Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
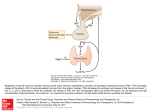
M I N I R E V I E W Minireview: Thyrotropin-Releasing Hormone and the Thyroid Hormone Feedback Mechanism Maria Izabel Chiamolera and Fredric E. Wondisford Division of Metabolism, Departments of Pediatrics, Medicine, and Physiology, Johns Hopkins University Medical School, Baltimore, Maryland 21287 Thyroid hormone (TH) plays a critical role in development, growth, and cellular metabolism. TH production is controlled by a complex mechanism of positive and negative regulation. Hypothalamic TSH-releasing hormone (TRH) stimulates TSH secretion from the anterior pituitary. TSH then initiates TH synthesis and release from the thyroid gland. The synthesis of TRH and TSH subunit genes is inhibited at the transcriptional level by TH, which also inhibits posttranslational modification and release of TSH. Although opposing TRH and TH inputs regulate the hypothalamicpituitary-thyroid axis, TH negative feedback at the pituitary was thought to be the primary regulator of serum TSH levels. However, study of transgenic animals showed an unexpected, dominant role for TRH in regulating the hypothalamic-pituitary-thyroid axis and an unanticipated involvement of the thyroid hormone receptor ligand-dependent activation function (AF-2) domain in TH negative regulation. These results are summarized in the review. (Endocrinology 150: 1091–1096, 2009) S erum concentrations of T4 and its biologically active form T3 are maintained in vivo in a narrow range by the ability of thyroid hormone (TH) to limit its own production by negative feedback at the hypothalamic TSH-releasing hormone (TRH) neuron and pituitary thyrotroph. This feedback is critically dependent upon the presence of normal TH receptors (TRs), which bind to the promoters of TRH and TSH subunit genes and regulate their expression (1–5). In the presence of its ligand, T3, TRs mediate ligand-dependent repression of the transcription of these genes, and in the absence of T3, the transcription rate is not simply returned to baseline, but ligand-independent activation is observed (6 – 8). Although it is still commonly stated that the major locus of TH regulation of the hypothalamic-pituitary-thyroid (HPT) axis is the pituitary, new findings in mouse models suggest otherwise. TH Action on the HPT Axis Early studies using primary cell cultures of mouse thyrotropic tumor cells demonstrated that TH treatment suppressed transcription of TSH subunit genes, and as a consequence, TSH synthesis was reduced (9). At about the same time, TH was shown to ISSN Print 0013-7227 ISSN Online 1945-7170 Printed in U.S.A. Copyright © 2009 by The Endocrine Society doi: 10.1210/en.2008-1795 Received December 29, 2008. Accepted January 13, 2009. First Published Online January 29, 2009 suppress prepro-TRH mRNA levels from certain neurons in the hypothalamus (10). Finally, it was shown that TRH affected the bioactivity of TSH by altering its glycosylation pattern (11). Thus, TH could act at the pituitary, hypothalamic, or both levels to regulate TSH synthesis, which in turn would control TH production by the thyroid. Generation of mouse models where TRs were either deleted or mutated helped to define better TH feedback action on the HPT axis. These mouse lines were designed to model a human disorder referred to as resistance to thyroid hormone (RTH) where a dominantly inherited mutation in the -isoforms of the TR was found (12). The molecular basis of this disorder resides in the dominant inhibition of endogenous TRs by a mutant receptor, which results in elevation of both serum TSH and TH levels. To confirm that the pituitary was an important locus of negative feedback by TH, a transgenic mouse expressing a pituitary-specific mutant TR (⌬337T) was generated (13). Transgenic mice developed profound pituitary RTH, as demonstrated by markedly elevated baseline and non-T3-suppressible serum TSH and pituitary TSH- mRNA levels, and as expected, hypothalamic prepro-TRH mRNA levels were suppressed. Surprisingly, however, serum T4 levels were only marginally elevated in Abbreviations: CoA, Coactivator; CoR, corepressor; CART, cocaine- and amphetamineregulated transcript; CREB, cAMP response element-binding protein; D2, type II deiodinase; DMN, dorsomedial nucleus; GR, glucocorticoid receptor; HPT, hypothalamic-pituitary-thyroid; KO, knockout; MCT, monocarboxylate transporter; NPY, neuropeptide Y; OATP, organic anion transporting polypeptide; PVN, paraventricular nucleus; RTH, resistance to thyroid hormone; Src-1, steroid receptor CoA-1; TH, thyroid hormone; TR, TH receptor; TRH, TSH-releasing hormone. Endocrinology, March 2009, 150(3):1091–1096 endo.endojournals.org 1091 1092 Chiamolera and Wondisford Minireview these mice. After TRH administration, T4 concentrations increased in both transgenic and wild-type animals, but transgenic animals had a sustained increase over 72 h. Hypothyroid transgenic mice also exhibited