Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Protein domain wikipedia , lookup
Rosetta@home wikipedia , lookup
Circular dichroism wikipedia , lookup
List of types of proteins wikipedia , lookup
Intrinsically disordered proteins wikipedia , lookup
Structural alignment wikipedia , lookup
Homology modeling wikipedia , lookup
Protein structure prediction wikipedia , lookup
Nuclear magnetic resonance spectroscopy of proteins wikipedia , lookup
Chemical Shift
Restraints
Tools and Methods
Andrea Cavalli
Overview
Overview
• Methods
Overview
• Methods
• Details
Overview
• Methods
• Details
• Results/Discussion
Methods
Methods
Cheshire
base
solid-state
Methods
Cheshire
base
solid-state
CamShift
new predictor
Monte Carlo/Molecular Dynamics
Methods
Cheshire
base
solid-state
CamShift
new predictor
Monte Carlo/Molecular Dynamics
CamDock
protein-protein docking
About
CHESHIRE: CHEmical SHifts REstraints
About
CHESHIRE: CHEmical SHifts REstraints
3D structure determination from NMR
chemical shifts.
About
CHESHIRE: CHEmical SHifts REstraints
3D structure determination from NMR
chemical shifts.
•
Chemical shifts are “easy” to measure
About
CHESHIRE: CHEmical SHifts REstraints
3D structure determination from NMR
chemical shifts.
•
•
Chemical shifts are “easy” to measure
Can be measured with great accuracy
About
CHESHIRE: CHEmical SHifts REstraints
3D structure determination from NMR
chemical shifts.
•
•
•
Chemical shifts are “easy” to measure
Can be measured with great accuracy
Contain a lot of structural informations (CSI, TALOS,...)
About
CHESHIRE: CHEmical SHifts REstraints
3D structure determination from NMR
chemical shifts.
•
•
•
•
Chemical shifts are “easy” to measure
Can be measured with great accuracy
Contain a lot of structural informations (CSI, TALOS,...)
In some cases they are the “only” available data
About
CHESHIRE: CHEmical SHifts REstraints
3D structure determination from NMR
chemical shifts.
•
•
•
•
Chemical shifts are “easy” to measure
Can be measured with great accuracy
Contain a lot of structural informations (CSI, TALOS,...)
In some cases they are the “only” available data
but ...
NOE-NMR vs CHESHIRE
NOE-NMR vs CHESHIRE
•
NOEs have a direct structural interpretation as distances
NOE-NMR vs CHESHIRE
•
•
NOEs have a direct structural interpretation as distances
Chemical shifts values are indirectly related to geometry (SHIFTX, CamShift)
NOE-NMR vs CHESHIRE
•
•
•
NOEs have a direct structural interpretation as distances
Chemical shifts values are indirectly related to geometry (SHIFTX, CamShift)
NOEs have long-range information
NOE-NMR vs CHESHIRE
•
•
•
•
NOEs have a direct structural interpretation as distances
Chemical shifts values are indirectly related to geometry (SHIFTX, CamShift)
NOEs have long-range information
Chemical shifts are local
NOE-NMR vs CHESHIRE
•
•
•
•
•
NOEs have a direct structural interpretation as distances
Chemical shifts values are indirectly related to geometry (SHIFTX, CamShift)
NOEs have long-range information
Chemical shifts are local
NOEs are redundant
NOE-NMR vs CHESHIRE
•
•
•
•
•
•
NOEs have a direct structural interpretation as distances
Chemical shifts values are indirectly related to geometry (SHIFTX, CamShift)
NOEs have long-range information
Chemical shifts are local
NOEs are redundant
There is only one chemical shift per atom
NOE-NMR vs CHESHIRE
•
•
•
•
•
•
•
NOEs have a direct structural interpretation as distances
Chemical shifts values are indirectly related to geometry (SHIFTX, CamShift)
NOEs have long-range information
Chemical shifts are local
NOEs are redundant
There is only one chemical shift per atom
Clear quality control (number of assigned NOEs, NOEs violation)
NOE-NMR vs CHESHIRE
•
•
•
•
•
•
•
•
NOEs have a direct structural interpretation as distances
Chemical shifts values are indirectly related to geometry (SHIFTX, CamShift)
NOEs have long-range information
Chemical shifts are local
NOEs are redundant
There is only one chemical shift per atom
Clear quality control (number of assigned NOEs, NOEs violation)
Weak Q-factor
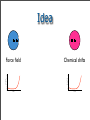
Idea
Idea
Force field
-920
Free Energy
-940
-960
-980
-1000
0
2
4
6
Cα-RMSD
8
10
Idea
Chemical shifts
-920
-420
-940
-440
Chemical Shift
Free Energy
Force field
-960
-460
-980
-480
-1000
-500
0
2
4
6
Cα-RMSD
8
10
0
2
4
6
Cα-RMSD
8
10
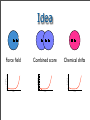
Idea
Combined score
Chemical shifts
-420
-420
-940
-440
-440
-960
Chemical Shift
-920
Chemical Shift
Free Energy
Force field
-460
-980
-460
-480
-1000
-480
-500
0
2
4
6
Cα-RMSD
8
10
-500
0
2
4
6
Cα-RMSD
8
10
0
2
4
6
Cα-RMSD
8
10
Idea
Combined score
Chemical shifts
-420
-420
-940
-440
-440
-960
Chemical Shift
-920
Chemical Shift
Free Energy
Force field
-460
-980
-460
-480
-1000
-480
-500
0
2
4
6
Cα-RMSD
8
10
-500
0
2
4
6
Cα-RMSD
8
10
0
2
4
6
8
10
Cα-RMSD
Structures have to be very close to the native one in order
to “feel” chemical shifts score.
