Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Water testing wikipedia , lookup
Thermodynamics wikipedia , lookup
History of molecular theory wikipedia , lookup
Physical organic chemistry wikipedia , lookup
Artificial photosynthesis wikipedia , lookup
Properties of water wikipedia , lookup
Freshwater environmental quality parameters wikipedia , lookup
Chemical thermodynamics wikipedia , lookup
Atomic theory wikipedia , lookup
Hypervalent molecule wikipedia , lookup
Chemical equilibrium wikipedia , lookup
Biochemistry wikipedia , lookup
Stability constants of complexes wikipedia , lookup
Water pollution wikipedia , lookup
Acid–base reaction wikipedia , lookup
Acid dissociation constant wikipedia , lookup
Water splitting wikipedia , lookup
Part I => CARBS and LIPIDS
§1.1 Thermodynamics
§1.1a General Principles
§1.1b Macromolecular Forces
§1.1c Acid-Base Equilibria
Section 1.1a:
General Principles
Synopsis 1.1a
- Thermodynamics is concerned with changes in heat and temperature as related to
energy exchange and work done by living systems—the major thermodynamic
parameters include changes in free energy (G), enthalpy (H), entropy (S), volume
(V), and heat capacity (Cp)
- The laws of thermodynamics provide general constraints that such systems must not
violate—of the four laws of thermodynamics (zeroth through third), the first and
second laws are of particular relevance to understanding living systems
- Natural processes are spontaneous (S > 0)—ie they are accompanied by an increase
in entropy (S) coupled with a decrease in free energy (G)—the energy available to do
useful work at a constant temperature, pressure, and pH
- Thermodynamic properties accompanying biochemical processes are usually quoted
under the so-called “standard state conditions” that are defined as:
Temperature (T) = 25°C (77°F / 298K)
Pressure (P) = 1 atm (105Pa)
pH = 7.0
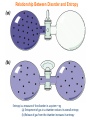
Relationship Between Disorder and Entropy
Entropy is a measure of the disorder in a system—eg
(a) Entrapment of gas in a chamber reduces its overall entropy
(b) Release of gas from the chamber increases its entropy
Energy Flow in the Biosphere—an Open System
- Living organisms are thermodynamically open systems that tend to maintain a “steady-state”
rather than reach equilibrium (G 0)—doing so would equate to death!
- “Steady-state” implies that the rates of synthesis and degradation of metabolic intermediates
within a cell are more or less equal such that their concentrations change little over time—eg
the mass of an organism generally remains more or less constant over time irrespective of how
much food and water are consumed!
Equilibrium vs Steady-State
Equilibrium (death)
- Consider the following reaction in progress:
A + B <=> C
- Let us assume that:
-d[A]/dt = rate of decay/breakdown of A (into C)
-d[C]/dt = rate of decay/breakdown of C (into A and B)
- At equilibrium, the forward reaction is exactly balanced by reverse reaction:
-d[A]/dt = -d[C]/dt
- Concentration of C stabilizes (reaches a constant) at equilibrium—the above reaction is @ equilibrium!
Steady-State (life)
- Consider the following reactions in progress:
A + B <=> C
C + D <=> E
- Let us assume that:
d[C1]/dt = rate of formation/synthesis of C (from A and B)
-d[C2]/dt = rate of decay/breakdown of C (into E)
- At steady-state, the rate of synthesis of C equals its rate of breakdown:
d[C1]/dt = -d[C2]/dt
- Concentration of C also stabilizes (reaches a constant) but under steady-state conditions—neither of
the above reactions is @ equilibrium!
