Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
History of manufactured fuel gases wikipedia , lookup
Spinodal decomposition wikipedia , lookup
Thermomechanical analysis wikipedia , lookup
Click chemistry wikipedia , lookup
Diamond anvil cell wikipedia , lookup
Chemical reaction wikipedia , lookup
Chemical potential wikipedia , lookup
Rate equation wikipedia , lookup
Computational chemistry wikipedia , lookup
Double layer forces wikipedia , lookup
Bioorthogonal chemistry wikipedia , lookup
Stability constants of complexes wikipedia , lookup
History of chemistry wikipedia , lookup
Thermodynamics wikipedia , lookup
Gas chromatography wikipedia , lookup
Vapor–liquid equilibrium wikipedia , lookup
Physical organic chemistry wikipedia , lookup
Bernoulli's principle wikipedia , lookup
Industrial gas wikipedia , lookup
Stoichiometry wikipedia , lookup
Determination of equilibrium constants wikipedia , lookup
Chemical thermodynamics wikipedia , lookup
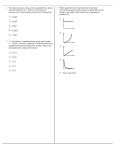
Unit 6: Physical chemistry of spectroscopy, surfaces and chemical and phase equilibria . 61 Chemical equilibrium and the kinetic theory of gases Equilibrium processes have a central importance to industrial chemistry. Although reactions are rarely allowed to reach equilibrium, knowledge of the factors that influence the position of equilibrium is critical for a chemical engineer. In this unit you will become familiar with various quantitative methods for solving problems connected with such equilibria. The unit begins with a study of the kinetic theory of gases because many of the processes that are used in industry involve gaseous reactants. The ideal gas equation, which is derived from kinetic theory, is important in allowing chemists to be able to analyse equilibrium systems involving gases. On successful completion of this topic: •• you will be able to apply the concept of chemical equilibrium (LO1). To achieve a Pass in this unit you should be able to: •• account for the relationship between the variables in the ideal gas equation (1.1) •• calculate terms in the ideal gas equation (1.2) •• write expressions for calculating chemical equilibrium constants (1.3) •• solve problems involving chemical equilibrium (1.4). 1 Unit 6: Physical chemistry of spectroscopy, surfaces and chemical and phase equilibria 1 The kinetic theory of gases Chemical engineers deal with a wide range of gases as components of the systems that they use in reactors. Knowledge of how these gases behave under different conditions of temperature and pressure is clearly going to be very important to a chemical engineer – and fortunately the behaviour of all gases is governed to a large extent by an equation known as the ideal gas law. You will see below how this law is derived from very simple principles. The kinetic model of gases This is a very simple model to describe the nature of gases, built on three key assumptions: •• gases consist of molecules in continuous random motion •• the size of these molecules is very much smaller than the distance between them •• there are no forces between gas molecules (except during a collision process). Collisions and distribution of speeds The total kinetic energy of a sample of gas at a given temperature will be constant, and therefore the average energy (and speed) of a gas molecule will also be constant. However, molecules do collide with each other and these collisions result in the transfer of momentum and kinetic energy. As a result, the speed of individual molecules within the gas varies from molecule to molecule. The distribution of these speeds is described as the Maxwell distribution and is most conveniently shown as in Figure 6.1.1, which is a plot of the fraction of molecules possessing particular ranges of speeds. Figure 6.1.1: The Maxwell distribution can be used to show how the distribution of molecular speeds changes at different temperatures. Relative abundance 273 K 373 K 473 K 0 200 6.1: Chemical equilibrium and the kinetic theory of gases 400 600 800 1000 1200 1400 Speed in m/s 2 Unit 6: Physical chemistry of spectroscopy, surfaces and chemical and phase equilibria Collisions and pressure Take it further For more information on how this equation is derived from the kinetic theory and on the idea of root mean square speed, you could consult a physical chemistry textbook such as Elements of Physical