Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Management of acute coronary syndrome wikipedia , lookup
Heart failure wikipedia , lookup
Mitral insufficiency wikipedia , lookup
Coronary artery disease wikipedia , lookup
Cardiac contractility modulation wikipedia , lookup
Myocardial infarction wikipedia , lookup
Quantium Medical Cardiac Output wikipedia , lookup
Hypertrophic cardiomyopathy wikipedia , lookup
Electrocardiography wikipedia , lookup
Heart arrhythmia wikipedia , lookup
Ventricular fibrillation wikipedia , lookup
Arrhythmogenic right ventricular dysplasia wikipedia , lookup
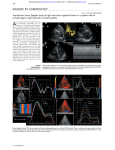
Journal of the American College of Cardiology © 2001 by the American College of Cardiology Published by Elsevier Science Inc. Vol. 38, No. 5, 2001 ISSN 0735-1097/01/$20.00 PII S0735-1097(01)01568-6 Cardiomyopathy Genotype-Phenotype Assessment in Autosomal Recessive Arrhythmogenic Right Ventricular Cardiomyopathy (Naxos Disease) Caused by a Deletion in Plakoglobin Nikos Protonotarios, MD,*† Adalena Tsatsopoulou, MD,* Aris Anastasakis, MD,† Elias Sevdalis, MD,† Godfrina McKoy, PHD,‡ Kostas Stratos, MD,† Kostas Gatzoulis, MD,† Kostas Tentolouris, MD,† Chara Spiliopoulou, MD,§ Demos Panagiotakos, PHD,† William McKenna, MD, FRCP, FACC,‡ Paulos Toutouzas, MD, FACC† Naxos and Athens, Greece; and London, United Kingdom The purpose of this study was to examine the genotype-phenotype relation with respect to penetrance, age and severity of expression, disease progression and prognosis in a recessively inherited arrhythmogenic right ventricular cardiomyopathy (ARVC). BACKGROUND Naxos disease is a recessively inherited ARVC caused by a mutation in the gene encoding plakoglobin (cell adhesion protein) in which the cardiac phenotype is associated with palmoplantar keratoderma and woolly hair. METHODS Twelve families with Naxos disease underwent cardiac and molecular genetic investigation. Serial cardiac assessment with annual resting 12-lead and 24-h ambulatory electrocardiogram (ECG) and two-dimensional echocardiography was performed during 1 to 16 years, median 7 ⫾ 6 years in all 78 surviving members. RESULTS Twenty-eight surviving members were homozygous and 40 were heterozygous for the mutation. All adults who were homozygous (n ⫽ 26) fulfilled the diagnostic criteria for ARVC, the youngest by the age of 13 years. In eight who were heterozygous, minor ECG or echocardiographic abnormalities were observed. Of the 26 subjects who were affected homozygotes, 92% showed ECG abnormalities, 92% ventricular arrhythmias, 100% right ventricular structural alterations and 27% left ventricular involvement. During follow-up (10 ⫾ 6 years), 16 (62%) developed structural progression, 12 (46%) arrhythmic events and 7 (27%) heart failure. The annual disease-related and sudden death mortality was 3% and 2.3%, respectively. CONCLUSIONS Autosomal recessive ARVC caused by a mutation in plakoglobin was 100% penetrant by adolescence. Affected subjects who were homozygous experienced progressive disease with adverse prognosis. A minority of subjects who were heterozygous showed minor ECG/ echocardiographic changes, but clinically significant disease did not develop. (J Am Coll Cardiol 2001;38:1477– 84) © 2001 by the American College of Cardiology OBJECTIVES Arrhythmogenic right ventricular cardiomyopathy (ARVC) is a heart muscle disorder causing life-threatening ventricular arrhythmias, heart failure and sudden cardiac death. The pathologic hallmark of the disease is the progressive replacement of myocardial cells by fat and fibrous tissue (1–3). Right ventricular involvement predominates, but left ventricular involvement may also occur with disease progression (4). In 1977 Guy Fontaine presented the first clinical characterization of ARVC under the name of “arrhythmogenic right ventricular dysplasia” (5). Since then, sporadic and familial forms of the disease have been described (6). Familial disease is usually autosomal dominant From the *Yannis Protonotarios Medical Center, Naxos, Greece; †Department of Cardiology, University of Athens, Athens, Greece; ‡Department of Cardiological Sciences, St. George’s Hospital Medical School, London, United Kingdom; and the §Department of Forensic Medicine and Toxicology, University of Athens, Athens, Greece. Supported by Yannis Protonotarios Medical Center, Ltd. Drs. McKoy and McKenna were supported by a grant from the British Heart Foundation. Manuscript received February 28, 2001; revised manuscript received June 11, 2001, accepted July 11, 2001. (7), and six gene loci have been identified (8 –13). Disease is claimed to be sporadic in up to 70% of patients (14). Published pedigrees, however, show 30% to 50% nonpenetrance, and, thus, without knowledge of disease-causing genes, accurate assessment of the proportion of sporadic versus familial ARVC is problematic. The long-term follow-up of 37 families with autosomal dominant disease has been reported (15). These reveal an overall favorable prognosis, though other reports emphasize the initial presentation of ARVC with sudden death, particularly in young, apparently healthy athletes (2,16,17). In 1986 we reported an autosomal recessive form of ARVC from the Aegean island of Naxos (18). In Naxos disease, the cardiac disorder was always associated with diffuse palmoplantar keratoderma and woolly hair and was apparent from infancy. In 1998 the gene locus for Naxos disease was mapped to 17q21 (19), and recently we identified the responsible gene mutation, a deletion in plakoglobin (20), which is an important constituent of the cell-to-cell junction. 1478 Protonotarios et al. Genotype-Phenotype Assessment in Naxos ARVC Abreviations and Acronyms ARVC ⫽ arrhythmogenic right ventricular cardiomyopathy CI ⫽ confidence interval ECG ⫽ electrocardiogram LBBB ⫽ left bundle branch block VT ⫽ ventricular tachycardia The identification of a disease-causing gene in autosomal recessive ARVC permits examination of the genotypephenotype relation with respect to penetrance, age and severity of expression, progression of disease and prognosis. METHODS Study population. Twelve Naxos disease families identified since 1984 were evaluated. All living family members underwent cardiac and molecular genetic investigation. Initial cardiac assessment included a detailed history of cardiac events, physical examination, resting 12-lead electrocardiogram (ECG), 24-h ambulatory ECG and twodimensional echocardiography. These investigations were performed every 12 months during a follow-up period of 7 ⫾ 6 years (range: 1 to 16 years). Follow-up was more frequent in those with documented or suspected arrhythmia or when structural progression was detected. Additionally, signal-averaged ECG (n ⫽ 34), electrophysiologic studies (n ⫽ 10), endomyocardial biopsies (n ⫽ 5) and surgical biopsies during antiarrhythmic surgery (n ⫽ 2) were performed. Postmortem cardiac biopsies were available in two patients with the informed consent of their families. The diagnosis of ARVC was based on European Society of Cardiology/International Society and Federation of Cardiology guidelines (14). Genetic study. Genomic DNA was extracted from blood samples; the procedure was followed by amplification of a 2 kilobase (kb) genomic region surrounding the deletion site (Pk2157del2, where the mutation appears by the deletion of two bases) using polymerase chain reaction with specifically designed primers (20). Because the mutation destroys a Bst01 restriction site, we could verify the presence of the mutation by restriction enzyme digestion. The results were confirmed in 1.5% agarose gel depending on the number and the molecular weight of the DNA bands that are produced by Bst01 cleavage. Electrocardiography. Standard 12-lead ECG was recorded at rest (25 mm/s, 10 mm/mV and 20 mm/mV). QRS complex duration was measured by a caliper. Epsilon waves were included in these measurements. The difference between the widest QRS complex in leads V1, V2 or V3 minus that in V6 was estimated (QRS dispersion). Extension of inverted T-waves (including flattened T) in the precordial leads was noted. A 24-h ambulatory ECG was recorded on an outpatient basis. The number of ventricular extrasystoles and the number, rate and duration of episodes JACC Vol. 38, No. 5, 2001 November 1, 2001:1477–84 of ventricular tachycardia (VT) (ⱖ3 consecutive ventricular complexes at a rate of ⱖ120 beats/min) were recorded. Signal-averaged ECG was performed for assessment of arrhythmic risk from a three-channel 24-h ambulatory ECG during sinus rhythm (21). Time-domain analysis was obtained in each patient using two band pass filters at 40 to 250 Hz and 25 to 250 Hz (22). Initial evaluations were performed off cardioactive medication. Echocardiography. Echocardiography with a 2.5 MHz transducer was performed by the same investigator (N. P.). Measurements of the right ventricular dimensions in twodimensional echocardiographic recordings were selected from the outflow tract on parasternal long-axis view and from the inflow tract on apical four-chamber view according to the protocol by Foale et al. (23). Normal limits (mean ⫾ 2 SD) were defined by echocardiographic