Laws of Thermodynamics
First law of Thermodynamics
Energy is neither created nor destroyed but only
conserved/exchanged —it is mathematically
expressed as:
U = q – w
U = Change in internal energy (of the system)
q = Heat exchanged/added (eg to generate steam)
w = Work done (eg piston movement)
Second law of Thermodynamics
Natural processes are spontaneous (S > 0), leading
to an increase in disorder or entropy (S)—it is
mathematically expressed as:
Suni = (Ssys + Ssur) > 0
S is the change in entropy of the universe (uni),
system (sys), and surroundings (sur)
Steam Engine
Gibb’s Free Energy (G)
- Biological manifestation of the first and second laws of
thermodynamics is given by the Gibb’s equation:
G = H - TS
where G = Change in free energy (cal/mol)
H = Change in enthalpy (cal/mol)
S = Change in entropy (cal/mol/K)
T = Absolute temperature (K)
- The sign denotes standard conditions: 25C, 1 atm, and pH 7
Willard Gibbs
(1839-1903)
- Gibb’s equation provides a measure of the thermodynamic potential of a biological
process to do useful work
- Biological processes are overall accompanied by a decrease in free energy—ie G < 0
- To satisfy the above thermodynamic constraint, endergonic processes (G > 0) are
coupled to exergonic reactions (G < 0)
- Similarly, endothermic processes (H > 0) are driven by an increase in entropy (TS > 0)—
ie they are under entropic control
Relationship Between H, S and G
When G=0 => T=H/S
In thermodynamic terms, thermodynamically favorable reactions (G < 0) are
described as being under:
(1) Enthalpic control
=>
H < 0 and TS < 0
(2) Entropic control
=>
H > 0 and TS > 0
(3) Enthalpic and entropic control
=>
H < 0 and TS > 0
Equilibrium Thermodynamics
- Consider the following reaction with an equilibrium dissociation constant (Kd) of 10M (10x10-6M):
A + B <=> C
- Change in free energy of the reaction (G) under non-equilibrium setting is given by:
G = G + RTlnKa = G - RTlnKd
[1]
where
G = change in free energy of all species under standard state (cal/mol)—ie @ equilibrium
R = Universal molar gas constant (2 cal/mol/K)
T = Absolute temperature (K)
Ka = Equilibrium association constant (M-1)
Kd = Equilibrium dissociation constant (M)
- Kd is defined as:
Kd = [A][B]/[C] = 1/Ka
where letters A-C in [ ] indicate corresponding concentration of each species @ equilibrium
- But, forward reaction equals reverse @ equilibrium—ie G = 0
- Thus, Eq [1] can be rewritten @ equilibrium as:
G = -RTlnKa
[2]
=>
G = RTlnKd
[3]
=>
G = (2 cal/mol/K).(298K).ln(10x10-6M) = (596 cal/mol).ln(10-5) = -(596 cal/mol).ln(105)
=>
G = -6862 cal/mol = -7 kcal/mol
Exercise 1.1a
-
Summarize the relationship between energy (U), heat (q), and
work (w)
-
State the first and second laws of thermodynamics
-
Explain why changes in both enthalpy (ΔH) and entropy (ΔS)
determine the spontaneity of a process
-
What is the free energy change for a reaction at equilibrium?
-
Write the equation showing the relationship between ΔG° and Kd
-
Write the equation showing the relationship between ΔG, ΔG°,
and the concentrations of the reactants and products
-
Explain how biochemists define the standard state of a solute
Section 1.1b:
Macromolecular Forces
Synopsis 1.1b
- TWO major attractive forces acting on biological molecules include:
(1) ionic interactions
(2) van der Waals forces (eg hydrogen bonding, dipolar interactions)
- Being polar (ie electrostatically polarized), water molecules form hydrogen
bonds with other molecules
- In terms of attractive forces, water can exist either in a liquid or crystalline
(ice) form depending on the nature of hydrogen bonding interactions
- The exclusion of nonpolar groups from polar surroundings so as to maximize
the entropy of water molecules is the basis of “hydrophobic effect”
- Atomic distances are measured in the units of Ångström (Å)
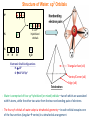
Structure of Water: van der Waals Envelope
Johannes van der Waals
(1837-1923)
- In chemical terms, water is dihydrogen monoxide (H2O)—wherein two hydrogen atoms are
covalently bonded to an oxygen atom
- While essential to life, dihydrogen monoxide is a lethal chemical (!) in that it can rapidly corrode and
destroy most materials!
