual) 8-5f Oscillating metallocene During the life time r and m conformers can be made Ex block type polymer can be prepared 8-5g Coinitiator 8-5g-1 MAO The most commonly used Lewis acid coinitiator (activator) → obtained by hydrolysis of TMA(trimethylaluminium), → n = 5 ~ 20 , mixtures of linear, cyclic , and 3D structures, 3D spherical cage like structure is desirable MAO contains TMA in two form; free TMA or associated TMA Free TMA decreases the initiator activity, removed by vacuum associated TMA can help the polymerization Large excess MAO is used (100-1000 to one metallocene) MAO is added first into the polymerization system to destroy deleterious impurities (which can lower the reactivity) before the addition of metallocene MAO does not have long term stability (low solubility in HC solvent) If the bottle is opened or exposed to air, the precipiation observed. the precipiate does not affect the reactivity. MMAO has improved stability prepared by hydrolysis of TMA and triisobutylaluminium Possible structures for the initiation Monometallic species Or bimetallic species → structure is not clear ! → more than one type structures might exists due two more than one type of MAO structures New type of coinitiators are desirable 1. To avoid the cost of large excess of MAO 2. Simpler system not various possible structures Boron type? 8-5g-2 Boron containing coinitiator ⇒ only one equivalent is needed to activate the initiator, Sometimes even smaller amount can be used ex) 2:1, initiator: conitiator system (possibly bimetallic system) Higher activity ascribed to the large and weakly coordiating anoins compared with MAO system Anions must be sufficiently week in coordiation to the cation not to compete with monomer for the coordination * Order of coordination ability [CH3MAO]- > [(CH3)B(C6F5)3]- > [B(C6F5)][B(C6H5)]- cannot be used because strong coordiation with cations Normally activity ↑ streoselectivit↓ but not always 8-5h Kinetics 8-5h-1 Rate of polymerization Zr + MAO I* +M M* + M K1 K2 kp I* M* M* If K2 is irreversible and fast, then [M*] = [I*] Rp = K1kp[Zr][MAO][M] If K2 is reversible and fast, then [M*] [I*] Rp = K1K2kp[Zr][MAO][M]2 K1 Zr + MAO I* +M K2 I* M* Using an isolated metallocenium borate (I*), there is no equilibium corresponding to K1 (K1 is irreversible) If K2 is irreversible and fast, then [M*] = [I*] Rp = kp[I*][M] If K2 is reversible and fast, then [M*] [I*] Rp = K2kp[I*][M]2 8-5h-2 Degree of Polymerization Same as Z-N system. Similar chain transfer and termination Spontaneous intramolecular β-hydride transfer Vinyilidene Chain transfer to monomer Chain transfer to the group I-III metal alkyl others Vinyil Double-bond end group composition varies Ex) 1-hexane by rac-Me2(H2Ind2) ZrCl2 /MAO Temp ↑ MW↓ transfer activation E is larger than that of polymerization At 0˚C, vinylene double bond constitute 85% of all double bond, less spontaneous termimation At 80˚C, vinylidene double bond constitute 75% of all double bond Monomer ↑ vinylidene content ↓ less spontaneous termination Metallocene ↑ vinylidene content ↑ 8-5h-3 Supported Metallocene Metallocene system follows the Z-N system called drop-in technology Mix MAO with silica first then add metallocene Supported metallocenes generally have lower activity than do homogeneous metall ocenes because of steric hindrance by the support. At the same time, less MAO is required because deactivation processes are also hindered. Polymer molecular weight and stereoselectivity are either unaffected or increased, depending on the specific reaction system. 