Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Hypothalamus wikipedia , lookup
Hormone replacement therapy (menopause) wikipedia , lookup
Androgen insensitivity syndrome wikipedia , lookup
Hormone replacement therapy (male-to-female) wikipedia , lookup
Hypothalamic–pituitary–adrenal axis wikipedia , lookup
Growth hormone therapy wikipedia , lookup
Hypopituitarism wikipedia , lookup
Hyperandrogenism wikipedia , lookup
Congenital adrenal hyperplasia due to 21-hydroxylase deficiency wikipedia , lookup
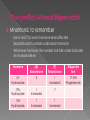
Marissa Hampton RN, BSN, SNNP University of Texas Medical Branch at Galveston School of Nursing NNP III/IV GNRS 5633/5434 Dr. Debra Armentrout PhD, RN, MSN, NNP-BC Dr. Leigh Ann Cates PhD, APRN, NNP-BC,RRT-NPS,CHSE Objectives › Define Congenital Adrenal Hyperplasia (CAH) › Define fetal pathophysiology › Describe clinical features of CAH › Discuss diagnostic evaluation of the neonate › Discuss therapeutic approach and treatment options for the infant with CAH Definition › A group of inherited disorders of the adrenal glands that cause improper production or synthesis of the hormones cortisol (glucocorticoid) and/or aldosterone (mineralcorticoid). Autosomal recessive disorder Cortisol helps the body respond to stress Infections Aldosterone helps body maintain normal electrolyte balance. Sodium and potassium regulation › In common forms of CAH, overproduction of androgens occur simultaneously. Leads to virilization (Merck Manuals, 2012) (Mayo Clinic, 2014) Definition › Common forms of CAH 90 % of CAH 21- Hydroxylase deficiency overall incidence is 1 in 16,000 Autosomal recessive Remaining 10 % of CAH: 17 Alpha- Hydroxylase deficiency 11 Beta- Hydroxylase deficiency 3 Beta- Hydroxylase deficiency 20, 22 Desmolase deficiency (Merck Manuals, 2012) (Mayo Clinic, 2014) Pathophysiology › Multiple steps in synthesis of cholesterol to cortisol › Defects in enzyme metabolism lead to impaired cortisol production Impaired cortisol production causes a secondary increase in Adrenocorticotropic hormone(ACTH) Elevated ACTH increases adrenocortical activity to normalize cortisol production. Impaired synthesis of adrenal hormones results from enzymatic blocks. Overproduction of steroids (Palmert& Dhams, 2013) Pathophysiology Simply: › Errors or deficiencies in metabolic pathways in the synthesis of cortisol from cholesterol cause: Increased hormone stimulation Improper hormone stimulation Mnemonic to remember › Use A and T for each hormone level affected › Separate each number under each hormone › Whichever hormone the number one falls under indicates an increased level Hormone (A) Aldosterone (T) Testosterone Diagnostic test 21Hydroxylase 2 1 increased 17-OH Progesterone 17AHydroxylase 1 Increased 7 11BHydroxylase 1 Increased 1 Increased Lets focus on the most common form of CAH 21- hydroxylase deficiency Pathophysiology Hormones: The adrenal glands (cortex) are responsible for the production of glucocorticoids, mineralcorticoids and androgens. › Cortisol: › Normal regulation of these hormones involves pituitary and adrenal glands. ACTH activates the adrenal glands to produce cortisol. ACTH and cortisol work to balance each other. Cortisol main function to maintain blood sugars, fluid and electrolyte status and protects the body from stress › Aldosterone: › Adrenal Androgens: Secreted under angiotensin (produced by renin in the kidneys) Regulates serum sodium, total body water and blood pressure in body. Secreted in both males and females prior to birth Production decreases after birth then resurges during puberty (Migeon & Wisniewski, 2014) Pathophysiology 21- Hydroxylase deficiency Caused by deletion or mutation in 21- hydroxylase gene (CYP21) › 2 Forms Classic Severe form Virilization form: partial enzyme deficiency Mild salt lost: Adrenal insufficiency does not occur Not life threatening Classic salt wasting: complete enzyme deficiency Life threatening form Mild form Presents later in life without salt wasting or virilization (Gomella, Eyal & Cunningham, 2013) (Thomas, 2014) Pathophysiology Simple virilizing CAH › Partial deficiency of 21- Hydroxylase enzyme Increased ACTH produced near normal levels of cortisol Increased production of 17- OHP as cortisol precursors Increased 17-OHP produces minor salt wasting Increased salt loss causes increased aldosterone levels Aldosterone levels able to compensate for salt loss Increased androgens lead to female virilization Ambiguous genitalia of female infants (Migeon & Wisniewski, 2014) Pathophysiology Salt wasting form of CAH › Total or near total deficiency of 21-Hydroxylase enzyme › Inability to produce cortisol and aldosterone Inability to produce cortisol May lead to hypoglycemia Increase in ACTH to regulate cortisol levels Build up of cortisol precursors due to improper metabolism 17-OHP Androgens › No aldosterone production Extreme serum sodium and water loss › Increased androgens lead to female virilization Ambiguous genitalia of female infants (Migeon & Wisniewski, 2014) Clinical Manifestations › Signs and symptoms of CAH have different clinical presentations Clinical presentation differs according to: Chromosomal sex of infant Form of CAH Classic form Salt wasting form Classic CAH (21-OH) › Females Severe form Excessive adrenal androgen secretion leads to prenatal virilization Ambiguous genitalia at birth Fusion of labioscrotal folds: Partial Phallic