- van der Waals envelope (or surface) is the approximate perimeter of a molecule as demarcated by
the outer boundary of the surrounding cloud of electrons—the distance from the center of the
molecule to the van der Waals envelope is called the “van der Waals radius”
- van der Waals radius (r) of water is 1.4Å—two water molecules cannot get closer to each other
more than 2r—ie the distance from the center of one molecule to the center of the other!
Structure of Water: sp3 Orbitals
2P
x y z
4 x SP3
hybridized
orbitals
2S
1S
H
H
O
H2O
Electronic Shell Configurations
H 1S1
O 1S2.2S22p4
Triangular face (x4)
Vertex/Corner (x4)
Edge (x6)
Tetrahedron
- Water is comprised of four sp3 hybridized (or mixed) orbitals—two of which are associated
with H atoms, while the other two arise from the two non-bonding pairs of electrons
- The four sp3 orbitals of water adopt a tetrahedral geometry—ie each orbital occupies one
of the four vertices (singular vertex) in a tetrahedral arrangement
Structure of Water: Hydrogen Bonding
- Electrons involved in mediating covalent bonds
between a pair of atoms are not equally distributed
but rather become slightly polarized toward one or
the other bonding partner (depending on their
+
relative electronegativity), thereby resulting in the
formation of diploes (or charge separation)
-
+
-
- Under such polarization in the context of a covalent
bond between a pair of atoms, one atom carries a slightly negative charge (-) while
the other a slightly positive charge (+)—interactions between such oppositely charged
ends of dipoles are broadly termed “van der Waals” forces—an umbrella term!
+
+
- If such dipole-dipole interactions occur between an electropositive H atom (bonded to an highly
electronegative atom such as O or N) and another highly electronegative atom, the resulting van der
Waals forces are called “hydrogen bonding”—ie hydrogen bonding (or H-bonding) is a special case of
van der Waals forces due to its rather strong nature coupled with its ubiquity in biological systems
- Hydrogen bonding—represented by a dotted or dashed line—is the supreme attractive force that
renders water a liquid at room temperature
- Changes in hydrogen bonding pattern impart upon water the ability to exist in a liquid or crystalline
(ice) form
- Because of charge separation or polarization of electronic clouds of H and O atoms, water is
described as being a highly “polar” molecule—such polarity of water enables it to act both as Hbond donor as well as an H-bond acceptor in biochemical processes
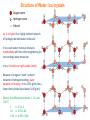
Structure of Water: Ice crystals
Oxygen atom
Hydrogen atom
----- H-bond
- Ice is a crystal of an highly ordered network
of hydrogen-bonded water molecules
- In ice, each water molecule interacts
tetrahedrally with four other neighboring (or
surrounding) water molecules
- In ice, H-bonds are highly stable (static)
- Because of a regular “open” ordered
network of hydrogen bonding, water
expands on freezing—ie ice (0.92 g/ml) has a
lower density than liquid water (1.00 g/ml)
- What is the difference between 1, 1.0, and
1.00?!