assessment of 80 unrelated healthy subjects (40 men, 40 women) from Naxos island with a mean age of 39 years (range: 13 to 73 years). Abnormalities including hypokinetic, akinetic or dyskinetic areas, diastolic bulging and trabecular disarrangement were documented (24,25). Definitions. Syncope was defined as a sudden and brief loss of consciousness associated with a loss of postural tone, from which recovery is spontaneous (26). Ventricular tachycardia was defined as sustained when lasting more than 30 s. Ventricular extrasystoles were noted on resting 12-lead ECG and were characterized as frequent when they were more than 1,000/24 h on 24-h ambulatory ECG. Structural abnormalities of the right ventricle were classified as major: severe dilation (more than 3 SD from normal values), diffuse hypokinesia and localized aneurysms, and minor: mild dilation (2 to 3 SD from normal values) and regional hypokinesia (14). Structural progression was defined as progressive alteration in ventricular dimensions and/or wall motion abnormalities during serial echocardiographic evaluation. Severe heart failure was defined as New York Heart Association functional class III or IV accompanied by jugular venous engorgement, hepatomegaly or ascites. Sudden cardiac death was defined as unexpected death occurring in ⬍1 h from the onset of symptoms. Cardiovascular collapse occurring in the context of severe heart failure was not regarded as sudden death. Statistical analysis. All values from quantitative variables are presented as mean ⫾ 1 SD and from qualitative variables as relative frequency and counts. To screen out significant covariates related to sudden cardiac death, univariate analysis was applied on all those who were homozygous gene carriers on the assumption that they were potentially at risk for the adverse outcome. The relation between each factor and sudden death was evaluated using contingency tables, logistic regression analysis and nonparametric methods. Kaplan-Meier curve was produced to reveal the survival as well as the event-free rate. Reported p values are two-sided, exact and compared to a significant level of 5%. Statistical calculations were performed using STATA 6 (STATA Corp., College Station, Texas). JACC Vol. 38, No. 5, 2001 November 1, 2001:1477–84 Figure 1. Section of right ventricular apex from the heart of a 20-year-old girl with Naxos disease who died suddenly. Low magnification (⫻2.5) demonstrates massive replacement of mediomural and subepicardial layers by fatty tissue. Subendocardial layers are infiltrated by fibrosis. RESULTS Genotype and diagnosis of ARVC. Seventy-eight living family members belonging to 12 nuclear families (7 ⫾ 4 members per family, range: 1 to 12) were identified. Systematic questioning for premature cardiovascular symptoms or death identified two female probands with the cutaneous Naxos disease phenotype who were asymptomatic before sudden death at the ages of 14 and 52 years. Molecular genetic investigation revealed 28 subjects who were homozygous for the plakoglobin mutation (14 men, 14 women) age 33 ⫾ 19 years (range: 1 to 74 years), 40 carriers who were heterozygous (21 men, 19 women) age 42 ⫾ 19 years (range: 7 to 86 years) and 10 who were homozygous for the normal allele (5 men, 5 women) age 33 ⫾ 22 years (range: 8 to 80 years). All of the subjects who were homozygous had diffuse palmoplantar keratoderma and extremely woolly hair apparent from infancy. Noninvasive cardiac assessment demonstrated abnormalities, which fulfilled the established diagnostic criteria for ARVC in 26 (affected subjects who were homozygous). In particular, 13 subjects fulfilled one major plus at least two minor criteria while the other 13 fulfilled at least two major criteria. Their age at diagnosis of ARVC was 36 ⫾ 17 years (range: 13 to 74 years). One of the subjects who was seven years old at initial examination, did not meet the diagnostic criteria until the age of 18. Diagnosis was supported histologically in eight subjects: from endomyocardial biopsies in five subjects, from surgical biopsies in two subjects and from autopsy in one subject (Fig. 1). Two children did not fulfill the criteria for diagnosis. One of the children, a five-year-old child, had frequent ventricular extrasystoles of left bundle branch block (LBBB) pattern on the 24-h ambulatory ECG but resting 12-lead ECG; signal-averaged ECG and two-dimensional echocardiography were within normal limits. The other Protonotarios et al. Genotype-Phenotype Assessment in Naxos ARVC 1479 child, a one-year-old, showed neither arrhythmias, electrocardiographic