Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Baker Heart and Diabetes Institute wikipedia , lookup
Coronary artery disease wikipedia , lookup
Jatene procedure wikipedia , lookup
Management of acute coronary syndrome wikipedia , lookup
Heart failure wikipedia , lookup
Mitral insufficiency wikipedia , lookup
Cardiac contractility modulation wikipedia , lookup
Electrocardiography wikipedia , lookup
Myocardial infarction wikipedia , lookup
Quantium Medical Cardiac Output wikipedia , lookup
Hypertrophic cardiomyopathy wikipedia , lookup
Heart arrhythmia wikipedia , lookup
Ventricular fibrillation wikipedia , lookup
Arrhythmogenic right ventricular dysplasia wikipedia , lookup
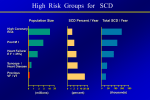
Article "Development of the European Network in Orphan Cardiovascular Diseases" „Rozszerzenie Europejskiej Sieci Współpracy ds Sierocych Chorób Kardiologicznych” Title: Arrhythmogenic right ventricular cardiomyopathy – still a diagnostic and therapeutic challenge. RCD code: III-4A Author: Prof. Piotr Podolec, MD, PhD Affiliation: Department of Cardiac and Vascular Diseases, John Paul II Hospital in Cracow, Poland Date: BACKGROUND Arrhythmogenic right ventricular cardiomyopathy (ARVC), previously known as arrhythmogenic right ventricular dysplasia (ARVD) is a rare myocardial disease caused by replacement of cardiomyocytes by fibrous and fatty tissue. This pathological process leads to dilation of the right ventricle (RV), thinning of its wall, hemodynamic dysfunction and electrical instability. Patients with ARVC usually complain of heart palpitations, dizziness, and recurrent syncopes. DISCUSSION ARVC is regarded as a distinct morphological and functional cardiomyopathy, which can be subclassified into familial (genetic) and nonfamilial types. The prevalence of ARVC is estimated to be 1:5000/10 000 in the general adult population and it mostly affects men. It is considered a frequent cause of otherwise unexplained sudden cardiac death (SCD) which in John Paul II Hospital in Kraków Jagiellonian University, Institute of Cardiology 80 Prądnicka Str., 31-202 Kraków; tel. +48 (12) 614 33 99; 614 34 88; fax. +48 (12) 614 34 88 e-mail: [email protected] www.crcd.eu those patients is strongly related to exercise (e.g. in athletes). It is probably triggered by increased exertional RV stress and high levels of catecholamines. In at least half of the reported cases, ARVC is found among members of a patient’s family and is typically autosomal dominant with variable penetrance. The majority of causative mutations affect the genes encoding desmosomal proteins, others concern genes for cardiac ryanodine receptor and transforming growth factor‑β3. So far at least nine different genetic types of ARVC have been distinguished. Fibro-fatty degeneration of RV myocardium is a reparative mechanism for necrosis and apoptosis of myocytes. The regions affected are those prone to increased hemodynamic stress (the so called “triangle of dysplasia” which includes the RV inflow and outflow tracts and its apex). A tendency to ventricular arrhythmias is best explained by the micro- and macroreentry circuits that form in the borders between the normal and altered ventricular wall. The disease usually manifests itself in adolescence and early adulthood. The main symptoms include heart palpitations, dizziness, syncope or even SCD. In most cases, ARVC is a progressive disorder, often consisting of three stages: (1) latent phase (discrete morphological changes, normal ECG, absent/asymptomatic arrhythmias); (2) electrical instability phase (regional RV wall motion abnormalities, symptomatic ventricular arrhythmias); (3) dilation phase (RV dilation and failure). The 2010 ESC Task Force algorithms encompass six categories such as: (1) dysfunction and structural alternations of RV, (2) RV wall histology, (3) ECG repolarization abnormalities, (4) ECG depolarization/conduction abnormalities, (5) arrhythmias, (6) family history. There are major and minor criteria within each category. Diagnosis is made on the basis of following tests’ results: electrocardiogram, echocardiography, cardiac magnetic resonance imaging, RV angiography, endomyocardial biopsy and electrophysiological study with electro-anatomic mapping. Due to evident association between SCD and exercise, persons diagnosed with ARVC should not be engaged in any professional sports. The goal in the management of ARVC is to prevent SCD. Several