1
=> 0.5-1.4
1.0 => 0.95-1.04
1.00 => 0.995-1.004
Structure of Water: Liquid
3-mer
4-mer
5-mer
- Liquid water consists of a rather “loose” network of hydrogen-bonded water molecules—ie
water molecules rapidly fluctuate and tumble on a picosecond (ps) timescale (1ps = 10-12s)
- Unlike ice, liquid water thus harbors an highly disordered and irregular structure
- In liquid water, H-bonds are highly unstable (dynamic)
- Nevertheless, water molecules transiently engage in rings of three (3-mer), four (4-mer), or
five (5-mer) molecules in liquid
- Because of their irregularity in liquid, water molecules can pack together much more tightly
than in ice, thereby rendering water (1.00 g/ml) more dense than ice (0.92 g/ml)—cf highlyordered rows of people (ice) versus a random crowd (water)
Typical Bond Energies in Biomolecules
- Non-covalent forces underlying intermolecular interactions between biological molecules
(or biomolecules) can be divided into TWO major categories:
(1) Ionic interactions —eg between oppositely charged ions such as Na+ and Cl(2) van der Waals forces —interactions due to dipoles
- Hydrogen bonding is a type of van der Waals interaction—albeit of a major significance
- The term “electrostatic interactions” is ambiguous and must be avoided at all costs
Van der Waals Forces: Dipole-Dipole Interactions
Van der Waals forces are dipole-dipole
interactions that can be divided into three
major categories:
polar
polar
(a) Dipole-dipole interactions—interactions
between permanent dipoles such as a C=O group (H-bonding is a special case
of such dipolar interactions)
(b) Dipole-induced-dipole interactions—
permanent dipoles in groups such as –
C=O can also induce a dipole moment in
a neighboring group (eg -CH3) by virtue
of their ability to distort the distribution
of its electronic cloud
(c) London dispersion forces—these arise
from the fact that the electronic cloud of
nonpolar groups such as –CH3 is not
“static” but rather experiences rapidly
fluctuating motions and, in so doing,
generates a small transient dipole
polar
nonpolar
nonpolar
nonpolar
Hydration: Solvation of Ionic Substances
Cations are shielded by
electronegative O atoms
Anions are shielded by
electropositive H atoms
- Water is often described as a “universal solvent” due to the fact that its polar character
renders it an excellent solvent for hydrophilic substances—eg those with polar or ionic
character
- The ability of water to dissolve polar substances—such as NaCl—arises from the fact that
its dipolar character enables it to weaken attractive forces between oppositely charged
Na+ and Cl- ions
- Multiple water molecules can surround each ion and neutralize its charge in a
phenomenon referred to as “hydration”—or solvation in generic terms
Hydration: Solvation of Polar Substances
Hydroxyl
Keto
Carboxylate
Amino
- Water is also an excellent solvent for
polar substances for the same reason
that it is for ionic substances
- The dipolar character of water enables
it to engage in H-bonding with other
polar groups such as hydroxyl (OH),
carboxylate (O=CO-), keto (C=O) and
amino (NH3+)
Hydrophobic Effect: An Entropic Phenomenon
- Apolar (or nonpolar) molecules such as oils and lipids aggregate when in contact with
water—ie they tend to “stick” together rather than dissolve in water—why?!
- Such ability of apolar molecules to minimize contact with water or vice versa is termed
the “hydrophobic effect”—what thermodynamic force drives the hydrophobic effect?
Enthalpic (H) or entropic (TS)?
- H accompanying the transfer of apolar substances from water to an apolar solvent is
unfavorable (H >= 0), while TS is consistently favorable (TS > 0)
- Thus, the hydrophobic effect is largely driven by an entropic force in that the ability of
apolar substances to aggregate confers upon surrounding water molecules an entropic
advantage—ie exclusion of water molecules enables them to move and tumble freely in
lieu of being “locked” or “entrapped” in an ordered manner with apolar neighbors
Hydrophobic Effect: Orientation of Water Molecules
- Maintenance of intramolecular
H-bonding network is critical to the
random motion of water molecules
- Intrusion of apolar solute into water
disrupts such extensive network due
to its inability to engage in H-bonding
interactions
- Accordingly, water molecules orient
away from the surface of the apolar
solute to engage in H-bonding network
with bulk water molecules—the surrounding
water molecules that are not in direct contact
with the solute
- Such orientation constitutes an ordering of the water structure (as their degree of
freedom or the number of ways in which they can hydrogen bond becomes restricted)
- In order to minimize such ordering of water molecules, apolar molecules aggregate so as
to minimize their surface area in contact with water and thereby maximize the overall
entropy of the system
Hydrophobic Effect: Aggregation of Apolar Substances
Individual hydration of
apolar substances increases their surface
area in contact with water—thereby
resulting in greater loss of entropy
Aggregation of apolar substances
minimizes their surface area in
contact with water—thereby
resulting in lesser loss of entropy
Hydrophobic effect is central to many (bio)physicochemical phenomena:
- Separation of oil and water
- Membrane bilayer integrity
- Folding of proteins in water and lipid bilayer
Exercise 1.1b
- Sketch a diagram of a water molecule and indicate the ends that
bear partial positive and negative charges
- Compare the structures of ice and water with respect to the
number and geometry of hydrogen bonds
- Describe the nature and relative strength of covalent bonds,
ionic interactions, and van der Waals forces
- Explain why polar substances dissolve in water while nonpolar
substances do not
- What is the role of entropy in the hydrophobic effect?
