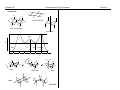
Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Homoaromaticity wikipedia , lookup
Ring-closing metathesis wikipedia , lookup
Aromaticity wikipedia , lookup
Marcus theory wikipedia , lookup
Organosulfur compounds wikipedia , lookup
Woodward–Hoffmann rules wikipedia , lookup
Ene reaction wikipedia , lookup
Petasis reaction wikipedia , lookup
Asymmetric induction wikipedia , lookup
Hydroformylation wikipedia , lookup
Strychnine total synthesis wikipedia , lookup
Chapter 1-1 What is Organic Chemistry? Organic Chemistry What is it? Why is it a two semester course? Why is it important to careers in health care? Organic chemistry is essential to the understanding of the intricate details of life The interactions and reactions of organic molecules are what define living systems. One needs to understand bonding, structure, properties, reactions, and synthesis to understand natural systems. CHAPTER 1 INTRODUCTION Organic Chemistry: the study of carbon compounds... organic compounds contain the elements C, H, N, O, S, Cl, Br, etc. Organic compounds were originally thought to come only from living organisms...thus the term organic (until about 1830) Inorganic compounds were all those which came from non-living sources Scientists thought a vital force was necessary to produce organic compounds and that they could not be synthesized in the lab. In 1828 Fredreich Woeller synthesized urea, an organic compound excreted as waste, from ammonium cyanate, and vitalism slowly died out. Organic compounds range from methane , CH4 (natural gas) to DNA, the genetic coding material, and taxol, a plant derived substance which is a potential anticancer agent. Organic chemistry is fundamental to many scientific disciplines...biochemistry, polymer chemistry, microbiology, botany, pharmacy, medicine...since living systems are composed primarily of organic compounds and water. Chem 61 Electronic Structure of Carbon Chapter 1-2 STRUCTURAL THEORY AND BONDING Carbon is intermediate in electronegativity, therefore it neither completely donates or completely accepts electrons. As a result it forms covalent bonds to itself and other atoms. It can bond to itself to form chains and rings...catenation. This allows the formation of a staggering number of organic compounds. 95% of all known compounds are organic. ATOMIC STRUCTURE It is important to understand molecular structure to understand reactivity of organic compounds. Molecular structure depends on atomic structure. Atomic structure of carbon C: 1s22s22p2 that is, carbon has 2 electrons in the lowest energy level, the 1s orbital 2 electrons in the next energy level, the 2s orbital 2 electrons in the third energy level, the 2p orbital 1s, 2s, 2p orbitals 1s is spherical with the same phase throughout 1s 2s is spherical with a node node is where ψ2 = 0 node 2s 2p... three orbitals of equal energy (degenerate) 2px 2py 2pz Chem 61 Chapter 1-3 Rules for Electronic Configuration Pauli Exclusion Principle: Maximum of two electrons per orbital. Electrons have a spin of +1/2 or -1/2 which gives rise to a small magnetic field since electrons are charged. Repulsion between the electrons is reduced if they have opposite spins and thus opposite magnetic moments. If two electrons occupy the same orbital they must have opposite spins or be paired. Aufbau Principle: orbitals are filled from lower to higher energy 4s 3p 3s 2p 2s 1s __ __ __ __ __ __ __ __ order of filling of atomic orbitals __ __ THUS: Li 1s22s1 Hund's Rule: orbitals of equal energy (degenerate) receive one electron until there is one electron in each orbital: then pairing of electrons begins. THUS: C 1s22s22px12py1 Chem 61 Chapter 1-4 Atomic Radius and Electronegativity ATOMIC RADIUS Atomic Radius: distance between the nucleus and the outermost electrons (valence electrons) or one half the bondlength of a diatomic molecule H—H bond length is 0.74Å, atomic radius is 0.37Å Atomic radius increases with increasing number of electron shells within an atom and decreases with the increase in the number of protons within an atom Thus atomic radius decreases from left to right within the same row of the periodic table (increasing number of protons in the nucleus) and increases from top to bottom within a group of the periodic table (increasing number of electronic shells) ELECTRONEGATIVITY Electronegativity: a measure of an atom's