a TSH response that was only 30% of the response observed in wild-type animals. These findings indicate that pituitary expression of this mutant TR impairs both T3-independent activation and T3-dependent suppression of TSH subunit gene expression in vivo. The discordance between basal TSH and T4 levels and the reversal of these findings with TRH administration demonstrates that resistance at the level of both the thyrotroph and the hypothalamic TRH neuron are required to elevate TH levels in patients with RTH (13). Although hypothalamic TRH is the major stimulator of TSH synthesis and release from the anterior pituitary (14, 15), TH negative feedback at the pituitary was believed to be the most important physiological regulator of serum TSH levels (9). Recently, the central role for TRH in normal TH feedback of the HPT axis was demonstrated. Mice that lack either TRH [TRH knockout (KO)], the -isoforms of TH receptors (TR KO), or both (double KO) were studied. As previously reported, TR KO mice have significantly higher TH and TSH levels compared with wild-type mice. In contrast, double KO mice had reduced TH and TSH levels compared with control animals. Unexpectedly, hypothyroid double KO mice also failed to mount a significant rise in serum TSH levels, and pituitary TSH immunostaining was markedly reduced compared with all other hypothyroid mouse genotypes. This impaired TSH response, however, was not due to a reduced number of pituitary thyrotrophs because thyrotroph cell number, as assessed by TSH-immunopositive cell number, was restored after chronic TRH treatment. Thus, the TRH neuron is absolutely required for both TSH and TH synthesis and appears to be the locus of the set-point in the HPT axis (16). FIG. 1. Endocrinology, March 2009, 150(3):1091–1096 TRH and the Hypophysiotropic TRH Neuron TRH is a tripeptide amide (pyro-Glu-His-Pro-NH2) derived from a large precursor protein, prepro-TRH (ppTRH), by posttranslational processing (prohormone convertase enzymes PC1, -2, and -3) (17). The rat prepro-TRH is a 29-kDa polypeptide composed of 255 amino acids. The rat precursor contains an N-terminal 25-amino-acid leader sequence, five copies of the TRH progenitor sequence Gln-His-Pro-Gly flanked by paired basic amino acids (Lys-Arg or Arg-Arg), four non-TRH peptides lying between the TRH progenitors, an N-terminal flanking peptide, and a C-terminal flanking peptide (18, 19). Rats and mice have five Gln-His-Pro-Gly TRH progenitor sequences, whereas humans have six TRH sequences (19). Serum TH levels can affect the processing of pro-TRH by altering the prohormone convertases; low TH levels stimulate TRH and prohormone convertase expression in the paraventricular nucleus (PVN) (20, 21). The hypothalamic PVN, a triangular shaped nucleus located at the dorsal limits of the third ventricle (22–24), consists of a periventricular parvocellular part containing neurosecretory neurons (hypophysiotropic neurons) that release their hormones into the hypophyseal portal circulation in the median eminence, and a magnocellular part that contains neurosecretory cells projecting to the posterior pituitary, which release oxytocin and vasopressin (25). Studies in rats showed an inverse relationship between serum TH levels and the expression of prepro-TRH mRNA in the PVN during experimentally induced hypo- and hyperthyroidism confirming an essential role for these neurons in this classical endocrine negative feedback loop (10). This regulation was confined to a small population of TRH neurons located within the PVN. Three main neuronal groups mediate the effects of other physiological stimuli on hypophysiotropic TRH neurons (17) (Fig. 1). First, adrenergic input from the medulla is believed to mediate the stimulatory effects of cold exposure on the TRH neuron (26, 27). Catecholamines are believed to increase the set-point for inhibition of TRH gene expression by T3, thereby permitting high circulating levels of TH to contribute to increased thermogenesis. Catecholamines act on TRH neurons primarily through ␣1 adrenergic receptors (26) that can induce the phosphorylation of the cAMP response element-binding protein (CREB) (28). CREB activates the TRH promoter by binding to a CREB response element in the promoter, which overlaps with a TR binding site (29). It is hypothesized that cold exposure increases phosphorylated CREB, which then competes with TR for binding to the promoter region of TRH (17). Adrenergic fibers in contact with TRH neurons also contain at least two neuropeptides: cocaine- and amphetamine-regulated transcript (CART) and neuropeptide Y (NPY) (30, 31). CART exerts a stimulatory effect on Physiological pathways for regulation of hypophysiotropic TRH neurons. the synthesis and release of TRH (22) and may Endocrinology, March 2009, 150(3):1091–1096 potentiate the action of epinephrine on the TRH neurons during cold exposure. In contrast, NPY exerts a potent inhibitory effect on the transcription of the TRH gene (32) through inhibition of the cAMP-CREB second messenger pathway (33). NPY may play a role in antagonizing the increased release of epinephrine in the PVN in several physiological or pathological situations (34). A second input to TRH neurons arises from peptidergic neurons in the arcuate nucleus; these neurons are believed to mediate leptin changes in the HPT axis during fasting (35). Fasting reduces leptin secretion, which results in an increased appetite, energy conservation, and alterations in neuroendocrine axes (36, 37). The HPT axis is affected by fasting resulting in reduced prepro-TRH mRNA synthesis in the PVN, and as a consequence, lower TSH and TH serum levels (38). Two separate leptin-responsive neuronal groups in the arcuate nucleus, with opposing function, send projections to TRH neurons. These neurons signal through either the anorectic peptides CART and ␣-MSH or the orexigenic peptides NPY and agouti-related protein (AGRP) (35). The balance between the effects of both neuronal groups may also be important in establishing the TRH neuronal setpoint for TH feedback inhibition. Finally, the hypothalamic dorsomedial nucleus (DMN) works as a metabolic sensor for hypophysiotropic TRH neurons. The arcuate nucleus sends axon terminals containing ␣-MSH to the DMN and then the DMN send projections to TRH neurons (39). Direct arcuate-PVN and indirect arcuate-DMN-PVN signaling to the TRH neuron may represent alternative pathways by which leptin acts to regulate this neuron (18). In summary, TRH can be synthesized and secreted in many regions of the brain (40, 41), but the hormone synthesized in the hypophysiotropic TRH neurons is the only one regulated by TH (10). In addition to TH, stress, cold, and nutrition can affect TRH expression. TRs TR isoforms are members of the nuclear receptor superfamily of ligand-modulated transcriptional factors (42). Alternative splicing and transcription initiation of two genes produce all known ligand-binding TR isoforms: TR␣1, TR1, TR2, and TR3. The expression and regulation of the TRs vary with isoform and tissue type (5, 43, 44). The immunocytochemical localization of TR isoforms in the adult rat brain was reported in several areas including the hypothalamus (45). More intense TR expression was found in the PVN, the arcuate nucleus, and median eminence of the adult rat (46), and TR2 was found in high abundance in the PVN (5). Indeed, the restricted expression of TR2 (thyrotroph, TRH neurons of PVN, developing ear, and developing retina) contrasts with the more ubiquitous expression of TR␣1 and TR1 isoforms (42). A study using siRNA delivery to the mouse hypothalamus showed that the siRNA directed against TR1 blocks both T3-independent activation and T3-dependent modulation of TRH transcription. In contrast, siRNA directed against TR2 abrogated only T3 repression of transcription (47). In addition to these findings, the study of TR2-null mice (animals lacking the TR2 isoform) demonstrated that basal endo.endojournals.org 1093 prepro-TRH expression was increased in TR2-null mice to levels seen in hypothyroid wild-type mice, but expression did not change significantly in response to either hypothyroidism or T3 treatment. In contrast, the suppression of prepro-TRH mRNA expression in response to fasting was preserved in TR2-null mice. Thus, TR2 is the key TR isoform responsible for T3mediated negative-feedback regulation by hypophysiotropic TRH neurons (48). Deiodinase and MCT8 The intracellular concentration of T3 is an essential determinant of TRH regulation. The intracellular concentration of T3 is determined by cellular uptake as well as T3 production and degradation in the central nervous system. The two most important transporter families that are involved in the TH transport in the brain are the organic anion transporting polypeptide (OATP) and the monocarboxylate transporter (MCT). Among these, MCT8 shows particularly high activity toward T3 (49). One member of the OAT family, OATP 14, is expressed in the PVN, but this is not the higher-affinity TH transporter. On the other hand, MCT8 is expressed in many tissues, including the brain, where it is predominantly localized in neurons. MCT8 plays an important role in the transport of T3 in neurons and mutations in MCT8 interfere with the action and metabolism of T3 in these cells (49). Mice lacking MCT8 have normal TSH levels despite having high T3 levels. Furthermore, mice lacking the MCT8 have low cerebral T3 levels consistent with an inability to transport T3 into neurons (50). In humans, mutations in the MCT8 gene, located on the X chromosome, result in males with neurological abnormalities, including global developmental delay, central hypotonia, spastic quadriplegia, dystonic movements, rotary nystagmus, and impaired gaze and hearing. The endocrine findings include elevated T3 and decreased T4 levels in the presence of a normal TSH secretion (51). T3 production and degradation occurs through T4 deiodination by two