Section 1.1c:
Acid-Base Equilibria
Synopsis 1.1c
- The acidity of a solution is expressed in terms of a pH value
- An acid is a compound that can donate a proton
- A base is a compound that can accept a proton
- The Henderson–Hasselbalch equation relates the pH of a
solution of a weak acid to the pK and the concentrations of
the acid and its conjugate base
- Biological molecules that harbor ionizable groups are
sensitive to changes in pH
Proton Jumping Occurs Rapidly
- Water is a neutral molecule with a high
tendency to ionize into corresponding
hydrogen (H+) and hydroxide (OH-) ions:
HOH <=> H+ + OH- The hydrogen (H+) ion is more commonly referred to
as a “proton”
- In essence, the proton (H+) largely exists
as an hydronium ion (H3O+) in solution—ie the
H+ is associated with another H2O molecule rather
than roaming around as a free agent!
- The H+ of H3O+ ion is not static but highly dynamic in that
it can jump from one H2O molecule to another and so
on virtually in an infinite (or endless) manner in a
phenomenon referred to as “proton jumping”
- Owing to proton jumping, H+ and OH- ions exhibit much higher mobilities in bulk water
compared to other ions—accordingly, acid-base reactions (involving exchange of H+) rank
among the fastest processes occurring in water
- For simplicity, the H+ is often considered as one of the two dissociative products of ionization of
water in lieu of the more complex H3O+ ion
pH Values: Measure of the acidity of a solution
- Consider the ionization of water:
HOH <=> H+ + OH-
- In pure water @ 25C, the concentration of H+ is close to 10-7M (100nM):
[H+] = [OH-] = 10-7M = 100nM
[H+] + [OH-] = 2x10-7M = 200nM
- A solution is described as:
Neutral
Acidic
Alkaline/Basic
=>
=>
=>
if [H+] = 10-7M
if [H+] > 10-7M
if [H+] < 10-7M
=> 1M = 1mol/L
Soren Sorensen
(1868-1939)
- The measure of acidity (or alkalinity/basicity) of an aqueous solution is defined by the concentration of
H+ ions expressed in terms of a quantity known as “pH”
- First introduced by Sorensen in 1909, the pH of a solution is defined as:
pH = -log[H+]
where [H+] must be in the units of molar (M)—ie moles per liter (or litre in Imperial English)
- Thus, the pH is the negative log (to base 10) of the concentration of H+ ions in solution
- The pH of pure water is:
pH = -log[10-7M] = 7
pH Values: Relationship of pH, [H+], and [OH-]
pH Values: pH of Common Substances
pKa Values: Measure of the strength of an acid
- Consider the dissociation of an acid HA into its constituent components in hydrogen ion
(H+) and the conjugate base (A-):
HA <=> H+ + A-
- The equilibrium dissociation constant of the acid (Ka) is defined as:
Ka = [H+][A-]/[HA]
[1]
- The strength an acid in aqueous solution is expressed in terms of a quantity called the
“pKa“, which is analogous to pH!
