Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
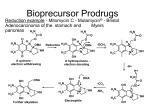
Gene Therapy (2000) 7, 224–231 2000 Macmillan Publishers Ltd All rights reserved 0969-7128/00 $15.00 www.nature.com/gt ACQUIRED DISEASES RESEARCH ARTICLE Secreted human -glucuronidase: a novel tool for gene-directed enzyme prodrug therapy D Weyel1, H-H Sedlacek2, R Müller1 and S Brüsselbach1 1 Institute of Molecular Biology and Tumor Research (IMT), Philipps-University Marburg; and 2Hoechst Marion Roussel Deutschland, Marburg, Germany A major problem of tumor gene therapy is the low transduction efficiency of the currently available vectors. One way to circumvent this problem is the delivery of therapeutic genes encoding intracellular enzymes for the conversion of a prodrug to a cytotoxic drug which can then spread to neighboring non-transduced cells (bystander effect). One possibility to improve the bystander effect could be the extracellular conversion of a hydrophilic prodrug to a lipophilic, cell-permeable cytotoxic drug. Toward this end, we have used a secreted form of the normally lysosomal human -glucuronidase (s-Gluc) to establish an extracellular cytotoxic effector system that converts an inactivated glucuronidated derivative of doxorubicin (HMR 1826) to the cytotoxic drug. We demonstrate that s-Gluc-transduced tumor cells convert HMR 1826 to doxorubicin which is taken up by both transduced and non-transduced cells. s-Gluc in combination with HMR 1826 efficiently induces tumor cell killing both in cell culture and in vivo. This effect is mediated through a pronounced bystander effect of the generated cytotoxic drug. Most notably, this gene therapeutic strategy is shown to be clearly superior to conventional chemotherapy with doxorubicin. Gene Therapy (2000) 7, 224–231. Keywords: -glucuronidase; prodrug; HMR 1826; doxorubicin; GDEPT Introduction A major limitation of conventional chemotherapy is its lack of selectivity for tumor cells resulting in dose-limiting, toxic side-effects and the generation of multidrug resistant tumor cells due to insufficient drug concentrations at the tumor site.1 One possibility to overcome these problems is the use of non-toxic forms of cytotoxic compounds (prodrugs) that are converted to the toxic drug selectively at the tumor site. Strategies using tumorspecific antibodies to guide the enzyme to the tumor (antibody-enzyme conjugates) have been replaced to a large extent by gene-directed approaches. Unfortunately, the vectors currently in use for gene therapy show a very low transduction efficiency in vivo and lead to the expression of the therapeutic gene in only a small fraction of the target tissue. For this reason it is necessary to use an effector mechanism which targets not only the transduced cell itself, but the neighboring cells as well (bystander effect). Most cytotoxic effector systems available at present – such as the herpes simplex virus thymidine kinase (HSVtk),2 the E. coli cytosine deaminase (EcCD)3,4 and the E. coli nitroreductase (Ec-NR)5 – act intracellularly, ie convert the respective prodrug to the active drug within the cell. One way to improve the bystander effect could be the extracellular conversion of a prodrug, which is easily taken up by surrounding cells, as exemplified by prokaryotic carboxypeptidase G2 displayed on Correspondence: R Müller, Institute of Molecular Biology and Tumor Research (IMT), Philipps-University Marburg, Emil-Mannkopff-Strasse 2, D-35033 Marburg, Germany Received 16 June 1999; accepted 13 September 1999 the surface of the transduced cells.6 We have chosen the human -glucuronidase to establish an extracellular cytotoxic effector system, converting a hydrophilic non-cellpermeable prodrug (glucuronide prodrug of doxorubicin, HMR 1826) to the lipophilic, membrane-permeable drug doxorubicin. -Glucuronidase seems to be a particularly suitable candidate as a prodrug-converting enzyme, because the endogenous enzyme is located in lysosomes and therefore not available for prodrug conversion under normal circumstances. In addition, -glucuronidase leaking out of cells is rapidly internalized via the mannose-6-phosphate (M6P) receptor on the cell surface as discussed in detail below. The physiological function of -glucuronidase is the degradation of glucuronic acid containing glucosaminoglycans (like heparan sulfate, chondroitin sulfate, and dermatan sulfate).7 In higher eukaryotes, the enzyme undergoes a series of post-translational modifications.8 Individual subunits of 651 amino acids are synthesized on membrane-bound ribosomes and translocated into the endoplasmic reticulum (ER), where several post-translational events occur. These include signal sequence cleavage, N-linked glycosylation, disulfide bond formation, and