separate enzymes, type II (D2) and type III (D3) deiodinase (52). D2 activates thyroid hormone by converting T4 to T3, whereas D3 inactivates thyroid hormone by converting T3 into T2 and T4 into reverse T3. In some studies, hypothyroidism induced only a moderate increase in D2 mRNA in the hypothalamus and no increase in D2 activity (53). In the same way, when hypothalamic cells were analyzed in association with iodine deficiency, there was no increase in D2 activity in the hypothalamus contrary to what was observed in other regions in the brain (54). Taken together, these studies suggest that maintenance of a constant T3 tissue level may not be the main function of D2 in the hypothalamus. In contrast, T3 produced by tanycytes, a unique glial cell type that lines the third ventricle, may be the primary source of T3 for feedback regulation of TRH neurons. Tanycytes express high concentration of D2 mRNA and produce T3 from peripheral circulating T4 (55). T3 can then diffuse into the substance of the brain to reach the hypothalamic PVN (55) or may be released into the median eminence and transported by axon terminals to the hypophysiotropic TRH neurons (24, 56 –59). The D2 activity in the tanycytes under different circulating TH levels seems to contribute to the negative feedback regulation of the HPT axis 1094 Chiamolera and Wondisford Minireview perhaps because it allows the hypophysiotropic TRH neurons to sense any changes in T4 output by the thyroid gland. D2-KO mice demonstrated the critical importance of local T3 production to control of the HPT axis. These animals have low brain T3 levels associated with elevated serum T4 and TSH levels, indicating the presence of central resistance to TH due to inadequate central production of T3 (60). The Thyroid Feedback Mechanism TH regulates TRH gene expression and production through a negative feedback mechanism; TRH expression is high when TH levels are low, and TRH expression is suppressed when TH levels are increased. As outlined earlier, TRH expression is regulated by TH in the PVN (10, 61). This cell-specific action of TH suggests that the TRH neurons in the PVN have all the elements necessary to sense and respond to circulating peripheral TH levels. As noted above, circulating T4 is converted to T3 by D2 in tanycytes. The balance of evidence suggests that T3 then gains access to the TRH neuron via the MCT8 transporter. After T3 enters TRH neurons in the PVN, regulation occurs at two levels: expression of the prepro-TRH transcript and processing of proTRH into the mature TRH peptide. The regulation of TRH gene expression by T3 occurs mostly through TR2, presumably via a direct mechanism. It is also possible that T3 acts in the signaling pathway of TRH gene expression via other hypothalamic nuclei because TR2 is also expressed in the arcuate and ventromedial nuclei, and both nuclei can alter the set-point of TRH expression to fasting. Clearly, the basal level of TRH gene expression is important in determining the set-point for regulation by TH through either a direct or indirect mechanism. What determines the basal level of TRH gene transcription is a matter of much debate. In in vitro studies, the unliganded TR-2 has been shown to activate the TRH gene via its unique amino-terminal domain (62). This finding is consistent with the critical in vivo role of TR-2 in regulating the HPT axis (48). Moreover, it does not appear to be possible to dissociate T3-independent properties of the TR, which activate genes like TRH, from its T3-dependent activities that result in TH inhibition of gene expression. One of the first steps toward understanding TRH regulation by TH was to map TH response elements in the promoter. Deletional analysis of the TRH gene identified a region in the proximal promoter, named site 4, that contained two structurally different negative TH response elements. This region is highly conserved both in murine and human species (2, 63). The core sequence of site 4 (TGACCTCA) is similar to a CREB response element, suggesting a mechanism for cAMP and TH cross talk at the promoter (64). Although this site is important for cAMP and T3 regulation of the TRH gene in vitro, the physiological importance of this region in vivo has yet to be proven. Using transgenic knock-in mouse models, the mechanism of TH negative regulation has begun to be explored in vivo. In one model, two amino acids within the P box of the DNA binding domain of TR- were mutated to those residues found in the glucocorticoid receptor (GR). This mutation (GS125) in vitro Endocrinology, March 2009, 150(3):1091–1096 completely abolished TR DNA binding while preserving T3 binding and cofactor interactions with TR. In functional assays, the mutant displayed defective trans-activation on both positively and negatively regulated promoters (TRH, TSH␣, and TSH). However, the mutant GS125 TR bound to a composite TR/GR-response element and was fully functional on this hybrid TR/GR-response element. Mice