8-6 Cycloalkenes 8-6a 1,2-disubstituted alkenen, Cycloalkenes Normally 1,2-disubstituted alkenens are not polymerized except cycloalkene Or isomerization is involved in the polymerization of 1,2-disubstituted alkenens Ex) 2-butene yields poly(1-butene) from Z-N system Cycloalkene can be polymerized due to the relief of ring strain Two possible polymerization for cyclobutene Four possible different isomers See p632 cis or trans Ring-opening olefin metathesis • Vanadium (V) based catalyst ⇒ polymerization through double bond • Tungsten (W), Ti, Ru ⇒ Ring-opening olefin metathesis 8-6b styrene Slighlty polar compared with olefins. Isoselective polymerization is possible from Z-N systems Synditactic polymerization is possible from Metallocene system Partial syndiotactic polymerization form anionic and cationic initiators 8-7 copolymerization See p 685 Table 8-7 Reactivity: ethene > propene > 1-butene > 1-hexane Reactivity when using the same initiator system 8-10 polymerization of dienes 8-10a radical polymerization General trend: 1,4-poly. > 1,2-poly. & 3,4-poly. Why 1,4-polymerization ① Steric effect trans-1,4-addition > cis-1,4-addition ~CH2-CH=CH-CH2· ↕ · ~CH2-CH-CH=CH2 1,4-addition 1,2-addition ② Stability of the final product • Temp ↑ cis↑ (1,4-addition) • 1,2-addition or 3,4-addition ⇒ do not change much 8-6 Anionic and Anionic coordination Polymerization No clear explanation! In polar solvent (THF) → polymerization via free ion or solvent separated ion pair favors 1,2-addition for butadiene Anionic center at carbon 2 is not extensively delocalized because the double bond is not strong ewg. 3,4-addition for isoprene (due to steric effect, less hindered C is attacked) In nonpolar solvent (n-alkane) Strong coordination power favors 1,4-addition This effect is most pronounced in Li and also unstable cis polyperization is favored although trans product is more stable. 1. slow polymerization (enough time to have isomerization) 2. When Li is counter ion, mostly cis polymer because cis monomer is more reactive In polar solvent this carbon attacks unassocoated more free ion like More reactive C- can react More stable anion, so 1,4 in nonpolar solvent, assocoated more contact ion pair like The predominance of cis 1,4-polymerization over trans 1,4-polymerization has been ascribed to the greater reactivity of cis-LI, Cis 1,4 is predominant in non solvent system with low concentration of initiator Becaues isomerization of cis-LI to the more stable trans-LI increases faster than propagation of cis-LI with an increase in initiator concentration since propagation proceeds only through unassociated species while isomerization proceeds through both associated and unassociated species. In polar solvent this carbon attacks Unassocoated more free ion like More stable anion, so 1,4 in nonpolar Solvent, assocoated more contact ion pair like Effect of initiator 8-6 Cationic Polymerization Not practical Because low MW polymers with cyclized structures are obtained 8-12 Stereospecific polymerization of Polar vinyl monomers General rule ◎ Soluble (uncoordinated) system → propagating species are free syndiotacticity is increasingly favored w/ decreasing rxn temp. ⇒ radical & ionic poly. in highly solvating media ◎ poorly solvating media (coordinated) → isospecific polymerization can occur. 8-12a MMA In polar solvent (THF, pyridine) generally syndiotactic polymers ∵ generally free ion or solvent separated ion pair * the counter ion is removed from the vicinity at the propagating center and does not exert a stereoregulating influence on the entry of next monomeric unit * Syndiotacticity Li > Na > K ∵ Li+ (smaller ion) most highly solvated ! * Syndiotacticity ↑ Temp. ↓ In nonpolar solvent => generally isotactic polymer isotacticity ↑ Li > Na > K ↓ greater coordinating power isotacticity ↓ Temp. ↑ Effect of metallocene initiators 8-12b Vinyl ether, styrene Isotactic polymers were obtained from cationic and Z-N systems Syndiotactic polymers were obtained from Monomers having bulky groups 6-membered ring could be formed Aldehydes Isoselective for anioic polymerization using zinc and aluminium alkyls, Grignard reagent, and lithium alkoxide Caionic system ( Ex) BF3etherate) less isoselective max. 70%