homotetramerization. In the Golgi complex, the glycans are modified to expose M6P residues. In the trans-Golgi network these M6P residues serve as ligands for specific receptors, which transport lysosomal enzymes to a prelysosomal compartment. After pHdependent dissociation of the receptor-enzyme complex, the receptor relocates to the Golgi, and the enzyme stays in the developing lysosome. The active homotetramer has a Mr of 332 000. There are two active sites located in a large cleft at the interface of Extracellular prodrug conversion D Weyel et al two monomers. Each cleft has two catalytic centers, which means that there are four catalytic centers per tetramer.9 The pH-optimum of -glucuronidase is approximately 5.5 which corresponds to its natural acid environment in lysosomes. The prodrug used in this study, glucuronyl-doxorubicin [N-(4--glucuronyl-3-nitrobenzyloxycarbonyl)doxorubicin], has been successfully used for an experimental monotherapy of human xenografts in mice.10 The rational of this approach is the presence of large amounts of endogenous -glucuronidase in necrotic tumor areas. A problem associated with this strategy obviously is the fact that not all tumors contain necrotic areas, and that such regions of the tumor are particularly poorly penetrated by chemicals. An initial destruction of cells at the tumor edge due to the expression of transduced -glucuronidase could therefore improve the penetration of the prodrug into the tumor and result in an amplification of the cleavage reaction by endogenous -glucuronidase. In the present study, we demonstrate that tumor cells transduced with a secreted form of human -glucuronidase (s-Gluc) convert HMR 1826 to doxorubicin, and that the generated drug produces a strong bystander effect in cell culture. We also show that the secreted -glucuronidase in combination with HMR 1826 efficiently induces tumor cell killing in a human xenograft model in nude mice through a clear bystander effect, and that this gene therapeutic strategy is superior to conventional chemotherapy with doxorubicin. Results Secretion of enzymatically active human -glucuronidase Human -glucuronidase cDNA containing the signal sequence from the human IgG1 heavy chain was cloned into the pcDNA3 expression vector. Transiently transfected cells of the human choriocarcinoma cell line JEG3 were established and the expression of the construct was verified using a monoclonal antibody specific for human -glucuronidase. The immunofluorescence analysis in Figure 1A shows a staining pattern that is typical of proteins entering the secretory pathway, with a bright staining around the nuclear membrane and vesicles distributed in the cytoplasm. Expression of -glucuronidase was confirmed by the immunoprecipitation (Figure 2a). Analysis of the immunoprecipitate by denaturing polyacrylamide gel electrophoresis showed a strong band of monomeric -glucuronidase with the expected size of 83 kDa. In contrast, expression of endogenous -glucuronidase was not detectable by immunofluorescence (data not shown), and only very low amounts of -glucuronidase could be immunoprecipitated from untransfected cells (Figure 2a). For further biochemical studies and therapeutic experiments, stable clones were selected in G418-containing medium from a pool of transfected cells. These clones were analyzed for -glucuronidase expression by immunofluorescence analysis and immuno-precipitation. Clone JEG-3sG, which contains approximately 15% -glucuronidase positive cells, was used for all subsequent experiments. Secretion of -glucuronidase was verified by pulse-chase labeling of cells with 35S-methionine followed by immunoprecipitation of the cell lysate and supernatant. -Glucuronidase was detected in the supernatant as early as 3 h after withdrawal of the radioactively labeled methionine, although the synthesized enzyme was only partially secreted (Figure 2a). The reason for this lies presumably in the fact that secretion is brought about by endosomal overflow (see Discussion). In parallel the activity of lactate dehydrogenase (LDH) as a cytosolic marker enzyme was determined to assess the passive release of -glucuronidase from dying cells. No LDH activity was detectable in the culture medium during the course of the experiment (data not shown), indicating that the -glucuronidase in the culture supernatant was actively secreted. We will subsequently refer to this secreted form of -glucuronidase as s-Gluc. The enzymatic activity of the recombinant s-Gluc was demonstrated using two different approaches. JEG-3sG cells were stained with the chromogenic substrate X-gluc which is converted to a blue precipitate upon cleavage (Figure 1B, bottom panel). In agreement with the expression data obtained by immunofluorescence analysis (Figure 1A) 15% of the cells stained blue. The ‘negative’ cells in the transduced cell population showed a similarly weak staining as the parental non-transduced cells (Figure 1B, top panel), indicating that the staining of the former cells results from the expression of endogenous lysosomal -glucuronidase. In addition, the culture supernatant was analyzed for enzymatic activity under different pH conditions using pNPG as the substrate (Figure 2b). As expected, this analysis showed a pH-optimum around pH 5. Half-maximal -glucuronidase activity was observed at a pH of approximately 6. Under physiological pH conditions (pH approximately 7) the enzymatic activity was about 20% of the maximum. 