nor echocardiographic abnormalities. Eight of the subjects who were heterozygous carriers showed minor cardiac abnormalities; however, none of them fulfilled the criteria for diagnosis of ARVC. Four subjects had T-wave inversion in V1 and V2; three had mild right ventricular dilation, and one had T-wave inversion from V1 to V3 and mild right ventricular outflow tract dilation. Five of them also had the woolly hair phenotype. Palmoplantar keratoderma, ventricular arrhythmias and late potentials on signal-averaged ECG were not observed in any of the subjects who were heterozygous carriers. None of 10 who were homozygous for the normal allele had skin or hair abnormalities; one had T-wave inversion in V1 and V2, and another one had mild right ventricular inflow tract dilation. Late potentials were not observed in signal-averaged ECG (n ⫽ 5), and ventricular arrhythmias were not recorded in 24-h ambulatory ECG. During follow-up none of the subjects who were heterozygous carriers or those who were homogygous for the normal allele developed symptoms, arrhythmias and ECG or echocardiographic abnormalities. Cardiac phenotype in the 26 affected homozygotes. ELECTROCARDIOGRAPHY AND ECHOCARDIOGRAPHY. Resting ECG was abnormal in 24 of the 26 affected patients (92%). Twenty patients had inverted T waves in leads V1 to V3 or across the precordial leads; 19 had QRS complex prolongation (⬎110 ms) in leads V1, V2 or V3; 9 had complete or incomplete right bundle branch block, and 11 had epsilon waves (Fig. 2). QRS dispersion more than 20 ms existed in 17 patients (Table 1). Frequent ventricular extrasystoles of LBBB configuration were recorded in 24 patients (92%). All patients who were submitted to signal-averaged ECG (n ⫽ 10) fulfilled the criteria for late potentials. Right ventricular structural alterations were observed on two-dimensional echocardiography in all patients; there were minor alterations in seven (27%) and major alterations in 19 (73%) (Fig. 3). Right ventricular trabecular disarrangement and irregularly shaped echodense muscular bands were detected in 23 patients. Seven patients (27%) had left ventricular wall motion abnormalities; four of them showed regional hypokinesia (two apical, two posterior wall), and three showed diffuse hypokinesia of the left ventricle. During follow-up, eight patients (31%) showed ECG changes including prolongation of QRS complex (n ⫽ 6) and extension of T-wave inversion in contiguous precordial leads (n ⫽ 3). Serial echocardiographic studies revealed progression of the initially detected right ventricular abnormalities in 16 patients (62%). In 10 of the patients, appearance or worsening of left ventricular involvement was also observed (Table 1). Symptoms appeared in 18 of the 26 patients. The initial presenting symptom was syncope in 15, sustained palpitation in 2 and fatigue in 1 at age 31 ⫾ 18 years (range: 12 to 68 years) (Table 1). During follow-up (10 ⫾ 6 years, range: 1 to 16 years), arrhythmic events occurred in 12 of the 26 patients (46%). Oral CLINICAL EVENTS AND OUTCOME. 1480 Protonotarios et al. Genotype-Phenotype Assessment in Naxos ARVC JACC Vol. 38, No. 5, 2001 November 1, 2001:1477–84 32.71), p ⫽ 0.017. Other variables such as antiarrhythmic treatment (p ⫽ 0.171), QRS dispersion ⱖ40 ms (p ⫽ 0.178), episodes of sustained VT (p ⫽ 0.427) and family history of sudden death (p ⫽ 0.944) were not significantly related to sudden cardiac death. Kaplan-Meier curves for cumulative survival and event-free survival of all subjects who were homozygous are indicated in Figure 4. By the age of 35 years, the cumulative survival was 74% and the event-free survival 53%. DISCUSSION Figure 2. Recordings from baseline 12-lead electrocardiogram (precordial leads) in seven patients with Naxos disease. There is T-wave inversion from V1 to V3 or V4 or V5, QRS complex prolongation, complete or incomplete right bundle branch block pattern and epsilon waves (C4, C5, C18 and C20). Elevation (1 mm) of the ST segment in leads V1 to V4 is observed in Patient 3. Patient 5 has a pattern resembling Brugada’s syndrome. antiarrhythmic treatment was applied in 13 patients for 69 ⫾ 34% of their follow-up (Table 1). Two patients were operated on at the beginning of follow-up; an arrhythmogenic focus from the right ventricular free wall was successfully excised under endocardial and epicardial electrophysiologic mapping guidance. In three patients an antitachycardia pacemaker defibrillator was implanted. In two of those patients episodes of VT were terminated by antitachycardia pacing (n ⫽ 6) or low energy synchronized