CHESHIRE
Determination or
prediction?
Experiment
Theory
CHESHIRE
Determination or
prediction?
NMR
X-ray
Experiment
Theory
CHESHIRE
Determination or
prediction?
NMR
X-ray
ab initio
Experiment
Theory
CHESHIRE
Determination or
prediction?
NMR
X-ray
Experiment
Homology modeling > 50 %
Homology modeling < 50 %
ab initio
Theory
CHESHIRE
Determination or
prediction?
NMR
X-ray
Experiment
Homology modeling > 50 %
CHESHIRE
Homology modeling < 50 %
ab initio
Theory
CHESHIRE
Determination or
prediction?
Jigsaw puzzle
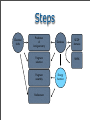
Steps
Chemical
shifts
Prediction
of
local geometry
Database
Fragment
selection
Fragment
assembly
Refinement
SCOP
domains
SHIFX
Energy
function
Local structure 1
Prediction
of
local geometry
Chemical
shifts
Database
Secondary structure prediction
P3(S1, S2, S3|AA1, AA2, AA3), Pcs(S|Hα, N,Cα,Cβ, AA)
N
N
i=1
i=1
E = − ∑ log P3(i) − Kcs ∑ log Pcs(i)
Secondary structure propensity
NS
P(S|A) =
N
Local structure 2
Chemical
shifts
Prediction
of
local geometry
Database
Torsion angle prediction
S(Φi, Ψi|A,CS) = Sym(B, A) + Sym(∆CSA, ∆CSB) + Sym(SA, SB)
Three best scoring cluster centers are taken as
prediction.
Fragment selection
Chemical
shifts
Fragment
selection
Database
Fragments of length 3 and 9 aa
N
N
i=1
i=1
E = ∑ Ecs(Ai, ∆CSA, Bi, ∆CSB) + Ktor ∑ Etar (Φi, Ψi, B)
Performance
Protein
3Pred
TOPOS
Ubiquitin
0.75
0.93
FF domain
0.90
0.86
Calbindin
0.85
0.95
HPR
0.87
0.86
Fold
Fragment
assembly
Energy
function
Fold
Fragment
assembly
Energy
function
Refinement 1
Chemical
shifts
Refinement
Energy
function
Energy function
Ere f = E f f / log(1 −Ccs)
where
Ccs =
∑
χ∈{Hα,N,Cα,Cβ}
Kχ(1 −Cχ), Cχcorrelation of CS type χ
Refinement 2
Refinement 2
• Structure with large Rg are
discarded
Refinement 2
• Structure with large Rg are
discarded
• Side-chains are added
Refinement 2
• Structure with large Rg are
discarded
• Side-chains are added
• Initial ranking
Refinement 2
Takes one structure
at random from the best-list.
New structure generated by
simulated annealing.