Chemistry (P. Atkins and J. de Paula, OUP, 2009). The derivation of this equation is covered on pages 23–24 and pages 37–38. What we usually call the pressure of a gas can be thought of as arising from the force exerted by molecules colliding with the walls of a container. The kinetic theory of gases allows us to calculate the pressure of a gas in terms of the speed of the molecules, c (more strictly this is the root mean square speed): nMc2 3V p= where n = number of moles of molecule M = molar mass of molecules c = root mean square speed V = volume of gas. The ideal gas law Gas laws The behaviour of gases when subjected to changes of pressure, volume or temperature can be summarised by three simple experimentally-derived equations: •• Boyle’s law: the pressure of a fixed mass of gas (at constant temperature) is inversely proportional to its volume. Hence if a gas has pressure P1 with volume V1, the new pressure P2 when the volume is changed to V2 is given by the relationship: (1) P1 V1 = P2V2 •• Charles’s law: the volume of a fixed mass of gas (at constant pressure) is proportional to its absolute temperature (that is, the temperature measured in Kelvin). (2) V1 T1 = V2 T2 •• Gay-Lussac’s law (or the pressure law): the pressure of a fixed mass of gas (at constant volume) is proportional to its absolute temperature. P1 P2 Activity (3) Show how equation (4) is derived. (Hint: start from equation (1) and use equation (2) to substitute in values of V1 and V2 in terms of temperature.) Using any two of these equations, they can be combined into one equation: (4) T1 = P1 V1 T1 T2 = P2 V2 T2 or PV T = a constant Avogadro’s law A further experimentally derived law states that the volume of a gas is proportional to the number of moles (n) of gas it contains. 1 mole of gas occupies 22.4 dm3 at 273 K and 1 atm pressure, so at 273 K: V = 22.4 n 6.1: Chemical equilibrium and the kinetic theory of gases 3 Unit 6: Physical chemistry of spectroscopy, surfaces and chemical and phase equilibria Equation of state: the ideal gas law The ideas from the gas laws and Avogadro’s law can be combined into the ideal gas law: Activity Calculate a value for R in units of atm dm–3 K–1 mol–1 by using data for the volume of 1 mole of a gas at 1 atm pressure and 273 K. For 1 mole of gas PV T = R, the gas constant For n moles of gas: PV T = nR, so (5) PV = nRT R is the ideal gas constant and has a value of 8.314 J K−1 mol−1. When the gas law is used, all the quantities must be in SI units. The ideal gas law is described as an equation of state, describing the properties of matter under a specific set of conditions. Conversion to SI units •• Pressure in Pa: 1 Pa = 9.871 × 10–6 atm = 105 bar = 0.1 Mbar. •• Volume in m3: 1 m3 = 106 dm3 = 106 litres. •• Temperature in K: temperature in K = temperature in °C + 273. The ideal gas law and kinetic theory Although the ideal gas law was first derived from experimental laws, it can be shown to be consistent with the expression for pressure obtained from the kinetic theory: nMc2 1 P= so PV = nMc2 3 3V Since temperature is a measure of the average kinetic energy of the molecules in the gas, ½Mc2, this equation is obviously related directly to the ideal gas equation above. Case study: Using the ideal gas equation Chemical engineers may need to be able to calculate the volumes required to store or collect specified amounts of gases (as products or reactants). The gases may be cooled (for storage) or heated (to meet the requirement for specific reaction conditions). The ideal gas equation enables a simple prediction of these volumes to be made. In different manufacturing locations, different units may be used for pressure, volume (and even temperature; for instance, °F is still in common use in the USA), so before the ideal gas equation can be used, these data must be converted into SI units. The following calculations will give practice in applying the ideal gas equation and/or the individual gas laws. In each case, assume that the gas is behaving ideally. R, the gas constant = 8.314 J K−1 mol−1. 1 The volume of a gas at 10 bar pressure is 36.4 dm3. Calculate the new volume if the pressure is increased to 200 bar (temperature remains constant). 2 The volume of a gas at 24 °C is 135 dm3. Calculate the new volume if the temperature is increased to 200 °C (note: think about the units that should be used in the calculation). The pressure remains constant. 