- pKa is defined as the negative log (to the base 10) of Ka:
pKa = -logKa
where Ka must be in the units of molar (M)
[2]
- The relationship between the pH of a solution and pKa of an acid can be derived as follows:
(1) Rearrange Eq [1] for [H+]:
[H+] = Ka[HA]/[A-]
(2) Take negative logarithm of each term: -log[H+] = -logKa - log{[HA]/[A-]}
(3) Substitute the quantities and rearrange:
pH = pKa + log{[A-]/[HA]}
[3]
- Eq[3] has come to be known as the “Henderson-Hasselbalch equation”
pKa Values: Ka and pKa of Common Acids @ 25C
Ka
pKa
pKa Values: Henderson-Hasselbalch Equation
pH = pKa + log{[A-]/[HA]}
- When [HA] = [A-], log{[A-]/[HA]} = 0
=>
pH = pKa
- In other words, the pH of a solution is equivalent to pKa of an acid @
50% dissociation
- Henderson-Hasselbalch equation does not take into account
ionization of water
Lawrence Henderson
(1878-1942)
- Thus, it is only useful for rationalizing the ionization of weak
acids/bases such as buffers and amino acid sidechain groups in
proteins
- A buffer with a pKa value of 8 would be most effective @ pH 8! Why?
- Since buffers resist changes in pH, they are most effective when the
concentration of its hydrogen ion (H+) and conjugate base (A-) are
equal—and when they are so, pH = pKa
Karl Hasselbalch
(1874-1962)
pKa Values: Titration Curves of Weak Acids
- A buffer is a mixture of a weak acid (HA)
and its conjugate base (A-):
HA <=> H+ + A- Thus, a buffer helps to maintain constant
pH. How?
- Addition of small amounts of OH- or H+ are
quickly mopped up with little changes in
solution pH:
OH- + H+ <=> H2O
H+ + A- <=> HA
- A buffer ONLY resists small changes in pH in
the region close to its pKa value:
pH = pKa 1
Consider acetate buffer (pKa ~ 5):
(1) When pH << 4, acetate largely exists as
HA (undissociated acid form) => thus no
good as a buffer!
(2) When pH >> 6, acetate largely exists as A(dissociated base form) => again no good
as a buffer!
pKa Values: Titration of a Polyprotic Acid
pK3
pK2
pK1
- Acids such as phosphoric acid (H3PO4) are called “polyprotic acids” due to the fact that they
can lose more than one proton upon successive ionizations
- Thus, a polyprotic acid has multiple pKa values—eg H3PO4 has three pKa values:
pK1 = 2.2
pK2 = 6.8
pK3 = 12.2
- Thus, phosphate buffer can be used to resist small changes in pH around pH 2.2, 6.8 and 12.2
Exercise 1.1c
- What are the products of ionization of water? How are their
concentrations related?
- Describe how to calculate pH from the concentration of H+ or OH- Define acid and base
- What is the relationship between the strength of an acid and its
pKa value?
- What must a buffer solution include in order to resist changes in
pH on addition of acid or base?
- Why is it important to maintain biological molecules in a buffered
solution?
Debunking Alkaline Diet—an Alternative Fact
Rather Than Alternative Medicine!
- Alkaline diet postulates that the consumption of certain fruits and vegetables helps to alkalinize the body
fluids to a pH of around 7.4—a pH that cellular homeostasis achieves regardless of alkaline diet!
- Thus, alkaline diet is believed to offset acidosis resulting from a typical Western diet, thereby keeping
diseases such as cancer at bay—indeed, many cancers cells thrive under acidic conditions!
- But, the enigma is that the proponents of alkaline diet cannot explain how it helps to alkalinize the body
fluids—do all foods not go through the stomach?!
- If so, then all foods are churned in the stomach @ a pH of around 2! Does it really matter what the
acidity/alkalinity of the intake food is? It represents a small drop in a big ocean—does it not?
- What about thousands who swear to have benefited from having been on the super-scampensive alkaline
diet?—nothing but a coincidence and a placebo effect—correlation does not imply causation!