cardioversion (n ⫽ 7) while they were on amiodarone (200 to 400 mg daily). Seven patients (27%) developed heart failure; four of them suffered severe heart failure and were considered for heart transplantation (Table 1). Symptoms of heart failure were associated with severe right ventricular deterioration in two patients and biventricular deterioration in five patients. Eight patients died prematurely of a cardiac cause (six suddenly and two of heart failure). Two patients who died suddenly had discontinued amiodarone against medical advice. The mean age at death was 32 ⫾ 13 years (range: 17 to 53 years). The annual disease related and sudden death mortality was 3% and 2.3%, respectively. Survival analysis. The univariate risk ratios for the significant factors related to adverse outcome were: episodes of syncope 5.89 (95% confidence interval [CI]: 1.12 to 45.43), p ⫽ 0.037; left ventricular involvement 6.48 (95% CI: 1.94 to 45.74), p ⫽ 0.046; early (age ⬍35 years) onset of symptoms 6.87 (95% CI: 1.82 to 29.91), p ⫽ 0.036); early (age ⬍35 years) structural progression 8.45 (95% CI: 3.08 to The identification of the causative gene in these autosomal recessive ARVC families provided the opportunity to examine the genotype-phenotype relation with respect to penetrance, age and degree of expression, progression of disease and prognosis. Penetrance and clinical expression of ARVC. In published pedigrees of families affected by dominant ARVC, approximately 50% of family members are affected (15). However, without knowledge of disease causing genes, estimation of penetrance is problematic. In this study, all subjects who were homozygous for the mutation presented the skin and hair phenotype from early infancy, while the diagnostic cardiac abnormalities were 100% penetrant by adolescence. The presence of the cutaneous phenotype enabled identification of children who would go on to develop ARVC. Children were usually asymptomatic showing only a few minor right ventricular abnormalities or asymptomatic ventricular arrhythmias by the age of five years yet not fulfilling the conventional criteria for clinical diagnosis. In the dominant form, childhood disease is recognized (15). The symptomatic presentation of Naxos ARVC was usually with syncope during adolescence. At the time of first event, ARVC presented with the simultaneous development of ECG and echocardiographic abnormalities fulfilling at least one major of the established criteria. In the dominant form, however, echocardiographic abnormalities appeared to be of greater diagnostic sensitivity than changes on resting ECG, while 48% of patients fulfilled only minor criteria (15). Among the carriers who were heterozygous, some showed minor phenotypic features of Naxos disease consisting of woolly hair, minor ECG abnormalities and mild right ventricular dilation, not fulfilling the criteria for ARVC. Progression and prognosis of ARVC. Arrhythmogenic right ventricular cardiomyopathy is known to be a progressive heart disease. In this study, structural progression on echocardiography was detected in 62% of patients, of whom 50% showed ECG progression. Echocardiography was more sensitive in detecting the progression of heart disease (27). Progression was usually accompanied by the development of clinical events. When the heart disease involved both ventricles, ARVC was difficult to differentiate clinically from dilated cardiomyopathy (28). Heart failure was observed in 27% of patients—a phenomenon—which is similarly common in most long-term follow-up studies (28 –31). JACC Vol. 38, No. 5, 2001 November 1, 2001:1477–84 Table 1. Clinical Details of 28 Homozygotes With Naxos Disease Initial Evaluation Case Age (yrs) QRSD (ms) T-Wave Inversion M M F M M F F M M F M F F F M M M M F M F F F F M F F M 7 17 32 29 17 41 36 43 35 32 74 61 56 49 40 60 26 19 16 47 45 15 17 58 13 5 39 1 0 40 0 40 40 20 20 0 40 40 40 20 20 60 20 40 40 40 40 60 20 40 40 60 40 0 40 20 V1–V3 V1–V6 V1–V4 V1–V4 V1–V3 V1–V5 V1 0 V1–V4 V1–V3 V1–V5 V1–V4 V1–V3 V1–V6 V1–V5 V1–V4 V1–V6 V1–V6 0 V1–V4 V1–V3 V1–V2 V1–V3 V1–V6 V1–V4 V1–V2 V1–V2 V1–V2 17 42 29 18 48 (SYNC) (SYNC) (SYNC) (SYNC) (SYNC) 32 (SPT) 48 (fatigue) 68 68 43 29 18 (SYNC) (SYNC) (SYNC) (SYNC) (SYNC) 21 (SYNC) 21 (SPT) 15 (SYNC) 20 (SYNC) 15 (SYNC) 12 (SYNC) Follow-Up Clinical Events Antiarrhythmic Treatment 0 RVT, HF (FC IV) RVT, VT/ICD RVT, VT/ICD, HF (FC II) RVT, HF (FC III) SYNC/NSVT, HF (FC III) 0 RVT, HF (FC II) Fatigue, chest pain 0 0 SYNC/NSVT Recurrent SYNC/VES RVT Recurrent SYNC/VES 0 0 RVT, HF (FC IV) HF (FC II) 0 0 SYNC/VES 0 0 0 (VT on EP) 0 