3 Calculate the volume of 2.0 moles of gas at 400 K and 20 bar pressure. 6.1: Chemical equilibrium and the kinetic theory of gases 4 Unit 6: Physical chemistry of spectroscopy, surfaces and chemical and phase equilibria Real gases Although the ideal gas equation is an adequate description of the behaviour of gases at atmospheric conditions of temperature and pressure, considerable deviation from ideal behaviour occurs at high pressure and low temperature. This is most easily seen by looking at Figure 6.1.2, which shows how the quantity PV PV RT varies over a range of temperatures and pressures. If the gas behaved ideally, RT would have a value of 1, regardless of the conditions. PV RT Figure 6.1.2: Deviations from ideal gas behaviour occur at high pressures and low temperatures. 3 200 K 500 K 2 1000 K Ideal gas 1 0 0 300 600 900 P (atm) These variations occur because two of the assumptions used in deriving the ideal gas equation theoretically are not justified at high pressure: •• the size of the molecules becomes significant, compared to their separation, when a gas is compressed •• there is significant interaction (‘intermolecular forces’) between molecules when a gas is compressed. Van der Waals equations These deviations from ideal behaviour can be corrected for by adding some extra terms into the ideal gas equation. This yields alternative equations of state, such as the Van der Waals equation: [P + an2 ](V – nb) = nRT V2 This represents the reduction in pressure due to the attraction between molecules. This represents the reduction in ‘available volume’ due to the space occupied by the molecules themselves. In this equation a and b are constants that vary for different gases; they are evaluated experimentally and the larger the value the more deviation from ideal behaviour will be observed. Compressibility and the virial equation PV Figure 6.1.3 shows the values of RT for a range of gases at 273 K. PV RT is known as the compressibility factor of a gas. For an ideal gas it would have the value 1, but for non-ideal gases it is described by the virial equation. 6.1: Chemical equilibrium and the kinetic theory of gases 5 Unit 6: Physical chemistry of spectroscopy, surfaces and chemical and phase equilibria For 1 mole of gas: PV B C RT = (1 + V + V2 …) where B and C are temperature dependent coefficients that have different values for each gas. B C Figure 6.1.3: Different gases show different levels of deviation from ideal behaviour. PV RT When V is large (as at low pressure), the terms V + V2 in the bracket become negligible, and so the gas behaves like an ideal gas. 2.0 1.5 Ideal gas 1.0 N2 CH4 H2 CO2 0.5 0 0 200 400 600 800 1000 P (atm) Take it further Chemical engineers may need to use equations such as the Van der Waals or virial equations frequently in their calculations of pressure and volume of reacting gases. Solving equations like this is mathematically tricky and chemical engineers will have access to suitable software packages to enable rapid solutions to be obtained. A simplified version of one of these types of packages is available online at http://www.webqc.org/van_der_waals_gas_law.html, enabling you to apply the Van der Waals equation to a range of situations. Portfolio activity (1.1, 1.2) Hydrogen is widely used as a reactant in important industrial processes, such as the Haber process. Along with helium, it is one of the gases whose behaviour most closely matches the behaviour of an ideal gas. Use ideas about ideal gas behaviour to carry out the following calculations: 1 A sample of hydrogen occupies 14 m3 at 140 K. Calculate the volume of this sample at 300 K (assume the pressure remains constant). 2 The volume of a sample of hydrogen at 298 K is 23.6 dm3 and 1 bar pressure. The pressure is increased to 50 bar, but the temperature is kept constant at 298 K. Calculate the new volume of the gas. 3 Calculate the volume in dm3 of 5 moles of hydrogen gas at a temperature of 500 °C and 2 × 106 bar pressure. In your answer: •• state any equations you use, explaining the meaning of the terms •• describe how the equations used to describe the behaviour of ideal gases are consistent with the ideas of the kinetic theory of gases •• (Distinction) discuss why hydrogen might be expected to show little deviation from ideal behaviour under these conditions. 