carrying this mutation in the germline of both alleles displayed abnormal T3 regulation of the HPT axis identical to phenotypic abnormalities previously observed in TR KO mice. TR- DNA binding, therefore, is essential for negative feedback regulation of the HPT axis by TH (65, 66). A second transgenic knock-in mouse model was constructed to determine whether TR cofactor interactions were essential for TH negative regulation. In vitro studies have demonstrated that TR activity is regulated by binding to both corepressor (CoR) and coactivator (CoA) proteins on TH positively regulated genes. TH stimulation is thought to involve dissociation of CoRs, such as nuclear receptor corepressor (NCoR) and silencing mediator for retinoic acid and TR (SMRT) (67), from the transcriptional complex and recruitment of CoAs, such as steroid receptor CoA-1 (Src-1), to the liganded TR. The physiological role of CoAs bound to TRs, however, had yet to be defined in vivo. A TR knock-in mouse was generated using the E457A TR mutation; this mutation completely abolished CoA recruitment in vitro while preserving normal T3 binding and CoR interactions. As expected, mice bearing this allelic mutation displayed abnormal TH-stimulated gene expression. Interestingly, however, these animals also displayed abnormal regulation of the HPT axis. Serum TH, TSH level, and pituitary TSH subunit mRNA levels were inappropriately elevated compared with those of wild-type animals, and T3 treatment failed to suppress serum TSH and pituitary TSH subunit mRNA levels. These data illustrate the importance of an intact CoA-binding surface for both positive and negative regulation by TH in vivo (68). CoAs are proteins that can remodel chromatin via enzymatic acetylation of histone tails or via regulation of transcriptional complex assembly at the promoter by interactions with RNA polymerase and general transcription factors (69). One of the best studied CoA proteins is Src-1, a member of the p160 class of transcriptional factors, first identified as steroid receptor coactivator (70) and then characterized as a coactivator of other nuclear receptors, including the TR (71, 72). Src-1 KO mice showed mild resistance to TH (73), suggesting that this CoA may be critical in TH regulation of the HPT axis in the E457A mouse. How recruitment of a CoA to the liganded TR results in inhibition of gene expression in the HPT axis remains a puzzle. Perhaps binding of this cofactor to the TR AF-2 domain recruits other corepressor proteins to the transcriptional complex, which then repress gene expression. In conclusion, the mechanism of TH negative feedback control of the HPT axis has been clarified by recent studies. Experimental models demonstrated the critical role of hypothalamic TRH in the control of HPT axis and in establishing the set-point of the axis. The realization that the TR-2 isoform primarily regulates the HPT axis defined a mechanistic difference between TH inhibition and stimulation. Further studies are directed at Endocrinology, March 2009, 150(3):1091–1096 understanding other unique features of TH inhibition at the transcriptional level. Finally, TRH gene expression is also regulated by temperature, food intake, and stress. Thus the TRH neuron is well positioned to integrate information about the environment as well as circulating TH levels and ultimately affect metabolism in response to these physiological changes. Acknowledgments Address all correspondence and requests for reprints to: Fredric E. Wondisford, Division of Metabolism, Departments of Pediatrics, Medicine, and Physiology, Johns Hopkins University Medical School, Baltimore, Maryland 21287. E-mail: [email protected]. Disclosure Statement: The authors have nothing to disclose. References 1. Wondisford FE, Farr EA, Radovick S, Steinfelder HJ, Moates JM, McClaskey JH, Weintraub BD 1989 Thyroid hormone inhibition of human thyrotropin -subunit gene expression is mediated by a cis-acting element located in the first exon. J Biol Chem 264:14601–14604 2. Hollenberg AN, Monden T, Flynn TR, Boers ME, Cohen O, Wondisford FE 1995 The human thyrotropin-releasing hormone gene is regulated by thyroid hormone through two distinct classes of negative thyroid hormone response elements. Mol Endocrinol 9:540 –550 3. Madison LD, Ahlquist JA, Rogers SD, Jameson JL 1993 Negative regulation of the glycoprotein hormone ␣ gene promoter by thyroid hormone: mutagenesis of a proximal receptor binding site preserves transcriptional repression. Mol Cell Endocrinol 94:129 –136 4. Pennathur S, Madison LD, Kay TW, Jameson JL 1993 Localization of promoter sequences required for thyrotropin-releasing hormone and thyroid hormone responsiveness of the glycoprotein hormone ␣-gene in primary cultures of rat pituitary cells. Mol Endocrinol 7:797– 805 5. Lechan RM, Qi Y, Jackson IM, Mahdavi V 1994 Identification of thyroid hormone receptor isoforms in thyrotropin-releasing hormone neurons of the hypothalamic paraventricular