attraction for outer bonding electrons Electronegativity increases with increasing charge on the nucleus and with decreasing distance between the nucleus and the electrons Thus electronegativity increases from left to right within a row of the periodic table and from bottom to top within a group in the periodic table Chem 61 Chemical Bonding Chapter 1- 5 CHEMICAL BONDING G.N. Lewis put forth the first explanation of the nature of the chemical bond. Ionic Bond..one or more electrons is completely transferred from one atom to another creating ions: a cation (positively charged) and an anion (negatively charged). the ions are held together by electrostatic attractions ionic bonds generally occur between atoms of highly different electronegativies. e.g. Nao – e– Na+ + e– Clo + e– Cl – Nao + Clo Na+Cl – Covalent Bond...one or more electrons is shared by atoms...atomic orbitals merge into shared or molecular orbitals covalent bonds are usually formed between atoms of similar electronegativities since carbon is intermediate in electronegativity it usually forms covalent bonds Generally...atoms not having the noble gas configuration tend to form bonds such that each atom may obtain the stable noble gas electronic configuration [OCTET RULE] 4H + :C: H H C H H H C::O H single bond double bond H:C:::C:H triple bond Atoms of the elements of organic compounds can form a fixed number of bonds. .. O .. N .. C H Chem 61 Chemical Bonding Chapter 1- 6 COVALENT BONDS Bond lengths: the distance between two covalently bonded nuclei Bond angle: the angle formed between two covalent bonds Bond dissociation energy: the energy required for homolytic cleavage of a bond; Cl Cl H 3C H . Cl + Cl . . H 3C + H . two radicals Heterolytic cleavage: cleavage of a bond to give a cation and an anion; one atom which is part of the covalent bond retains both electrons from the bond Cl+ + Cl – Cl Cl H 3C H H 3C + cation + H– anion ∆H (change in enthalpy) for homolytic cleavage of many covalent bonds has been determined H3C—H H—H Br—Br H—OH 104 kcal/mole 104 kcal/mole 46 kcal/mole 110 kcal/mole CH3—CH3 CH2=CH2 HC≡CH 88 kcal/mole 163 ckal/mole 230 kcal/mole Chem 61 Chapter 1-7 Polar Covalent Bonds POLAR COVALENT BONDS covalent bonds between atoms of similar electronegativities are said to be nonpolar bonds since each atom shares the electrons of the bond approximately equally H—H CH3—CH3 if atoms of different electronegativities form a covalent bond, the more electronegative atom will have a stronger attraction for the electrons and polarize the bond giving a polar covalent bond in which one atom is partially negatively (δ–)charged and one atom is partially positively charged (δ+) δ+ δ− H3C—Cl CH3—NH2 δ+ δ− δ+ δ− H—Cl δ+ δ− H2C=O δ+ δ− H3C—OH If any entire molecule has an overall dipole, then the molecule is said to be polar. CCl4...nonpolar, polar covalent bonds cancel each other because of tetrahedral symmetry HCCl3...polar H2O, NH3...very polar Cl _ C Cl δ_ Cl Cl Cl dipoles cancel δ Cl δ_ + C δ H δ_ Cl δ_ δ+ dipoles add δ_ O δ+ H H δ+ dipoles add δ+ Chem 61 Chapter 1-8 Polar Covalent Bonds ATTRACTIONS BETWEEN MOLECULES ion-ion forces: the attraction between oppositely charged ions and repulsion between like charges very strong...not often encountered in organic compounds van der Waals forces: all dipole-dipole forces are both repulsive and attractive... the distance at which the repulsive forces are minimized and the attractive forces are maximized is called the van der Waals radius Induced dipole-dipole interactions or London forces: small temporary dipoles occur and induce dipoles in another molecule due to small uneven distribution of electron density. Dipole-dipole interactions: permanent dipoles in molecules attract or repel Hydrogen bonding: a specific type of dipole-dipole interaction; very strong occur only between a hydrogen atom bonded to an electronegative atom O, N, S and a lone pair on O, N, S O H :N 7 kcal /mole N H :N 3 kcal/mole N H :O 2 kcal/mole O H :O 5 kcal/mole Intermolecular hydrogen bonds increase the boiling points of compounds and increase their solubility in water. Hydrogen bonds also can influence the shapes of biomolecules by internal hydrogen bonding as well as hydrogen bonding between molecules. Chem 61 Formulas and Formal Charge Chapter 1-9 CHEMICAL FORMULAS empirical formula: gives the types and ratios of atoms in a molecule molecular