225 Conversion of a doxorubicin prodrug and toxicity in cell culture We next investigated whether s-Gluc is able to cleave the glucuronide prodrug of doxorubicin, HMR 1826. In addition, we addressed the question to which extent the activated drug is taken up by cells not expressing the prodrug-converting enzyme in order to assess a potential bystander effect. For this purpose, we made use of the strong autofluorescence of doxorubicin with an emission maximum at 600 nm.11 HMR 1826 is hydrophilic and cannot pass cell membranes. After cleavage of the prodrug, the generated hydrophobic doxorubicin can cross cell membranes, intercalates into DNA and thereby leads to a red nuclear fluorescence. This experiment was carried out with a transiently transfected cell population containing 2% s-Gluc expressing cells. Two days after transfection the cells were incubated with the prodrug for 24 h. Transfected cells were visualized by indirect immunofluorescence to detect -glucuronidase expression (green fluorescence in Figure 1C, panel b). In comparison to untransfected cells, where weak staining was observed (Figure 1C, panel a), all the nuclei stained strongly positive for doxorubicin in the transfected dish (Figure 1C, panel c) despite the low number of s-Gluc expressing cells. The weak staining of untransfected cells (Figure 1C, panel a) is due to a low level of generated doxorubicin rather than autofluorescence of the cells (data not shown). Cytotoxic effects of the s-Gluc/HMR 1826 system in cell culture were analyzed using the crystal violet assay. As can be seen in Figure 3, the JEG-3sG clone containing Gene Therapy Extracellular prodrug conversion D Weyel et al 226 A C a B b c Figure 1 (A) Immunostaining of s-Gluc transfected JEG-3 cells. (B) Detection of enzyme activity with X-gluc as substrate. (C) Prodrug conversion by JEG-3 cells transiently transfected with s-Gluc. Two days after transfection the cells were incubated with 10 m prodrug for 24 h and formalinfixed. -Glucuronidase expressing cells were detected by indirect immunofluorescence using a DTAF-labeled secondary antibody (green; panel b) and prodrug conversion was visualized by the red nuclear staining of doxorubicin intercalated into DNA (panel c). Untransfected cells did not show any prodrug conversion (panel a). Arrows point to s-Gluc expressing cells. 15% s-Gluc expressing cells was highly sensitive to the prodrug (IC50 = 1.8 m). This IC50 value is considerably higher than that for doxorubicin (34 nm) which is due to an incomplete conversion of the prodrug to doxorubicin under the cell culture conditions used (eg dilution of s-Gluc by the large volume of culture medium). In contrast, control JEG-3 cells (con JEG-3) were affected by the prodrug only at high concentrations (IC50 ⬎ 8 m). In vivo functionality of the s-Gluc/HMR 1826 system Since the s-Gluc/HMR 1826 system was functional in cell culture we sought to study its effect in an in vivo tumor model. In order to verify expression and enzymatic activity of s-Gluc in the tumor tissue, s-Gluc expressing cells (JEG-3sG) and non-expressing control Gene Therapy JEG-3 cells were injected subcutaneously (s.c.) into nu/nu mice. When tumors had grown to a size of 100 mm2 the mice were killed, and cryosections of the tumors were prepared for enzyme histochemistry. In JEG-3sG xenografts -glucuronidase activity could be demonstrated as shown by the deposition of the water-insoluble red naphthol-neufuchsin dye complex (Figure 4a). Addition of an excess of a saccharolactone (D-saccharic-1,4-lactone), a competitive inhibitor of -glucuronidase, completely abrogated staining (data not shown). No enzymatic activity could be detected in control JEG-3 xenografts (Figure 4b). Tumor growth curves did not show any significant difference between s-Gluc expressing and control JEG-3 cells (Figure 5). The extent of staining in the tumor tissue was compatible with the expression of s-Gluc in 15% of the injected cell population (see above). Extracellular prodrug conversion D Weyel et al 227 Figure 3 Cytotoxic effect