nucleus. Endocrinology 135:92–100 6. Wondisford FE, Steinfelder HJ, Nations M, Radovick S 1993 AP-1 antagonizes thyroid hormone receptor action on the thyrotropin -subunit gene. J Biol Chem 268:2749 –2754 7. Sjoberg M, Vennstrom B 1995 Ligand-dependent and -independent transactivation by thyroid hormone receptor 2 is determined by the structure of the hormone response element. Mol Cell Biol 15:4718 – 4726 8. Tagami T, Madison LD, Nagaya T, Jameson JL 1997 Nuclear receptor corepressors activate rather than suppress basal transcription of genes that are negatively regulated by thyroid hormone. Mol Cell Biol 17:2642–2648 9. Shupnik MA, Chin WW, Habener JF, Ridgway EC 1985 Transcriptional regulation of the thyrotropin subunit genes by thyroid hormone. J Biol Chem 5:2900 –2903 10. Segerson TP, Kauer J, Wolfe HC, Mobtaker H, Wu P, Jackson IM, Lechan RM 1987 Thyroid hormone regulates TRH biosynthesis in the paraventricular nucleus of the rat hypothalamus. Science 238:78 – 80 11. Persani L 1998 Hypothalamic thyrotropin-releasing hormone and thyrotropin biological activity. Thyroid 10:941–946 12. Refetoff S, Weiss RE, Usala SJ 1993 The syndromes of resistance to thyroid hormone. Endocr Rev 14:348 –399 13. Abel ED, Kaulbach HC, Campos-Barros A, Ahima RS, Boers ME, Hashimoto K, Forrest D, Wondisford FE 1999 Novel insight from transgenic mice into thyroid hormone resistance and the regulation of thyrotropin. J Clin Invest 103:271–279 14. Harris AR, Christianson D, Smith MS, Fang SL, Braverman LE, Vagenakis AG 1978 The physiological role of thyrotropin-releasing hormone in the regulation of thyroid-stimulating hormone and prolactin secretion in the rat. J Clin Invest 61:441– 448 15. Steinfelder HJ, Hauser P, Nakayama Y, Radovick S, McClaskey JH, Taylor T, Weintraub BD, Wondisford FE 1991 Thyrotropin-releasing hormone regulation of human TSHB expression: role of a pituitary-specific transcription factor (Pit-1/GHF-1) and potential interaction with a thyroid hormone-inhibitory element. Proc Natl Acad Sci USA 88:3130 –3134 endo.endojournals.org 1095 16. Nikrodhanond AA, Ortiga-Carvalho TM, Shibusawa N, Hashimoto K, Liao XH, Refetoff S, Yamada M, Mori M, Wondisford FE 2006 Dominant role of thyrotropin-releasing hormone in the hypothalamic-pituitary-thyroid axis. J Biol Chem 281:5000 –5007 17. Lechan RM, Wu P, Jackson IM, Wolf H, Cooperman S, Mandel G, Goodman RH 1986 Thyrotropin-releasing hormone precursor: characterization in rat brain. Science 231:159 –161 18. Lechan RM, Wu P, Jackson IM 1986 Immunolocalization of the thyrotropinreleasing hormone prohormone in the rat central nervous system. Endocrinology 119:1210 –1216 19. Fekete C, Lechan RM 2007 Negative feedback regulation of hypophysiotropic thyrotropin-releasing hormone (TRH) synthesizing neurons: role of neuronal afferents and type 2 deiodinase. Front Neuroendocrinol 28:97–114 20. Perello M, Friedman T, Paez-Espinosa V, Shen X, Stuart RC, Nillni EA 2006 Thyroid hormones selectively regulate the posttranslational processing of prothyrotropin-releasing hormone in the paraventricular nucleus of the hypothalamus. Endocrinology 147:2705–2716 21. Espinosa VP, Ferrini M, Shen X, Lutfy K, Nillni EA, Friedman TC 2007 Cellular colocalization and coregulation between hypothalamic pro-TRH and prohormone convertases in hypothyroidism. Am J Physiol Endocrinol Metab 292:E175–E186 22. Ishikawa K, Taniguchi Y, Inoue K, Kurosumi K, Suzuki M 1988 Immunocytochemical delineation of thyrotrophic area: origin of thyrotropin-releasing hormone in the median eminence. Neuroendocrinology 47:384 –388 23. Merchenthaler I, Liposits Z 1994 Mapping of thyrotropin-releasing hormone (TRH) neuronal systems of rat forebrain projecting to the median eminence and the OVLT. Immunocytochemistry combined with retrograde labeling at the light and electron microscopic levels. Acta Biol Hung 45:361–374 24. Fekete C, Mihaly E, Luo LG, Kelly J, Clausen JT, Mao Q, Rand WM, Moss LG, Kuhar M, Emerson CH, Jackson IM, Lechan RM 2000 Association of cocaine- and amphetamine-regulated transcript-immunoreactive elements with thyrotropin-releasing hormone-synthesizing neurons in the hypothalamic paraventricular nucleus and its role in the regulation of the hypothalamicpituitary-thyroid axis during fasting. J Neurosci 20:9224 –9234 25. Swanson LW, Sawchenko PE 1983 Hypothalamic integration: organization of the paraventricular and supraoptic nuclei. Annu Rev Neurosci 6:269 –324 26. Arancibia S, Tapia-Arancibia L, Astier H, Assenmacher I 1989 Physiological evidence for ␣1-adrenergic facilitatory control of the cold-induced TRH release in the rat, obtained by push-pull cannulation of the median eminence. Neurosci Lett 100:169 –174 27. Arancibia S, Rage F, Astier H, Tapia-Arancibia L 1996 Neuroendocrine and autonomous mechanisms underlying thermoregulation in cold environment. Neuroendocrinology 64:257–267 28. Thonberg H, Fredriksson JM, Nedergaard J, Cannon B 2002 A novel pathway for adrenergic stimulation of cAMP-response-element-binding