6.1: Chemical equilibrium and the kinetic theory of gases 6 Unit 6: Physical chemistry of spectroscopy, surfaces and chemical and phase equilibria 2 Chemical equilibrium Equilibrium constants Link This section links to Unit 5: Chemistry for Applied Biologists, Topic guide 5.3. Kc From previous chemistry courses you will be aware that for any given chemical equation, an expression for the equilibrium constant can be written, in terms of the concentrations of the substances present in the equilibrium mixture: aA + bB ⇌ cC + dD (5) Kc (the equilibrium constant in terms of concentration) = [C]c . [D]d [A]a . [B]b (where [A] = concentration of substance A in mol dm−3 etc.) Kp Key terms Partial pressure: In a mixture of gases, the partial pressure of a single gas is the pressure that the gas would exert if it was the only gas present. Mole fraction: In a mixture of gases, the mole fraction of a single gas is the ratio of the number of moles of that gas divided by the total number of moles of gas. For a gas phase reaction, it is more convenient to express the equilibrium constant in terms of partial pressures. The partial pressure of a gas, A, can be calculated from its mole fraction: (6) XA (mole fraction of component A) = nA ntotal then PA (partial pressure of component A) = Ptotal XA so: (7) PA = Ptotal . nA ntotal Then, for the equilibrium system above: pCc . pDd (8) Kp (the equilibrium constant in terms of partial pressures) = pa . pb A B Activity The formation of ammonia from nitrogen and hydrogen (known as the Haber process) is an important industrial process. In one reaction vessel, equilibrium was set up at conditions of 400 °C and 200 bar pressure. The equation for the Haber process is: N2(g) + 3H2(g) ⇌ 2NH3 (g) The number of moles of N2, H2 and NH3 present at equilibrium in the reaction vessel were as follows: N2: 1.5 mol H2: 4.6 mol NH3: 3.9 mol. 1 2 3 4 Calculate the mole fraction of N2, H2 and NH3. Calculate the partial pressures of each of these gases. Write an expression for Kp in terms of the partial pressures of N2, H2 and NH3. Calculate a value for Kp for the reaction under the conditions specified. Give appropriate units. 6.1: Chemical equilibrium and the kinetic theory of gases 7 Unit 6: Physical chemistry of spectroscopy, surfaces and chemical and phase equilibria The relationship between pressure and concentration The ideal gas law shows you how partial pressure and concentration are related: PA = Ptotal . Activity Show how equation (9) can be derived from the equations for Kp (7) and Kc (5), along with the relationship between pressure and concentration (8). (Hint: Start by substituting the expressions for PA, PB etc. (equation (8)) into the expression for Kp (equation (7)).) nA ntotal But, from the ideal gas equation, Ptotal = (9) So PA = ntotalRT Vtotal ntotalRT nA n = A RT = [A] RT Vtotal ntotal Vtotal This also means that: (10) Kp = KcRT−Δn (where Δn = number of moles of product molecules in the equation − number of moles of reactant molecules in the equation = (c+d) − (a+b)). Kx For a liquid phase reaction, it is more convenient to express the equilibrium constant in terms of mole fractions: aA(l) + bB(l) ⇌ cC(l) + dD(l) XCc . XDd (11) Kx (the equilibrium constant in terms of mole fractions) = Xa . Xb Now, because from (6) PA = Ptotal . XA A B (12) Kp = Kx PtotalΔn where Δn = (c+d) − (a+b) There is a similar relationship between Kp and Kc: (13) Kp = Kc (RT)∆n If Kc is quoted in units of mol dm−3, then R = 0.0831 bar dm3 mol−1 K−1. Activity Activity (Distinction level) Show how equation (12) can be derived from the expressions for Kp (8), Kx (11) and the definition of mole fraction (6). (Hint: Start by substituting the expressions for PA, PB etc. (6) into the expression for Kp (8).) The Haber process was introduced in an earlier activity. The equation for the reaction occurring in the Haber process is: N2(g) + 3H2(g) ⇌ 2NH3(g) You wrote an expression for Kp in the earlier activity and calculated a value of Kp at 400 °C. In a second reaction vessel, with a total pressure of 150 atm and a higher temperature of 450 °C, Kp has a value of 4.9 × 10−5 bar−2. 1 Write expressions for Kc and Kx for the reaction happening in this second reaction vessel. 