urethra Clitoromegaly Mild form Often identified later on in life due to subtle clinical manifestations Precocious pubic hair Clitoromegaly Accelerated growth and skeletal maturation Also present with similar findings with 3- beta hydro: oligomenorrhea, hirsutism, infertility (Antal, Z. & Zhou, P, 2009) Classic CAH (21-OH) › Males External genitalia are unaffected Subtle penile enlargement Without testicular enlargement Early overt signs of puberty: May occur as early as age 2-3 Axillary/pubic hair Acne Body odor Continued surge in androgens cause accelerated excessive growth Final adult heights are reached early Shorter then average male due to premature epiphyseal bone closure (Antal, Z. & Zhou, P, 2009) (Stresing, 2014) Salt wasting CAH (21-OH) › Both Male and Female Salt wasting due to inadequate aldosterone levels Can lead to: Poor feeding Vomiting and Dehydration Hyponatremia Hyperkalemia Hypovolemia Long term effects of salt wasting CAH Learning disabilities Behavioral problems Death (Androgen Excess Society, 2012) (Stresing, 2014) (Van der Kamp & Wit, 2004) Diagnostic evaluation › Assessment Identify female infants with external masculine features Identify males with impalpable gonads Infants who develop signs of adrenal insufficiency or severe crisis Family history of CAH Unexplained death in infancy (Palmert & Dahms, 2013) Diagnostic evaluation › Newborn screening Conducted within 24-48 hours of birth before potential salt wasting crisis However may have false positive result if sampled in first 36 hours after birth Identifies increased 17 OHP concentration in blood Term newborns must exceed a 99% range of 17 OHP for a positive screen Preterm infant levels of 17 OHP are scaled based on birth weight and gestational age. Higher incidence of false positive results (Van der Kamp & Wit, 2004) Diagnostic evaluation Positive newborn screen for CAH › For confirmation, send: 17 OHP Serum/urine electrolytes Serum: Hyponatremia, Hyperkalemia Urine: Hypernatremia Patients with mild salt wasting CAH may have normal electrolytes without hypovolemia! (Palmert & Dahms, 2013) Treatment Goals of treatment include normalizing ACTH secretion and providing replacement of hormone deficiencies › Replacement of cortisol, aldosterone and androgens › Each form has different treatment options. (Palmert & Dahms, 2013) Treatment Simple Virilization CAH › NOT salt wasting › Goal is to normalize ACTH Replacement with cortisol Hydrocortisone PO in 3 divided doses Dose usually 15-25 mg/m2 Increase dose with period of stress usually 2-3x maintenance dose for moderate to severe disease Simultaneous use of mineralcorticoid therapy may reduce amount of cortisol used. Adequate replacement is monitored by serum 17OHP level (Palmert & Dahms, 2013) Treatment Salt wasting CAH › Normalizing ACTH levels includes management of Sodium Replacement therapy in first years of life Renal tubules unable to conserve sodium Supplementation: 1-5 mEq/kg daily PO Fluid status Monitor for hypovolemia Replacement cortisol Hydrocortisone Mineralcorticoid therapy (Palmert & Dahms, 2013) Treatment Acute salt wasting crisis › Fluid replacement Give NS bolus at 20 ml/kg PRN › Sodium replacement › Continuous glucose infusion Dextrose 10% initially › Glucocorticoid therapy Hydrocortisone at 3-6 x maintenance level IV › Mineralcorticoid therapy Florinef Acetate 0.1-0.2 mg PO Q 12- 24 hours › Also manage: Acidosis Hypoglycemia Hyperkalemia (Palmert & Dahms, 2013) Conclusion › Impaired synthesis of adrenal hormones may lead to multiple forms of CAH, two of which include virilization and salt wasting crisis. Androgen Excess Society. (2012). Congenital adrenal hyperplasia: Definition, treatment, symptoms, diagnosis. Retrieved from http://www/ae-ociety.org/congenital_adrenal_hyperplasia Antal, Z. & Zhou, P. (2009). Congenital adrenal hyperplasia: Diagnosis, evaluation and management. Pediatrics in Review, 30 (7), e 49-57. doi: 10.1542/pir.30-7-e49 Gomella, T., Cunningham, M. D., & Eyal, F. G. (2013). Neonatology: Management, procedures, on-call problems, diseases and drugs (7th ed.). New York, NY: McGraw- Hill Education LLC. Mayo Clinic (2014). Congenital adrenal hyperplasia. Retrieved from http://www.mayoclinic.org/diseases-conditions/congenital-adrenalhyperplasia/basics/definition/con-20030910 Merck Manuals (2012). Congenital adrenal hyperplasia. Retrieved from http://www.merckmanuals.com/professional/pediatrics/endocrine_disorders_in_c hildren/congenital_adrenal_hyperplasia.html Migeon, C. J. & Wisniewski, A. B. (2014). Congenital adrenal hyperplasia due to 21hydroxylase deficiency: A guide for patients and their families. Retrieved from http://www.hopkinschildrens.org/cah/printable.html Palmert, M. R. & Dhams, W. T. (2013). Metabolic and endocrine disorders: Abnormalities of sexual differentiation. In R. J. Martin, A. A. Fanaroff, M.C. Walsh(Eds.), Neonatal- Perinatal Medicine: Diseases of the fetus and the infant (9th ed., pp.1577-1600. Philadelphia, PA: Mosby Elsevier. Stresing, D. (2014). Congenital adrenal hyperplasia. Retrieved from http://pediatrics.med.nyu.edu/endocrinology/patient-care/congenital-adrenalhyperplasia Thomas, T. A. (2014). Congenital adrenal hyperplasia. Retrieved from http://emedicine.medscape.com/article/919218-overview Van de Kamp, H. & Wit, J. M. (2004). Neonatal screening for congenital adrenal hyperplasia. European Journal of Endocrinology, 151, (pg71-75).