protein (CREB) phosphorylation: mediation via ␣1-adrenoceptors and protein kinase C activation. Biochem J 364:73–79 29. Nillni EA, Vaslet C, Harris M, Hollenberg A, Bjorbak C, Flier JS 2000 Leptin regulates prothyrotropin-releasing hormone biosynthesis. Evidence for direct and indirect pathways. J Biol Chem 275:36124 –36133 30. Wittmann G, Liposits Z, Lechan RM, Fekete C 2002 Medullary adrenergic neurons contribute to the neuropeptide Y-ergic innervation of hypophysiotropic thyrotropin-releasing hormone-synthesizing neurons in the rat. Neurosci Lett 324:69 –73 31. Wittmann G, Liposits Z, Lechan RM, Fekete C 2004 Medullary adrenergic neurons contribute to the cocaine- and amphetamine-regulated transcriptimmunoreactive innervation of thyrotropin-releasing hormone synthesizing neurons in the hypothalamic paraventricular nucleus. Brain Res 1006:1–7 32. Fekete C, Kelly J, Mihaly E, Sarkar S, Rand WM, Legradi G, Emerson CH, Lechan RM 2001 Neuropeptide Y has a central inhibitory action on the hypothalamic-pituitary-thyroid axis. Endocrinology 142:2606 –2613 33. Sarkar S, Lechan RM 2003 Central administration of neuropeptide Y reduces ␣-melanocyte-stimulating hormone-induced cyclic adenosine 5⬘-monophosphate response element binding protein (CREB) phosphorylation in pro-thyrotropin-releasing hormone neurons and increases CREB phosphorylation in corticotropin-releasing hormone neurons in the hypothalamic paraventricular nucleus. Endocrinology 144:281–291 34. Agnati LF, Fuxe K, Benfenati F, Battistini N, Harfstrand A, Tatemoto K, Hokfelt T, Mutt V 1983 Neuropeptide Y in vitro selectivity increases the number of ␣2-adrenergic binding sites in membranes of the medulla oblongata of the rat. Acta Physiol Scand 118:293–295 35. Lechan RM, Fekete C 2006 The TRH neuron: a hypothalamic integrator of energy metabolism. Prog Brain Res 153:209 –235 36. Wiersinga WM, Boelen A 1996 Thyroid hormone metabolism in nonthyroidal illness. Curr Opin Endocrinol Diabetes 3:422– 427 1096 Chiamolera and Wondisford Minireview 37. De Groot LJ 1999 Dangerous dogmas in medicine: the nonthyroidal illness syndrome. J Clin Endocrinol Metab 84:151–164 38. Legradi G, Emerson CH, Ahima RS, Flier JS, Lechan RM 1997 Leptin prevents fasting-induced suppression of prothyrotropin-releasing hormone messenger ribonucleic acid in neurons of the hypothalamic paraventricular nucleus. Endocrinology 138:2569 –2576 39. Mihaly E, Fekete C, Legradi G, Lechan RM 2001 Hypothalamic dorsomedial nucleus neurons innervate thyrotropin-releasing hormone-synthesizing neurons in the paraventricular nucleus. Brain Res 891:20 –31 40. Fliers E, Noppen NW, Wiersinga WM, Visser TJ, Swaab DF 1994 Distribution of thyrotropin-releasing hormone (TRH)-containing cells and fibers in the human hypothalamus. J Comp Neurol 350:311–323 41. Guldenaar SE, Veldkamp B, Bakker O, Wiersinga WM, Swaab DF, Fliers E 1996 Thyrotropin-releasing hormone gene expression in the human hypothalamus. Brain Res 743:93–101 42. Lazar MA 1993 Thyroid hormone receptors: multiple forms, multiple possibilities. Endocr Rev 14:184 –193 43. Bradley DJ, Towle HC, Young 3rd WS 1994 ␣ and -thyroid hormone receptor (TR) gene expression during auditory neurogenesis: evidence for TR isoform-specific transcriptional regulation in vivo. Proc Natl Acad Sci USA 91:439 – 443 44. Hodin RA, Lazar MA, Wintman BI, Darling DS, Koenig RJ, Larsen PR, Moore DD, Chin WW 1989 Identification of a thyroid hormone receptor that is pituitary-specific. Science 244:76 –79 45. Puymirat J, Miehe M, Marchand R, Sarlieve L, Dussault JH 1991 Immunocytochemical localization of thyroid hormone receptors in the adult rat brain. Thyroid 1:173–184 46. Alkemade A, Friesema EC, Unmehopa UA, Fabriek BO, Kuiper GG, Leonard JL, Wiersinga WM, Swaab DF, Visser TJ, Fliers E 2005 Neuroanatomical pathways for thyroid hormone feedback in the human hypothalamus. J Clin Endocrinol Metab 90:4322– 4334 47. Guissouma H, Froidevaux MS, Hassani Z, Demeneix BA 2006 In vivo siRNA delivery to the mouse hypothalamus confirms distinct roles of TR isoforms in regulating TRH transcription. Neurosci Lett 406:240 –243 48. Abel ED, Ahima RS, Boers ME, Elmquist JK, Wondisford FE 2001 Critical role for thyroid hormone receptor 2 in the regulation of paraventricular thyrotropin-releasing hormone neurons. J Clin Invest 107:1017–1023 49. Visser TJ 2007 Thyroid hormone transporters. Horm Res 68(Suppl 5):28 –30 50. Trajkovic M, Visser TJ, Mittag J, Horn S, Lukas J, Darras VM, Raivich G, Bauer K, Heuer H 2007 Abnormal thyroid hormone metabolism in mice lacking the monocarboxylate transporter 8. J Clin Invest 117:627– 635 51. Dumitrescu AM, Liao XH, Best TB, Brockmann K, Refetoff S 2004 A novel syndrome combining thyroid and neurological abnormalities is associated with mutations in a monocarboxylate transporter gene. Am J Hum Genet 74:168 –175 52. Bianco AC, Salvatore D, Gereben B, Berry MJ, Larsen PR 2002 Biochemistry, cellular and molecular biology, and physiological roles of the