2 Use the value for Kp given above and equation (12) to calculate values for Kx and Kc in this second reaction vessel (R = 0.0831 bar dm3 mol−1 K−1). Take it further Derivations of the interconversions between Kc, Kp and Kx are discussed in more detail online at http://www.psci305.utep.edu/ch1305/Chapter12/gc12.pdf, which also discusses how these expressions can be used to help interpret Le Chatelier’s principle. 6.1: Chemical equilibrium and the kinetic theory of gases 8 Unit 6: Physical chemistry of spectroscopy, surfaces and chemical and phase equilibria Calculations using equilibrium constants Activity 1 Hydrogen can be manufactured from steam and carbon monoxide. The equation for the process is: H2O(g) + CO(g) ⇌ H2(g) + CO2(g) The Kp value for this reaction at 700 K is 8.13. The partial pressures (in bar) of H2O, CO and CO2 in an equilibrium reached in a reaction vessel at 700 K were: H2O: 0.72; CO: 1.21; CO2: 2.10. Calculate the partial pressure of H2. 2 Esters, used in the food industry as flavourings, are manufactured by reacting carboxylic acids and alcohols. The equilibrium for the formation of ethyl ethanoate, CH3COOC2H5, is represented by the equation below: CH3COOH(l) + C2H5OH(l) ⇌ CH3COOC2H5(l) + H2O(l) Assuming that the initial concentrations of the product molecules were both zero, what can you say about the values of [CH3COOC2H5] and [H2O] at equilibrium? 3 In a reaction to manufacture ethyl ethanoate, the concentration of CH3COOH at equilibrium (at a temperature of 298 K) was 0.24 mol dm−3 and that of C2H5OH was 0.58 mol dm−3. Kc for this reaction at 298 K = 4.1. Use this information and your answer to 2 to calculate a value for the concentration of ethyl ethanoate at equilibrium. Equilibrium and thermodynamic quantities You will probably be familiar with the use of thermodynamic quantities to evaluate the feasibility of reactions and, by extension, to make qualitative conclusions about equilibrium positions of these reactions. Link Thermodynamic quantities that are used in this way include: You can remind yourself of these ideas in Unit 5: Chemistry for applied biologists, Topic guide 5.2. ΔG (Gibbs energy): The standard Gibbs energy change, ΔGƟ, is negative for any feasible reaction. ΔStotal (total entropy change): ΔStotal is positive for any feasible reaction. The Gibbs energy change and equilibrium As reaction mixtures reach equilibrium, the Gibbs energy of a mixture tends to reach a minimum value, as shown in Figure 6.1.4. Figure 6.1.4: Gibbs energy is a minimum at equilibrium. ΔGӨ reactants ΔG° =–20 kJ mol–1 equilibrium position favours products 100% reactants 6.1: Chemical equilibrium and the kinetic theory of gases Δ = GӨ products 100% products 9 Unit 6: Physical chemistry of spectroscopy, surfaces and chemical and phase equilibria If the Gibbs energy of reactants and products are measured under standard conditions, then the difference in Gibbs energy between them, as shown on the graph in Figure 6.1.4, is simply ΔGƟ. In this case, ΔGƟ is negative so the reaction is described as feasible. As ΔGƟ is less negative than about −40 kJ mol−1 we would expect the reaction to reach an equilibrium, favouring the products. However, if we consider any point on the graph between the pure reactants and pure products, then conditions will be non-standard. Nevertheless it is still possible to make a conclusion about the driving force for the reaction (the value of the Gibbs energy change) at this point in the reaction. We can think of the reaction Gibbs energy change, ΔGr, at this point as being represented by the slope of the graph of Gibbs energy, G, vs composition: ΔGr = ΔG Δn The different values of ΔGr are shown in Figure 6.1.5. Figure 6.1.5: The variation in reaction Gibbs energy with composition. Gθ ΔGθ ΔGr –ve Δn 100% reactants Take it further The idea of the variation of Gibbs energy with reaction progress is an important and difficult idea in thermodynamics. A fuller and stricter explanation of its derivation can be found in level 4/5 physical chemistry textbooks, such as Elements of Physical Chemistry (Atkins and de Paula, 2009), pages 154–156. ΔGr = 0 ΔGr +ve 100% products A negative value for ΔGr indicates that the reaction will tend to progress in the forward direction and a positive value that the reaction will tend to progress in the backwards direction. Clearly, as Figure 6.1.5 shows, equilibrium will be reached when ΔGr = 0, since at this point there is no driving force causing the reaction to progress in either direction. At this equilibrium point, ΔGr is related to both the standard Gibbs energy change, ΔGƟ, and to the equilibrium constant K. (14) ΔGr = ΔGƟ + RTlnK But, since ΔGr = 0 at equilibrium, (15) ΔGƟ = −RTlnK R, the gas constant, has a value of 8.314 J K−1 mol−1. Notice that this means that ΔGƟ must be expressed in units of J mol−1. For a reaction involving gases, K is numerically equivalent to the equilibrium constant in terms of partial pressures, Kp, although the two constants may differ in terms of their units. 