of s-Gluc mediated prodrug conversion in cell culture. s-Gluc expressing cells (JEG-3sG) and non-expressing control JEG-3 cells (con JEG-3) were incubated with either HMR 1826 or doxorubicin for 24 h in microtiter plates, followed by staining with crystal violet and measuring the optical density at 540 nm 2 days later (as an indicator of cell number). Specific cytotoxicity of the prodrug in -glucuronidase expressing cells relative to non-expressing control JEG-3 cells is evident. The following IC50 values were calculated from the data: JEG-3sG cells + prodrug, IC50 = 1.8 m; JEG-3sG cells + doxorubicin, IC50 = 34 nm; control JEG-3 cells + prodrug, IC50 ⬎ 8 m; control JEG-3 cells + doxorubicin, IC50 = 28 nm. Figure 2 (a) Secretion of s-Gluc. JEG-3 cells were stably transfected with s-Gluc and labeled with 35S-methionine in a pulse-chase experiment. A clear enrichment of secreted -glucuronidase (arrow) was seen in the supernatant. M, marker proteins. Numbers at the top indicate hours after removal of 35S-methionine. (b) pH-dependence of s-Gluc. The supernatant of -glucuronidase secreting cells was incubated with 1 mm pNPG at different pH-values and the optical density 415 nm was determined. We then compared the sensitivity of established con JEG-3 and JEG-3sG xenografts with HMR 1826 or doxorubicin. At a tumor size of 10–110 mm2 prodrug or drug was injected into the tail vein and tumor growth was monitored. Prodrug treatment (100 mg/kg) of mice injected with con JEG-3 cells did not show any significant difference in tumor growth in comparison with untreated mice injected with con JEG-3 cells or JEG-3sG cells (Figure 5a and b). In contrast, tumors established with JEG-3sG cells immediately started to regress (Figure 5a and b). This effect was independent of the tumor size at the time of prodrug injection, since even the biggest tumor (110 mm2) regressed and actually showed a transient complete response (Figure 5b). In four out of five mice, tumor growth resumed after another 20 days, and one mouse has remained tumor-free for 7+ months. We also performed mixing experiments where equal numbers of JEG-3sG and con JEG-3 cells were coinjected. In this case (ie approximately 7% transduced cells) a clear therapeutic effect was also detectable, but no complete tumor regression was seen (Figure 5a). In addition, tumor-bearing mice were treated with doxorubicin (1.5 mg/kg), but only a marginal effect on tumor growth was detectable (Figure 5a). Since the IC50 value of HMR 1826 is 270-fold higher than that of doxorubicin (see above), this dose of doxorubicin would correspond to 740 mg/kg of HMR 1826 with respect to toxicity. However, the prodrug showed a dramatic therapeutic effect already at 100 mg/kg (ie one-seventh of the calculated equitoxic dose) confirming the superiority of the prodrug-converting system. Discussion A major problem of conventional cancer chemotherapy is the low tumor specificity of the currently available drugs. Severe side-effects limit the applicable dose and consequently the achievable drug concentration in the tumor. This in turn favors the acquisition of multidrug resistance and recurrent tumor growth. One way to prevent such an adjustment of the tumor cells to chemotherapy would be the development of strategies that allow for high local drug concentrations. In the present study, we demonstrate that the local expression of extracellularly active human -glucuronidase can be used to sensitize tumor cells to a doxorubicin prodrug. Since the maximal tolerable dose of prodrug is at least 130-fold higher than that of doxorubicin,10 considerably higher doxorubicin concentrations can be reached at the site of the tumor in the absence of an increased unspecific toxicity. A number of strategies have been developed in the past to sensitize tumor cells to specific prodrugs. Most cytotoxic effector systems available at present, such as Gene Therapy Extracellular prodrug conversion D Weyel et al 228 a b Figure 4 Activity of -glucuronidase in cryosections of tumors grown in nude mice after s.c. inoculation of s-Gluc expressing cells (a) or nonexpressing control JEG-3 cells (b). -Glucuronidase activity was visualized by the deposition of the water-insoluble red naphthol-neufuchsin dye complex. HSVtk/ganciclovir (GCV), Ec-CD/5-fluorocytosine (5FC) or Ec-NR/CB1954, convert a prodrug to an active drug intracellularly. The distribution of the activated drug beyond the transduced cell (bystander effect) depends on gap junction,12,13 phagocytosis of apoptotic bodies14 or diffusion.15 An extracellularly active prodrug converting system could circumvent these problems which lead to a limited bystander effect, at least in part. To date one such system has been described.6 In this case, bacterial