0 0 0 AMIO (EP) AMIO (EP), ICD SOT, AMIO (EP), ICD AS, AMIO (EMP) AMIO (EMP)† 0 AMIO (EP) 0 0 Quinidine (EP) AMIO (EMP) AMIO (EMP) SOT (EP) 0‡ 0 AMIO (EMP)† AS, AMIO (EMP) 0 0 0 0‡ ICD 0 AMIO (EP) 0 0 0 Structural Progression RV* RV* RV, LV RV, LV RV* RV, LV 0 RV, LV RV, LV 0 0 RV RV RV RV, LV 0 RV, LV* RV, LV* RV, LV* 0 0 RV, LV* 0 0 0 0 0 0 Outcome Duration (yrs) Remains ASYMP Death of HF ASYMP (last 1 year) Death of NCC Sudden death Sudden death Remains ASYMP HF (FC II) Sudden death Remains ASYMP ASYMP (last 13 years) ASYMP (last 7 years) ASYMP (last 8 years) Death of NCC ASYMP (last 1 year) Remains ASYMP Sudden death Death of HF Sudden death Remains ASYMP Remains ASYMP Sudden death ASYMP (last 6 years) Remains ASYMP ASYMP (last 3 years) Remains ASYMP Remains ASYMP Remains ASYMP 16 11 16 15 11 12 15 13 15 16 7 16 15 12 15 12 1 14 1 8 4 5 4 2 2 2 2 3 *Progression detected before the age of 35 years; †discontinued; ‡refused to follow treatment. AMIO ⫽ amiodarone; AS ⫽ antiarrhythmic surgery; ASYMP ⫽ asymptomatic; EMP ⫽ empirically; EP ⫽ electrophysiologic study; FC ⫽ NYHA functional class; HF ⫽ heart failure; ICD ⫽ implantable cardioverter defibrillator; LV ⫽ left ventricle; NCC ⫽ noncardiac cause; NSVT ⫽ nonsustained ventricular tachycardia; QRSD ⫽ QRS dispersion; RV ⫽ right ventricle; RVT ⫽ recurrent sustained ventricular tachycardia; SOT ⫽ sotalol; SPT ⫽ sustained palpitation; SYNC ⫽ syncope; VES ⫽ frequent ventricular extrasystoles; VT/ICD ⫽ sustained ventricular tachycardia terminated by implantable defibrillator; 0 ⫽ none. Protonotarios et al. Genotype-Phenotype Assessment in Naxos ARVC 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 Gender Age (yrs) at First Symptom 1481 1482 Protonotarios et al. Genotype-Phenotype Assessment in Naxos ARVC JACC Vol. 38, No. 5, 2001 November 1, 2001:1477–84 Figure 3. Two-dimensional echocardiographic images recorded from a patient with Naxos disease. (a) An aneurysm of right ventricular outflow tract (white arrows) is exhibited by a modified parasternal long-axis view. (b) An aneurysm of posterior wall of the right ventricle just below the tricuspid valve (white arrows) is exhibited by a subcostal four-chamber view. LA ⫽ left atrium; LV ⫽ left ventricle; RA ⫽ right atrium; RV ⫽ right ventricle; RVOT ⫽ right ventricular outflow tract; TV ⫽ tricuspid valve. In this study, the annual disease-related and sudden death mortality were 3% and 2.3%, respectively, which is higher than that reported in other ARVC series (15,28 –32). The worse prognosis in recessive ARVC is an analogous phe- nomenon to that observed in recessive forms of dilated cardiomyopathy (33). Risk stratification in ARVC remains a difficult task because of the small number of events, which limits multivariate regression analysis to assess factors re- Figure 4. Kaplan-Meier survival curves for cumulative survival (continuous line) and event-free survival (dotted line) among the subjects who were homozygous. JACC Vol. 38, No. 5, 2001 November 1, 2001:1477–84 lated to adverse outcome. In this study, the univariate analysis revealed that episodes of syncope, especially in the young; early structural progression (age ⬍35 years) and left ventricular involvement were the best predictors of sudden cardiac death. The adverse prognosis of ARVC in the young has also been described in the dominant form (34). Genotype and pathogenesis of ARVC. A deletion mutation in the gene encoding plakoglobin has been identified to underlie this recessive ARVC (20). A recent study demonstrated a ryanodine receptor mutation in a subgroup of patients with dominant ARVC responsible for exerciseinduced polymorphic VT and sudden death (35). Recently too, another cytoskeletal protein colocalizing with plakoglobin at the cardiac intercalated disk has been involved in pathogenesis of adult mice right ventricular cardiomyopathy (36). Previous ultrastructural studies on ARVC have revealed defects in myocardial cell-to-cell junctions including abnormalities in desmosomes and adherens junctions (37,38). Plakoglobin is an important protein common to the intracellular plaques of the adhesive junctions in heart and skin (20). The disruption of cell-cell adhesion caused by the mutant plakoglobin under conditions of mechanical stress leads to cell detachment and cell death within in vitro studies (39). Apoptotic myocardial cell death, which has been observed in ARVC (40,41), might exist in this case because plakoglobin is also involved in the pathways of apoptosis (42). A defect in the cell-adhesion complex may explain the pathogenesis of recessive ARVC. An