6.1: Chemical equilibrium and the kinetic theory of gases 10 Unit 6: Physical chemistry of spectroscopy, surfaces and chemical and phase equilibria Calculating K and Kp from thermodynamic quantities Equation (15) allows you to calculate K (and hence Kp) at given temperatures from ΔGƟ data. From earlier work on ΔG, you should also know how to express ΔGƟ in terms of ΔHƟ and ΔSƟ. (16) ΔGƟ = ΔHƟ − TΔSƟ So, alternatively, a calculation of K could start with ΔHƟ and ΔSƟ data. Case study Silver(I) carbonate decomposes reversibly to form silver(I) oxide and carbon dioxide. Ag2CO3(s) ⇌ Ag2O(s) + CO2(g) The position of equilibrium depends on the temperature; above about 220 °C decomposition is favoured, whereas below this temperature the reverse reaction is favoured. As a result, silver(I) oxide is used as a reversible absorber of carbon dioxide in environments such as spacecraft – it was used extensively in the space shuttle, for example. ΔHƟ for this reaction = +81.6 kJ mol−1 and ΔSƟsys= +168.5 J K–1 mol−1. 1 Calculate ΔGƟ at 25 °C (298 K). Comment on the answer you get in terms of the favourability of the process. 2 Calculate ΔGƟ at 493 K (220 °C), assuming that ΔHƟ and ΔSƟsys remain unchanged. 3 Use your answer to question 2 to calculate a value for Kp at 493 K. Comment on your answers for ΔGƟ and Kp at this temperature. 4 Suggest how silver(I) oxide could have been used as a reversible absorber of carbon dioxide in the space shuttle. Why is the reversibility of the process an advantage in this situation? Activity Lime (calcium oxide) is produced by the thermal decomposition of calcium carbonate: CaCO3(s) ➝ CaO(s) + CO2(g) As calcium oxide and calcium carbonate are solids, they do not appear in the Kp expression, which therefore has the form Kp = pCO 2 The decomposition is carried out at temperatures above 1200 K, at which point ΔGƟ becomes negative. 1 Calculate Kp at 1200 K, given that ΔGƟ at this temperature = −13.8 kJ mol−1. 2 Comment on the value of Kp at this temperature. The effect of temperature As you saw in the previous section, the favourability (as measured by the values of ΔGƟ and Kp) of a process varies with temperature. Van’t Hoff isochore How the favourability changes depends on the sign of ΔSƟ. If it has a negative value (because, for example, the number of moles of gas molecules increases) then ΔGƟ will be more negative at higher temperatures and the reaction will be more favourable. 6.1: Chemical equilibrium and the kinetic theory of gases 11 Unit 6: Physical chemistry of spectroscopy, surfaces and chemical and phase equilibria Because ΔGƟ is in turn related to the value of the equilibrium constant, K, an equation can be constructed showing how the value of K depends on ΔHƟ, ΔSƟ and T. Key term Isochore: An equation describing an isochoric process – i.e. one occurring in a closed system with constant volume. This is known as the Van’t Hoff isochore. (17) InK = – ΔHƟ ΔSƟ ΔHƟ 1 ΔSƟ + or InK = – . + RT R R T R (ΔHƟ and ΔSƟ are assumed to be constant over the temperature range being studied.) 1 A plot of InK vs T will therefore produce a straight line with slope: – ΔHƟ R ΔSƟ The y-intercept R can also be used to derive a value for ΔSƟ (although this may not always be easy to measure from a particular graph). Figure 6.1.6 shows a Van’t Hoff isochore plot for the reaction: In(Kp) Figure 6.1.6: The Van’t Hoff isochore suggests that a plot of ln(Kp) vs 1/T will produce a straight line graph, as shown here. C(s) + CO2(g) ⇌ 2CO(g) 4 3.5 3 2.5 2 1.5 1 0.5 0 0.00084 0.00086 0.00088 0.0009 0.00092 0.00094 1/T(K-1) Case study Iron is produced industrially by the reduction of iron(III) oxide to iron. At the temperatures used in the blast furnace, the reducing agent is carbon monoxide, generated by the reaction of carbon and carbon dioxide: C(s) + CO2(g) ⇌ 2CO(g) A plot of Kp for this process against 1/T is shown