carboxipeptidase G2 was endowed with a transmembrane domain to tether the enzyme to the cell surface. This enzyme was used in conjunction with prodrug that is converted by carboxipeptidase G2 to a mustard drug. This system was shown to exert a bystander effect in vitro and to be therapeutically efficacious in an experimental tumor model. The s-Gluc/HMR 1826 system differs from the carboxipeptidase system in several potentially relevant aspects, including the use of a human and thus non-immunogenic enzyme, the presence of an amplification effect in necrotic areas of the tumor and presumably the absence of enzyme leakage into the circulation. These aspects of the s-Gluc/HMR 1826 system are discussed in detail below. Human -glucuronidase is normally a lysosomal enzyme. Similar to secreted proteins all natural lysosomal Gene Therapy proteins have a leader sequence, but bind in the ER to the mannose-6-phoshate receptor which identifies these proteins for translocation to the lysosomes. If the expression of lysosomal proteins exceeds the capacity of this mechanism (as determined by the numbers of mannose-6-phoshate receptors in the ER) the protein will be secreted. This most likely applies to our system as well and explains the incomplete secretion of s-Gluc in Figure 2a. In order to achieve an optimal translocation of -glucuronidase to the ER we exchanged the endogenous signal sequence with the highly efficient leader of IgG. However, this sequence is removed from the protein upon processing in the ER with the consequence that the secreted s-Gluc is identical to wild-type enzyme. In the s-Gluc/HMR 1826 system introduced in the present study, a hydrophilic prodrug is cleaved in the intercellular space, and hydrophobic doxorubicin can enter the surrounding cells. In cell culture experiments, where s-Gluc can diffuse freely in the supernatant, 2% -glucuronidase-positive cells suffice to achieve a distribution of doxorubicin to all cells in the population, as can be seen by the positive nuclear staining. In vivo the situation is of course different. Diffusion of secreted -glucuronidase is restricted by tissue barriers and the secreted enzyme is internalized via M6P-receptors on the cell surface. On the one hand these mechanisms limit the bystander effect, but on the other hand they restrict the activation of the prodrug to the site of the tumor. This selectivity of prodrug activation is complemented by a site-restricted action of the generated cytotoxic drug, which is due to the specific chemical properties of the compounds. HMR 1826 is hydrophilic and can therefore hardly enter living cells which prevents its conversion by the normally lysosomal -glucuronidase. In contrast, doxorubicin is lipophilic and is rapidly taken up by surrounding cells, which strongly limits its leakage into the circulation. That the s-Gluc/HMR 1826 system is site-selective is indirectly shown in our in vivo experiments. Fifteen percent of s-Gluc expressing tumor cells were sufficient to achieve a complete macroscopic tumor regression in all mice treated with the prodrug showing the functionality of the system. On the other hand, the high concentrations of HMR 1826 that were applied systemically did not produce any visible toxic effects (unpublished observations). There are a number of other aspects that distinguish the s-Gluc system from other prodrug converting systems. First, the low pH-optimum of -glucuronidase might contribute to a preferential activation of the enzyme at the site of a tumor, because pH levels in certain tumors are often low.16,17 At physiological pH -glucuronidase activity is reduced, which limits prodrug conversion at distal sites, for example in case of the inadvertent expression of the recombinant enzyme in non-tumor cells. Second, HMR 1826 can also be converted to doxorubicin by the endogenous -glucuronidase when the enzyme is released from the cells. This happens in tumors containing necrotic areas where an efficient conversion of the prodrug has been shown, which leads to the suggestion of using HMR 1826 for monotherapy of necrotic tumors.10 A combination with a gene therapeutic approach should potentiate the effect. The tumor margins are normally well vascularized and therefore accessible by the vectors used for gene therapy, but these cells are Extracellular prodrug conversion D Weyel et al 229 Figure 5 Effect of the -Gluc/HMR 1826 system on tumor growth in vivo. s-Gluc expressing cells (JEG-3sG), non-expressing control cells (con JEG3) or a mixture (1:1) of both were injected s.c. into nude mice (106 cells per mouse). After 2 weeks tumors had reached a size of 20–110 mm2 and both groups of mice were divided into three subgroups each, either receiving 100 mg/kg of prodrug (+PD), 1.5 mg/kg of drug or no