• Structure with large Rg are
discarded
• Side-chains are added
• Initial ranking
Keeps a list
of the 100
best structures
Results
Results
The largest
The largest
2GW6, 123 aa 1.72 Å backbone RMSD
The smallest
The smallest
1PV0, 46 aa 1.37 Å backbone RMSD
Solid-State NMR
of protein G
Solid-State NMR
of protein G
Solid-State NMR
of protein G
Structure
RMSD
N (5.5 Å)
Q (RDC)
1P7F
0.40
0
0.03
3GB1
0.59
0
0.16
2GB1
0.97
1
0.37
2JU6
1.86
5
0.48
2K0P
1.04
3
0.40
Failures
Failures
0
S = 48.09-4.2458*NA, R=-0.9794
Score
-200
-400
-600
-800
60
80
100
120
140
Number of Amino Acids
160
180
200
Failures
1ZGG
-400
0
S = 48.09-4.2458*NA, R=-0.9794
-450
Refined Structures
Refined Native Structure
Expected Score
-200
Score
-400
-550
-450
-600
Score
Score
-500
-600
-650
-500
-550
0
-800
60
10
20
30
C!-RMSD
80
100
120
140
Number of Amino Acids
160
180
200
-700
0
5
10
15
20
C!-RMSD
25
30
35
40
Failures
1ZGG
-400
0
S = 48.09-4.2458*NA, R=-0.9794
-450
Refined Structures
Refined Native Structure
Expected Score
-200
Score
-400
-550
-450
-600
Score
Score
-500
-600
-650
-500
-550
0
-800
60
10
20
30
C!-RMSD
80
100
120
140
Number of Amino Acids
160
180
200
-700
0
5
10
15
20
C!-RMSD
25
30
35
Why?
Usually because the assembly stage does not
generate low RMSD models.
40
CamShift
CamShift
• Chemical shifts are
predicted using distances to
neighboring atoms
R
N
C
H
H
C
O
R
N
C
H
H
C
O
CamShift
• Chemical shifts are
predicted using distances to
neighboring atoms
• Accurate as ShiftX or
Sparta and orders of
magnitude faster
R
N
C
H
H
C
O
R
N
C
H
H
C
O
CamShift
• Chemical shifts are
predicted using distances to
neighboring atoms
• Accurate as ShiftX or
Sparta and orders of
magnitude faster
R
N
C
H
H
O
R
• CamShift with physical
force field and ReX
molecular dynamics
C
N
C
H
H
C
O
CamShift
• Chemical shifts are
predicted using distances to
neighboring atoms
• Accurate as ShiftX or
Sparta and orders of
magnitude faster
R
N
C
H
H
• ~ 1 Å from unfolded for
small proteins (1uzc,
1ubq, ..)
O
R
• CamShift with physical
force field and ReX
molecular dynamics
C
N
C
H
H
C
O
CamShift-MD
2jvw: 61 residues
Lowest Energy Structure
1.41Å RMSD
2jva: 108 residues
Lowest Energy Structure
1.98 Å RMSD
CamShift
Full
No Long range
Sparta
HN
0.53
0.61
0.57
HA
0.29
0.37
0.27
N
3.10
3.18
2.52
CA
1.18
1.20
0.98
CB
1.43
1.48
1.07
CO
1.16
1.27
1.08
Conclusions
Conclusions
•Protein structure determination with chemical shifts is possible...
Conclusions
•Protein structure determination with chemical shifts is possible...
•but difficult... very difficult...
Conclusions
•Protein structure determination with chemical shifts is possible...
•but difficult... very difficult...
•CHESHIRE works (at the moment) for proteins up to ~100 aa.
Conclusions
•Protein structure determination with chemical shifts is possible...
•but difficult... very difficult...
•CHESHIRE works (at the moment) for proteins up to ~100 aa.
•results are stable ~1.0-2.0 Å Cα RMSD.
Conclusions
•Protein structure determination with chemical shifts is possible...
•but difficult... very difficult...
•CHESHIRE works (at the moment) for proteins up to ~100 aa.
•results are stable ~1.0-2.0 Å Cα RMSD.
•self-consistent criterion to (maybe) detect failures of the
method.
Conclusions
•Protein structure determination with chemical shifts is possible...
•but difficult... very difficult...
•CHESHIRE works (at the moment) for proteins up to ~100 aa.
•results are stable ~1.0-2.0 Å Cα RMSD.
•self-consistent criterion to (maybe) detect failures of the
method.
•can be used
for complexes and with solid-state CS.
Conclusions
•Protein structure determination with chemical shifts is possible...
•but difficult... very difficult...
•CHESHIRE works (at the moment) for proteins up to ~100 aa.
•results are stable ~1.0-2.0 Å Cα RMSD.
•self-consistent criterion to (maybe) detect failures of the
method.
•can be used
for complexes and with solid-state CS.
• http://www.open-almost.org
Acknowledgments
Michele Vendruscolo
Chris Dobson
Xavier Salvatella
Kai Kohlhof
Paul Robustelli
Danny Hsu
Rinaldo Wander Montalvao