in Figure 6.1.6. •• Calculate the gradient of this graph. •• Use this value and equation (13) to calculate a value for ΔHƟ, in J mol−1 (R = 8.314 J K−1 mol−1). Alternatively, this equation can be used to show how the equilibrium constants (K1 and K2) at two different temperatures, T1 and T2, are related: ) ΔH 1 1 Or (19) InK = InK + R (T – T ) (18) In ( K2 ΔHƟ 1 1 = K1 R T1 – T2 Ɵ 2 6.1: Chemical equilibrium and the kinetic theory of gases 1 1 2 12 Unit 6: Physical chemistry of spectroscopy, surfaces and chemical and phase equilibria Activity Water vapour reacts with carbon to form hydrogen and carbon monoxide: H2O(g) + C(s) ⇌ H2(g) + CO(g) The equilibrium constant, Kp, has a value of 66 bar at 1200 K. Calculate a value for Kp at 800 K (ΔHƟ = +145.4 kJ mol−1; assume ΔHƟ remains constant between these temperatures). Use equation (18) or (19) to do this; let K2 represent the value of Kp at this new temperature (T2 ), and let K1 represent the value of Kp at T1 (1200 K). Portfolio activity (1.3, 1.4 part 1) The Haber process is represented by the equation N2(g) + 3H2(g) ⇌ 2NH3(g) •• Write expressions to demonstrate the meanings of the three different types of equilibrium constant (Kp, Kc and Kx). •• Use research to obtain values of ΔHƟ and ΔSƟ for this reaction and explain how these are used to calculate ΔGƟ and hence the equilibrium constant K for this reaction at a given temperature. •• Explain how Kc and Kx are related to the value for Kp for this reaction, and use the mathematical relationships obtained to calculate values for Kc and Kx. Portfolio activity (1.4 part 2) The oxidation of sulfur dioxide is a critical step in the manufacture of sulfuric acid. The reaction is represented by the equation SO2(g) + ½O2(g) ⇌ SO3(g) Data for the equilibrium constant, Kp, (numerically equal to K) is given in Table 6.1.1. •• Use a suitable graph and the Van’t Hoff isochore to explain how the sign and value of ΔHƟ may be deduced from this data. •• By using the graph or by calculation, find the value of Kp at 298 K. Kp (bar–½) Temperature (K) 400 397.0 500 48.1 600 9.53 700 2.63 800 0.915 900 0.384 1000 0.185 6.1: Chemical equilibrium and the kinetic theory of gases Table 6.1.1. 13 Unit 6: Physical chemistry of spectroscopy, surfaces and chemical and phase equilibria Checklist At the end of this topic guide you should be familiar with the following ideas: the behaviour of gases is described by the ideal gas equation, pV = nRT deviations from ideal behaviour can occur at high pressure and low temperature and equations exist to describe this non-ideal behaviour equilibrium systems are characterised by the value of the equilibrium constant at a particular temperature equilibrium constants can be written in terms of concentration, partial pressures or mole fractions the Gibbs energy change of a reaction is a measure of the feasibility or favourability of a process Gibbs energy changes are related to equilibrium constant values by the equation ΔG = −RTlnK equations can also be written which relate ΔH and/or ΔS values to equilibrium constant values. Further reading Much of the content of this topic guide is covered in greater detail in level 4/5 textbooks of physical chemistry. An excellent example is Elements of Physical Chemistry (Atkins and de Paula, 2009). This not only provides a clear background to many of the ideas encountered in the topic guide, but also takes the concepts much further, particularly in the derivation and application of the key mathematical equations encountered. Chapters 1, 7 and 8 are particularly relevant to this topic guide. For a more general (and less mathematical) treatment of the key ideas of chemical equilibrium and Gibbs energy, Why Chemical Reactions Happen (Keeler and Wothers, 2003) is a useful resource. It is designed as an introductory text to a level 4 chemistry course. Chapters 2 and 8 are most relevant to this topic guide. Acknowledgements The publisher would like to thank the following for their kind permission to reproduce their photographs: Corbis: David Sutherland All other images © Pearson Education Every effort has been made to trace the copyright holders and we apologise in advance for any unintentional omissions. We would be pleased to insert the appropriate acknowledgement in any subsequent edition of this publication. 6.1: Chemical equilibrium and the kinetic theory of gases 14