treatment. Tumor growth was recorded every 2–3 days. (a) Tumor size (area) plotted against time after prodrug/drug treatment. Values represent averages of five to eight mice. One mouse of the JEG-3sG/HMR 1826 group showed a lasting complete response (see also b) and was therefore not included in the calculations beyond day 15. (b) Graphs displaying the data for individual JEG-3sG mice and used for the calculation of average values in panel a (marked # or *). not necrotic and therefore do not release endogenous -glucuronidase. In contrast, the cells in the center of the tumor are hardly accessible by vectors due to the lack of intact blood vessels, but release -glucuronidase owing to their necrotic nature. This should lead to a scenario, where large areas in the tumor contain extracellular -glucuronidase available for prodrug cleavage. Third, an important aspect is the fact that s-Gluc can in principle be used in combination with any cytotoxic drug that can be inactivated by glucuronidation.18,19 This is relevant in view of the fact that even highly toxic compounds could be used that could normally not be used as chemotherapeutics because of their small therapeutic window. An important factor in this context is the ability to kill non-proliferating cells. Tumors often contain a fraction of ‘resting’ malignant cells, which are not eliminated by conventional radiation or chemotherapy. The most widely used prodrug converting system, the HSVtk/ganciclovir combination, lacks the ability to target non-proliferating cells. Although doxorubicin has some cell cycle independent cytotoxic effects it should be possible to use compounds for prodrug synthesis, which work much more efficiently with non-cycling cells. In addition, the use of non-conventional drugs could also be important to circumvent common problems associated with chemotherapy, such as multidrug resistance, p53 dependence or the expression of anti-apoptotic genes. In this context it may be particularly appealing to employ combinations of different prodrugs to achieve synergistic effects with respect to tumor cell killing. Fourth, immunologic aspects deserve some consideration. Previously established prodrug converting systems use xenogenic enzymes with unique substrate specificities to avoid unspecific prodrug activation by endogenous enzymes. However, foreign proteins provoke an immune response, which can limit the expression of the transgene20,21 and the action of its encoded product. The use of a human enzyme in human patients should circumvent this problem. On the other hand, activation of the immune system might enhance drug induced tumor regression. In the case of HSVtk/GCV a bystander effect has been described, which works in syngeneic mice only.22 Tumorigenicity was shown to depend on the induction of heat shock proteins which have been suggested to function through the cross-priming of antigen-presenting cells by antigens released from tumor cells and may provide an immunostimulatory signal in vivo to break tolerance to tumor Gene Therapy Extracellular prodrug conversion D Weyel et al 230 antigens.23 In agreement with this finding, the treatment of HSVtk transduced brain tumors with GCV2,24 and EcCD transduced colon carcinoma with 5-FC25 protected mice against the rechallenge with untransfected tumor cells. In the same way the ‘biochemical’ bystander effect of the s-Gluc/HMR 1826 system might cooperate with an ‘immunological’ component in syngeneic animals, a question we will investigate in future experiments. Materials and methods Plasmid constructs The human -glucuronidase c-DNA26 was cloned into the pcDNA3 expression vector, containing the neomycin resistance gene. The IgG signal sequence (coding for the first 19 amino acids)27 was cloned 5⬘ to the human -glucuronidase into the first PstI restriction site. For cloning of the signal sequence an oligonucleotide was synthesized, containing a Kozak sequence in front of the start codon to improve translation initiation. Plasmids were amplified in E. coli and purified according to a standard protocol (Qiagen, Hilden, Germany). Cloned oligonucleotide sequence was verified by DNA sequencing. Cell culture The human choriocarcinoma cell line JEG-3 (obtained from A Wellstein, Georgetown University, Washington DC), was cultured in Dulbecco-Vogt modified Eagle’s medium (DMEM; BioWhittaker, Verviers, Belgium), supplemented with 10% fetal bovine serum (FBS; BioWhittaker) and 2 mm l-glutamine (BioWhittaker). Cells were passaged 1:8 by trypsinization (0.05% trypsin, 0.02% EDTA; BioWhittaker) and grown at 37°C in 5% CO2. Cell culture experiments, where -glucuronidase activity was necessary (X-gluc staining, crystal violet assays, see below), cells were cultured in DMEM pH 7. JEG-3 cells were