increase in wall stress, known to be inversely proportional to wall thickness, might predominantly affect the cell adhesions in the thin-walled right ventricle. The disease is not expressed until adolescence, possibly due to the ability of cardiac tissue to compensate during the first two decades of life (43). The alterations in myocardial cell-to-cell junction, which precede the fibrofatty changes, have been recognized in areas of myocardium remote from the characteristic histologic changes (37) and might be one of the causes of rhythm disturbances that are characteristic of this cardiomyopathy (38). The severity of the disease in the young, as well as in athletes, could be attributed to conditions of increased mechanical stress acting as an environmental factor modifying disease progression and phenotypic expression. Conclusions. The genotype-phenotype assessment in this study revealed a 100% penetrance of autosomal recessive ARVC by adolescence. The disease is initially manifested with syncope and expressed with ECG and echocardiographic abnormalities. Affected individuals experienced progressive disease with adverse prognosis. Structural progression in the young, syncope and left ventricular involvement were the best predictors of adverse outcome. Reprint requests and correspondence: Dr. Nikos I. Protonotarios, Yannis Protonotarios, Medical Centre of Naxos, Hora Naxos 84 300, Greece. E-mail: [email protected]. Protonotarios et al. Genotype-Phenotype Assessment in Naxos ARVC 1483 REFERENCES 1. Marcus FI, Fontaine GH, Guiraudon G, et al. Right ventricular dysplasia: a report of 24 adult cases. Circulation 1982;65:384 –98. 2. Thiene G, Nava A, Corrado D, Rossi L, Pennelli N. Right ventricular cardiomyopathy and sudden death in young people. N Engl J Med 1988;318:129 –33. 3. Burke AP, Farb A, Tashko G, Virmani R. Arrhythmogenic right ventricular cardiomyopathy and fatty replacement of the right ventricular myocardium. Are they different diseases? Circulation 1998;97: 1571– 80. 4. Corrado D, Basso C, Thiene G, et al. Spectrum of clinicopathologic manifestations of arrhythmogenic right ventricular cardiomyopathy/ dysplasia: a multicenter study. J Am Coll Cardiol 1997;30:1512–20. 5. Fontaine G, Guiraudon G, Frank R, et al. Stimulation studies and epicardial mapping in ventricular tachycardia. Study of mechanisms and selection for surgery. In: Kulbertus HE, editor. Reentrant Arrhythmias. Lancaster, PA: MTP Press, 1977:334 –50. 6. Fontaine G, Fontaliran F, Frank R. Arrhythmogenic right ventricular cardiomyopathies: clinical forms and main differential diagnoses. Circulation 1998;97:1532–5. 7. Nava A, Thiene G, Canciani B, et al. Familial occurrence of right ventricular dysplasia: a study involving nine families. J Am Coll Cardiol 1988;12:1222– 8. 8. Rampazzo A, Nava A, Danieli GA, et al. The gene for arrhythmogenic right ventricular cardiomyopathy maps to chromosome 14q23q24. Hum Mol Genet 1994;3:959 – 62. 9. Rampazzo A, Nava A, Erne P, et al. A new locus for arrhythmogenic right ventricular cardiomyopathy (ARVD2) maps to chromosome 1q42-q43. Hum Mol Genet 1995;4:2151– 4. 10. Severini GM, Krajinovic M, Pinamonti B, et al. A new locus for arrhythmogenic right ventricular dysplasia on the long arm of chromosome 14. Genomics 1996;31:193–200. 11. Rampazzo A, Nava A, Miorin M, et al. ARVD 4, a new locus for arrhythmogenic right ventricular cardiomyopathy, maps to chromosome 2 long arm. Genomics 1997;45:259 – 63. 12. Ahmad F, Li D, Karibe A, et al. Localization of a gene responsible for arrhythmogenic right ventricular dysplasia to chromosome 3p23. Circulation 1998;98:2791–5. 13. Li D, Ahmad F, Gardner MJ, et al. The locus of a novel gene responsible for arrhythmogenic right ventricular dysplasia characterized by early onset and high penetrance maps to chromosome 10p12-p14. Am J Hum Genet 2000;66:148 –56. 14. McKenna WJ, Thiene G, Nava A, et al. Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Br Heart J 1994;71:215– 8. 15. Nava A, Bauce B, Basso C, et al. Clinical profile and long-term follow-up of 37 families with arrhythmogenic right ventricular cardiomyopathy. J Am Coll Cardiol 2000; 36:2226 –33. 16. Furlanello F, Bertoldi A, Dallago M, et al. Cardiac arrest and sudden death in competitive athletes with arrhythmogenic right ventricular dysplasia. Pacing Clin Electrophysiol 1998;21:331–5. 17. Tabib A, Miras A, Taniere P, Loire R. Undetected cardiac lesions cause unexpected sudden cardiac death during occasional sport activity. A report of 80 cases. Eur Heart J 1999;20:900 –3. 