iodothyronine selenodeiodinases. Endocr Rev 23:38 – 89 53. Diano S, Naftolin F, Goglia F, Horvath TL 1998 Fasting-induced increase in type II iodothyronine deiodinase activity and messenger ribonucleic acid levels is not reversed by thyroxine in the rat hypothalamus. Endocrinology 139: 2879 –2884 54. Serrano-Lozano A, Montiel M, Morell M, Morata P 1993 5⬘-Deiodinase activity in brain regions of adult rats: modifications in different situations of experimental hypothyroidism. Brain Res Bull 30:611– 616 55. Tu HM, Kim SW, Salvatore D, Bartha T, Legradi G, Larsen PR, Lechan RM 1997 Regional distribution of type 2 thyroxine deiodinase messenger ribonucleic acid in rat hypothalamus and pituitary and its regulation by thyroid hormone. Endocrinology 138:3359 –3368 56. Diano S, Naftolin F, Goglia F, Csernus V, Horvath TL 1998 Monosynaptic pathway between the arcuate nucleus expressing glial type II iodothyronine Endocrinology, March 2009, 150(3):1091–1096 57. 58. 59. 60. 61. 62. 63. 64. 65. 66. 67. 68. 69. 70. 71. 72. 73. 5⬘-deiodinase mRNA and the median eminence-projective TRH cells of the rat paraventricular nucleus. J Neuroendocrinol 10:731–742 Fekete C, Legradi G, Mihaly E, Huang QH, Tatro JB, Rand WM, Emerson CH, Lechan RM 2000 ␣-Melanocyte-stimulating hormone is contained in nerve terminals innervating thyrotropin-releasing hormone-synthesizing neurons in the hypothalamic paraventricular nucleus and prevents fasting-induced suppression of prothyrotropin-releasing hormone gene expression. J Neurosci 20:1550 –1558 Legradi G, Lechan RM 1998 The arcuate nucleus is the major source for neuropeptide Y-innervation of thyrotropin-releasing hormone neurons in the hypothalamic paraventricular nucleus. Endocrinology 139:3262–3270 Legradi G, Lechan RM 1999 Agouti-related protein containing nerve terminals innervate thyrotropin-releasing hormone neurons in the hypothalamic paraventricular nucleus. Endocrinology 140:3643–3652 Schneider MJ, Fiering SN, Pallud SE, Parlow AF, St Germain DL, Galton VA 2001 Targeted disruption of the type 2 selenodeiodinase gene (DIO2) results in a phenotype of pituitary resistance to T4. Mol Endocrinol 15:2137–2148 Dyess EM, Segerson TP, Liposits Z, Paull WK, Kaplan MM, Wu P, Jackson IM, Lechan RM 1988 Triiodothyronine exerts direct cell-specific regulation of thyrotropin-releasing hormone gene expression in the hypothalamic paraventricular nucleus. Endocrinology 123:2291–2297 Langlois MF, Zanger K, Monden T, Safer JD, Hollenberg AN, Wondisford FE 1997 A unique role of the -2 thyroid hormone receptor isoform in negative regulation by thyroid hormone. Mapping of a novel amino-terminal domain important for ligand-independent activation. J Biol Chem 40:24927–24933 Satoh T, Yamada M, Iwasaki T, Mori M 1996 Negative regulation of the gene for the preprothyrotropin-releasing hormone from the mouse by thyroid hormone requires additional factors in conjunction with thyroid hormone receptors. J Biol Chem 271:27919 –27926 Harris M, Aschkenasi C, Elias CF, Chandrankunnel A, Nillni EA, Bjoorbaek C, Elmquist JK, Flier JS, Hollenberg AN 2001 Transcriptional regulation of the thyrotropin-releasing hormone gene by leptin and melanocortin signaling. J Clin Invest 107:111–120 Shibusawa N, Hollenberg AN, Wondisford FE 2003 Thyroid hormone receptor DNA binding is required for both positive and negative gene regulation. J Biol Chem 278:732–738 Shibusawa N, Hashimoto K, Nikrodhanond AA, Liberman MC, Applebury ML, Liao XH, Robbins JT, Refetoff S, Cohen RN, Wondisford FE 2003 Thyroid hormone action in the absence of thyroid hormone receptor DNA-binding in vivo. J Clin Invest 112:588 –597 Lazar MA 2003 Thyroid hormone action: a binding contract. J Clin Invest 112:497– 499 Ortiga-Carvalho TM, Shibusawa N, Nikrodhanond A, Oliveira KJ, Machado DS, Liao XH, Cohen RN, Refetoff S, Wondisford FE 2005 Negative regulation by thyroid hormone receptor requires an intact coactivator-binding surface. J Clin Invest 115:2517–2523 Fondell JD, Guermah M, Malik S, Roeder RG 1999 Thyroid hormone receptor-associated proteins and general positive cofactors mediate thyroid hormone receptor function in the absence of the TATA box-binding proteinassociated factors of TFIID. Proc Natl Acad Sci USA 96:1959 –1964 Oñate SA, Tsai SY, Tsai MJ, O’Malley BW 1995 Sequence and characterization of a coactivator for the steroid hormone receptor superfamily. Science 5240:1354 –1357 Jeyakumar M, Tanen MR, Bagchi MK 1997 Analysis of the functional role of steroid receptor coactivator-1 in ligand-induced transactivation by thyroid hormone receptor. Mol Endocrinol 6:755–767 Feng W, Ribeiro RC, Wagner RL, Nguyen H, Apriletti JW, Fletterick RJ, Baxter JD, Kushner PJ, West BL 1998 Hormone-dependent coactivator binding to a hydrophobic cleft on nuclear receptors. Science 5370:1747–1749 Weiss RE, Xu J, Ning G, Pohlenz J, O’Malley BW, Refetoff S 1999 Mice deficient in the steroid receptor co-activator 1 (SRC-1) are resistant to thyroid hormone. EMBO J 18:1900 –1904