transfected at 70% confluence in six-well plates using the calcium-phosphate method.28 One day after transfection cells were diluted 1:3, and the next day selection with 750 g/ml G418 (Life Technology, Eggenstein, Germany) was started. Growing clones were analyzed for -glucuronidase expression by immunofluorescence. Immunostaining Cells were fixed with 3.7% formaldehyde and permeabilized with 0.2% Triton. Indirect immunostaining was performed using a monoclonal antibody against human -glucuronidase (hybridoma supernatant 2118/157, obtained from HMR, Marburg, Germany, diluted 1/10) and a Cy3(red)- or DTAF(green)-labeled secondary antimouse antibody (Dianova, Hamburg, Germany). Between each step cells were washed with PBS two to three times. Immunoprecipitation JEG-3sG cells, grown on 6 cm dishes, were labeled for 3 h with 0.2 Ci/ml 35S-methionine (Amersham Pharmacia Biotech, Freiburg, Germany) in DMEM + 10% dialyzed FBS. Afterwards the supernatant was disposed and cells were further incubated with 2 ml DMEM + 10% FBS. At the indicated time-points supernatant was collected and cells were lysed in 700 l RIPA buffer (10 mm Tris pH 8, 150 mm NaCl, 0.25% SDS, 1% sodium deoxycholate, Gene Therapy 1% Nonidet P40, 1 mm DTT, protease inhibitors: 0.5 mm PMSF, 50 g/ml Aprotinin, 50 g/ml Leupeptin). For immunoprecipitation 1/3 of supernatant, buffered with RIPA, or 1/3 of cell lysate was incubated with the -glucuronidase antibody (100 l) for 1 h on ice. Immune complexes were precipitated with protein A sepharose (Amersham Pharmacia Biotech) and washed five times with RIPA buffer (without protease inhibitors). Proteins were separated by SDS-PAGE (10%). The dried gel was exposed for 3 days. Enzymatic activity X-gluc: Cells were fixed with 0.1% glutaraldehyde in PBS and stained for 6 h with 0.08% X-gluc (5-bromo-4-chloro3-indolyl--D-glucuronide-cyclohexylammonium salt; Boehringer Ingelheim, Ingelheim, Germany), 3 m potassium ferricyanide, and 3 m potassium ferrocyanide in 0.1 m acetate buffer pH 5. Afterwards the cells were fixed with 3.7% formaldehyde and mounted in moviol (Calbiochem, Bad Soden, Germany). pNPG: Substrate reaction buffer were prepared by mixing 65 mm KH2PO4 and 65 mm Na2HPO4 in different amounts to obtain buffers ranging from pH 5 to 8. One mm pNPG (p-nitrophenyl--D-glucuronide; Sigma, Deisenhofen, Germany) was dissolved freshly before the experiment. Supernatant of transfected cells was collected, centrifuged, mixed with equal amounts of substrate-buffer, and incubated for 12–24 h at 37°C. The reaction was stopped by adding 2.5 m 2-amino-2-methyl-1,3propandiol (Sigma) and the extinction was measured at a wavelength of 415 nm. Controls were incubated in the presence of the specific inhibitor D-saccharic-1,4-lactone (Sigma) in phosphate buffer pH 5. Prodrug conversion Two days after transfection cell were incubated with 10 m prodrug [N-(4--glucuronyl-3-nitrobenzyloxycarbonyl)doxorubicin] for 24 h (obtained from HMR). Expression of transfected -glucuronidase was detected by indirect immunofluorescence with a DTAF labeled secondary antibody (Dianova, Hamburg, Germany). Converted prodrug was seen as red autofluorescence of the nuclei, due to DNA-intercalating doxorubicin. Crystal violet assay One day before drug application cells were seeded in 96well flat-bottomed microtiter plates (10 000 cells per well). Cells were incubated for 24 h with doxorubicin (Sigma) or prodrug (HMR), applied as serial 1:2 dilutions (100 l per well) ranging from 500 m down to 238 pm. Three days after drug application cell viability was assessed by the crystal violet assay. After removal of medium attached cells were incubated with 100 l crystal violet solution (0.5% crystal violet, Sigma, in 20% v/v methanol-H2O) for 15 min at room temperature. Unbound crystal violet was removed and retained dye was measured with an ELISA-reader at 540 nm. In vivo studies Nude mice (strain CD-1 nu/nu; five to eight animals per group) were injected s.c. with 106 control JEG-3 or JEG3sG cells (in 300 l PBS) using a 21 G needle. After 1–2 weeks tumors were visible. At a size of 20–110 mm2 pro- Extracellular prodrug conversion D Weyel et al drug (100 mg/kg) or doxorubicin (1.5 mg/kg) was injected i.v., and tumor growth was measured regularly. Histology -Glucuronidase activity was determined on cryosections following the procedure described by Bosslet et al.29 Controls were incubated in the presence of 10 mm D-saccharic-1,4-lactone monohydrate (Sigma) in 0.1 m acetate buffer pH 5. Acknowledgements We are grateful to Klaus Bosslet for useful discussions, to Claudia Cybon and Tina Stroh for excellent technical assistance, to A Wellstein for the JEG-3 cell line and to M Krause for synthesis of oligonucleotides. References 1 Ford JM, Hait WN. Pharmacology of drugs that alter multidrug resistance in cancer. Pharmacol Rev 1990; 42: 155–199. 