18. Protonotarios N, Tsatsopoulou A, Patsourakos P, et al. Cardiac abnormalities in familial palmoplantar keratosis. Br Heart J 1986;56: 321– 6. 19. Coonar AS, Protonotarios N, Tsatsopoulou A, et al. Gene for arrhythmogenic right ventricular cardiomyopathy with diffuse nonepidermolytic palmoplantar keratoderma and woolly hair (Naxos disease) maps to 17q21. Circulation 1998;97:2049 –58. 20. McKoy G, Protonotarios N, Crosby A, et al. A deletion in plakoglobin causes arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease). Lancet 2000; 355:2119 –24. 21. Cain M, Anderson J, Arnsdorf M, Mason J, Scheinman M, Waldo A. Signal averaged electrocardiography. J Am Coll Cardiol 1996;27:238 – 49. 22. Nava A, Folino AF, Bauce B, et al. Signal-averaged electrocardiogram in patients with arrhythmogenic right ventricular cardiomyopathy and ventricular arrhythmias. Eur Heart J 2000;21:58 – 65. 23. Foale R, Nihoyannopoulos P, McKenna W, et al. Echocardiographic measurements of the normal adult right ventricle. Br Heart J 1986;56: 33– 44. 1484 Protonotarios et al. Genotype-Phenotype Assessment in Naxos ARVC 24. Nava A, Scognamiglio R, Thiene G, et al. A polymorphic form of familial arrhythmogenic right ventricular dysplasia. Am J Cardiol 1987;59:1405–9. 25. Blomstrom-Lundqvist C, Beckman-Suurkula M, Wallentin A, Jonsson R, Olsson SB. Ventricular dimensions and wall motion assessed by echocardiography in patients with arrhythmogenic right ventricular dysplasia. Eur Heart J 1988;9:1291–302. 26. Kapoor WN. Syncope. N Engl J Med 2000;343:1856 – 62. 27. Metzger JT, de Chillou C, Cheriex E, Rodriguez LM, Smeets JL, Wellens HJ. Value of the 12-lead electrocardiogram in arrhythmogenic right ventricular dysplasia, and absence of correlation with echocardiographic findings. Am J Cardiol 1993;72:964 –7. 28. Pinamonti B, Sinagra G, Salvi A, et al. Left ventricular involvement in right ventricular dysplasia. Am Heart J 1992;123:711–24. 29. Blomstrom-Lundqvist C, Sabel KG, Olsson SB. A long-term follow-up of 15 patients with arrhythmogenic right ventricular dysplasia. Br Heart J 1987;58:477– 88. 30. Leclercq JF, Coumel P. Characteristics, prognosis and treatment of the ventricular arrhythmias of right ventricular dysplasia. Eur Hear J 1989;10 Suppl D:61–7. 31. Marcus FI, Fontaine GH, Frank R, Gallagher JJ, Reiter MJ. Longterm follow-up in patients with arrhythmogenic right ventricular disease. Eur Hear J 1989;10 Suppl D:68 –73. 32. Lemery R, Brugada P, Janssen J, Cheriex E, Dugernier T, Wellens HJJ. Nonischemic sustained ventricular tachycardia: clinical outcome in 12 patients with arrhythmogenic right ventricular dysplasia. J Am Coll Cardiol 1989;14:96 –105. 33. Mestroni L, Rocco C, Gregori D, et al. Familial dilated cardiomyopathy: evidence for genetic and phenotypic heterogeneity. J Am Coll Cardiol 1999;34:181–90. JACC Vol. 38, No. 5, 2001 November 1, 2001:1477–84 34. Daliento L, Turrini P, Nava A, et al. Arrhythmogenic right ventricular cardiomyopathy in young versus adult patients: similarities and differences. J Am Coll Cardiol 1995;25:655– 64. 35. Tiso N, Stephan DA, Nava A, et al. Identification of mutations in the cardiac ryanodine receptor gene in families affected with arrhythmogenic right ventricular cardiomyopathy type 2 (ARVD2). Hum Mol Genet 2001;10:189 –94. 36. Pashmforoush M, Pomies P, Peterson KL, et al. Adult mice deficient in actinin-associated LIM-domain protein reveal a developmental pathway for right ventricular cardiomyopathy. Nat Med 2001;7:591–7. 37. Guiraudon CM. Histological diagnosis of right ventricular dysplasia: a role for electron microscopy? Eur Heart J 1989;10 Suppl D:95– 6. 38. Roncali L, Nico B, Locuratolo N, Bertossi M, Chiddo A. Right ventricular dysplasia: an ultrastructural study. Eur Heart J 1989;10 Suppl D:97–9. 39. Schnittler HJ, Puschel B, Drenckhahn D. Role of cadherins and plakoglobin in interendothelial adhesion under resting conditions and shear stress. Am J Physiol 1997;273:H2396 – 405. 40. Mallat Z, Tergui A, Fontaliran F, Frank R, Durigon M, Fontaine G. Evidence of apoptosis in arrhythmogenic right ventricular dysplasia. N Engl J Med 1996;335:1190 – 6. 41. Valente M, Calabrese F, Thiene G, et al. In vivo evidence of apoptosis in arrhythmogenic right ventricular cardiomyopathy. Am J Pathol 1998;152:479 – 84. 42. Brancolini C, Sgorbissa A, Schneider C. Proteolytic processing of the adherens junctions components -catenin and ␥-catenin/plakoglobin during apoptosis. Cell Death Differ 1998;5:1042–50. 43. James TN. Normal and abnormal consequences of apoptosis in the human heart. From postnatal morphogenesis to paroxysmal arrhythmias. Circulation 1994;90:556 –73.