randomized controlled trials unequivocally confirmed benefits of implantable cardioverter-defibrillator (ICD) in both primary and secondary SCD prevention in those patients. Unfortunately ICD implantation complications may add up to those specific John Paul II Hospital in Kraków Jagiellonian University, Institute of Cardiology 80 Prądnicka Str., 31-202 Kraków; tel. +48 (12) 614 33 99; 614 34 88; fax. +48 (12) 614 34 88 e-mail: [email protected] www.crcd.eu to ARVC, for instance resulting in perforation of the thin RV wall. Moreover, progressive and extensive fibro-fatty myocardial degeneration may compromise effectiveness of ICD interventions. Although antiarrhythmic medications (sotalol, amiodarone), and radiofrequency (RF) ablation can decrease the number of ventricular arrhythmias, these methods do not prevent SCD and therefore cannot be considered an alternative to ICD but rather an adjunvant therapy. Increasing awareness of ARVC and advancement in therapeutic methods (particularly ICD application), have led to a substantial decrease in SCD statistics. However, severe RV failure leading to death has been not unfrequently observed in the recent years. CONCLUSION Arrhythmogenic right ventricular cardiomyopathy is a rare but very dangerous entity which may cause dangerous ventricular arrhythmias (including electrical storm) associated with sudden cardiac death and right ventricular failure. It should always be suspected in young, active adults with history of syncope and recurrent ventricular arrhythmias. There are specific diagnostic algorithms for ARVC. Early diagnosis is crucial and ICD implantation along with adjuvant therapy may prevent SCD and allow the patients to live a full normal life. REFERENCES 1. Maron BJ, Towbin JA, Thiene G, et al. Contemporary defintions and classification of the cardiomyopathies. Circulation 2006; 113: 1807–1816. 2. Norman M, Simpson M, Mogensen J, et al. Novel mutation in desmoplakin causes arrhythmogenic left ventricular cardiomyopathy. Circulation 2005; 112: 636–642. 3. Elliot P, Andersson B, Arbustini E, et al. Classification of the cardiomyopathies: a position statement from the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J 2007; 29: 270–277. 4. Gemayel C, Pelliccia A, Thompson PD. Arrhythmogenic right ventricular cardiomyopathy. J Am Coll Cardiol 2001; 38: 1773. 5. Tabib A, Loire R, Chalabreysse L, et al. Circumstances of death and gross and microscopic observations in a series of 200 cases of sudden death associated with arrhythmogenic right ventricular cardiomyopathy and/or dysplasia. Circulation 2003; 108: 3000–3005. John Paul II Hospital in Kraków Jagiellonian University, Institute of Cardiology 80 Prądnicka Str., 31-202 Kraków; tel. +48 (12) 614 33 99; 614 34 88; fax. +48 (12) 614 34 88 e-mail: [email protected] www.crcd.eu 6. Corrado D, Fontaine G, Marcus FI, et al. Arrhythmogenic right ventricular dysplasia/cardiomyopathy: need for an international registry. Study Group on Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy of the Working Groups on Myocardial and Pericardial Disease and Arrhythmias of the European Society of Cardiology and of the Scientific Council on Cardiomyopathies of the World Heart Federation. Circulation 2000; 101: E101. 7. Corrado D, Basso C, Schiavon M, et al. Screening for hypertrophic cardiomyopathy in young athletes. N Engl J Med 1998; 339: 364. 8. Hamid MS, Norman M, Quraishi A, et al. Prospective evaluation of relatives for familial arrhythmogenic right ventricular cardiomyopathy/dysplasia reveals a need to broaden diagnostic criteria. J Am Coll Cardiol 2002; 40: 1445. 9. Jacoby D, McKenna WJ. Genetics of inherited cardiomyopathy. Eur Hear J 2012; 33: 296–304. 10. Marcus FI, McKenna WJ, Sherrill D, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the Task Force criteria. Circulation 2010; 121: 1533 John Paul II Hospital in Kraków Jagiellonian University, Institute of Cardiology 80 Prądnicka Str., 31-202 Kraków; tel. +48 (12) 614 33 99; 614 34 88; fax. +48 (12) 614 34 88 e-mail: [email protected] www.crcd.eu John Paul II Hospital in Kraków Jagiellonian University, Institute of Cardiology 80 Prądnicka Str., 31-202 Kraków; tel. +48 (12) 614 33 99; 614 34 88; fax. +48 (12) 614 34 88 e-mail: [email protected] www.crcd.eu














![[INSERT_DATE] RE: Genetic Testing for Arrhythmogenic Right](http://s1.studyres.com/store/data/001678387_1-c39ede48429a3663609f7992977782cc-150x150.png)