2 Culver KW et al. In vivo gene transfer with retroviral vectorproducer cells for treatment of experimental brain tumors (see comments). Science 1992; 256: 1550–1552. 3 Mullen CA, Kilstrup M, Blaese RM. Transfer of the bacterial gene for cytosine deaminase to mammalian cells confers lethal sensitivity to 5-fluorocytosine; a negative selections sytem. Proc Natl Acad Sci USA 1992; 89: 33–37. 4 Huber BE et al. In vivo antitumor activity of 5-fluorocytosine on human colorectal carcinoma cells genetically modified to express cytosine deaminase. Cancer Res 1993; 53: 4619–4626. 5 Bridgewater JA et al. Expression of the bacterial nitroreductase enzyme in mammalian cells renders them selectively sensitive to killing by the prodrug CB1954 (published erratum appears in Eur J Cancer 1996; 32A: 186). Eur J Cancer 1995; 31A: 2362–2370. 6 Marais R et al. A cell surface tethered enzyme improves efficiency in gene-directed enzyme prodrug therapy. Nature Biotech 1997; 15: 1373–1377. 7 Paigen K. Mammalian beta-glucuronidase: genetics, molecular biology, and cell biology. Prog Nucleic Acid Res Mol Biol 1989; 37: 155–205. 8 Rosenfeld MG et al. Biosynthesis of lysosomal hydrolases: their synthesis in bound polysomes and the role of co- and post-translational processing in determining their subcellular distribution. J Cell Biol 1982; 93: 135–143. 9 Jain S et al. Structure of human beta-glucuronidase reveals candidate lysosomal targeting and active-site motifs. Nat Struct Biol 1996; 3: 375–381. 10 Bosslet K, Czech J, Hoffmann D. A novel one-step tumor-selective prodrug activation system. Tumor Targetting 1995; 1: 45–50. 11 Barabas K, Sizensky JA, Faulk WP. Transferrin conjugates of adriamycin are cytotoxic without intercalating nuclear DNA. J Biol Chem 1992; 267: 9437–9442. 12 Fick J et al. The extent of heterocellular communication mediated by gap junctions is predictive of bystander tumor cytotoxicity in vitro. Proc Natl Acad Sci USA 1995; 92: 11071–11075. 13 Mesnil M et al. Bystander killing of cancer cells by herpes simplex virus thymidine kinase gene is mediated by connexins. Proc Natl Acad Sci USA 1996; 93: 1831–1835. 14 Freeman SM et al. The ‘bystander effect’: tumor regression when a fraction of the tumor mass is genetically modified. Cancer Res 1993; 53: 5274–52783. 15 Bridgewater JA et al. The bystander effect of the nitroreductase/CB1954 enzyme/prodrug system is due to a cellpermeable metabolite. Hum Gene Ther 1997; 8: 709–717. 16 Vaupel P, Kallinowski F, Okunieff P. Blood flow, oxygen and nutrient supply, and metabolic microenvironment of human tumors: a review. Cancer Res 1989; 49: 6449–6465. 17 Tannock IF, Rotin D. Acid pH in tumors and its potential for therapeutic exploitation. Cancer Res 1989; 49: 4373–4384. 18 Lougerstay-Madec R et al. Synthesis of self-immolative glucuronide-based prodrugs of a phenol mustard. Anticancer Drug Des 1998; 13: 995–1007. 19 Bakina E, Wu Z, Rosenblum M, Farquhar D. Intensely cytotoxic anthracycline prodrugs: glucuronides. J Med Chem 1997; 40: 4013–4018. 20 Abina MA et al. Lac Z gene transfer into tumor cells abrogates tumorigenicity and protects mice against the development of further tumors. Gene Therapy 1996; 3: 212–216. 21 Van Ginkel FW et al. Intratracheal gene delivery with adenoviral vector induces elevated systemic IgG and mucosal IgA antibodies to adenovirus and beta-galactosidase. Hum Gene Ther 1995; 6: 895–903. 22 Vile RG et al. Systemic gene therapy of murine melanoma using tissue specific expression of the HSVtk gene involves an immune component. Cancer Res 1994; 54: 6228–6234. 23 Melcher A et al. Tumor immunogenicity is determined by the mechanism of cell death via induction of heat shock protein expression. Nature Med 1998; 4: 581–587. 24 Barba D, Hardin J, Sadelain M, Gage FH. Development of antitumor immunity following thymidine kinase-mediated killing of experimental brain tumors. Proc Natl Acad Sci USA 1994; 91: 4348–4352. 25 Mullen CA, Coale MM, Lowe R, Blaese RM. Tumors expressing the cytosine deaminase suicide gene can be eliminated in vivo with 5-glurocytosine and induce protective immunity to wildtype tumor. Cancer Res 1994; 54: 1503–1506. 26 Oshima A et al. Cloning, sequencing, and expression of cDNA for human beta-glucuronidase. Proc Natl Acad Sci USA 1987; 84: 685–689. 27 Riechmann L, Clark M, Waldmann H, Winter G. Reshaping human antibodies for therapy. Nature 1988; 332: 323–327. 28 Graham FL et al. A new technique for the assay of infectivity of human adenovirus 5 DNA. Virology 1973; 52: 456–467. 29 Bosslet K, Czech J, Hoffmann D. Tumor-selective prodrug activation by fusion protein-mediated catalysis. Cancer Res 1994; 54: 2151–2159. 231 Gene Therapy