formula: gives the type and actual number of atoms structural formula: gives the type, number and attachment of atoms...the actual structure for example: hexane: C6H14 CH3CH2CH2CH2CH2CH3 molecular formula C3H7 empirical formula structural formula LEWIS STRUCTURES 1. draw the molecular skeleton 2. count the number of available valence electrons (be sure to account for any overall charge on the species) 3. draw covalent bonds between all the atoms giving as many as possible an octet (duet for hydrogen) 4. assign charges in the molecule FORMAL CHARGE formal charge on an atom = # valence electrons - # shared pairs of 3 electrons - # unshared electrons :O: .. .. .. 1 N:O:H HNO3 Lewis structure: .. .. :O: .. 2 N...5-4-0 = +1 O1...6-2-4 = 0 O2...6-1-6 = -1 .. O3...6-2-4 = 0 :O: .. .. .. H...1-1 = O H SO Lewis Structure -2 2 4 SO4 :O:S:O: .. .. .. :O: .. O...6-1-6 = -1 S...6-4-0 = +2 overall charge (+2) + 4(-1) = -2 Chem 61 Formulas and Formal Charge Chapter 1-10 Structural Formulas Lewis Structures: as described above using dots for electrons Line-bond formulas: a line is used to represent two electrons forming a bond (a shared pair) Condensed formulas: bonds are not always shown and atoms of the same type bonded to another atom are grouped together H .. .. .. .. H.. . .C::O . . H C O H line-bond H C C H line-bond H H C C H H line-bond .. .. H2C=O Lewis condensed H:C:::C:H Lewis HC≡CH condensed ..H H.. .C::C . .. H H Lewis CH2=CH2 condensed Polygon formulas: polygon formulas are often used to represent cyclic compounds for simplicity; each carbon is assumed to have enough additional hydrogens to give each carbon four bonds cyclohexane cyclopropane H H H H C C H H C H C C H C HH H H cyclobutane benzene cyclopentane decalin Chem 61 Chapter 1-11 Acids and Bases ACIDS AND BASES Bronsted-Lowry: acid: a proton donor base: a proton acceptor Strong acid: completely ionized or dissociated in water e.g., HCl, H2SO4, HNO3, HBr Weak acid: only partially dissociated in water: carboxylic acids are weak acid CH3CO2H acid + CH3CO2- H2O + conjugate base base H3O+ conjugate acid amines are weak bases CH3NH2 base + HO – CH3NH3+ + H2O acid conjugate acid conjugate base Generally: strong acids have weak conjugate bases and weak acids have strong conjugate bases that is, as acid strength increases, the basicity of the conjugate base decreases Thus the ability of the conjugate base to stabilize a negative charge determines the strength of an acid Conjugate Acids H 2O pKa HCN 15.75 CH3CO2H 6.37 H3PO4 HCl 2.12 -7 4.75 increasing acid strength Conjugate Bases HO – NC – CH3CO2– H2PO4 – C l – decreasing base strength Chem 61 Chapter 1-12 Acids and Bases Factors affecting Acidity: the electronegativity and the size of the atom which carries the negative charge influence its ability to stabilize the negative charge size of the atom H—F H—Cl pKa 3.45 -7 H—Br -9 H—I -9.5 increasing size of halogen, increasing acid strength electronegativity pKa (CH3)3C—H 50 (CH3)2N—H 35 CH3O—H 15.5 F—H 3.45 increasing electronegativity of atom, increasing acid strength ACIDITY CONSTANTS, Ka's for acetic acid CH3CO2H + H2O Ka = CH3CO2– + H3O+ [CH3CO2–] [H+] [CH3CO2H] since stronger acids are more ionized, the larger Ka, the stronger the acid pKa = -logKa, the lower the pKa, the stronger the acid also, the higher the pKa, the weaker the acid or the stronger the base the stronger the acid, the more stable the anion produced by ionization of the acid. Chem 61 Chapter 1-13 Lewis Acids and Bases LEWIS ACIDS AND BASES Lewis acid: electron pair acceptor: any species with an electron deficient atom BBr3, AlCl3, H3C+ Lewis base: electron pair donor; any species with an unshared pair of electrons .. .. H3N:, CH3CH2..OH, H2C=O: Chem 61 Quantum Mechanics Chapter 1-14 QUANTUM MECHANICS...Molecular Orbitals In 1923 Louis De Broglie postulated that electrons have properties of three dimensional waves Later a wave equation was developed. Solutions (Ψ) to this wave equation give the various electronic states known as atomic orbitals. Ψ = probability of finding an electron in a certain space = electron 2 probability density plots of Ψ give the familiar s,p,d...orbitals 2 WAVE PROPERTIES The amplitude of a wave may be above the resting state (positive) or below (negative)...no charge implied A node is a point at which the amplitude is zero Waves reinforce creating a wave of higher amplitude if they they are in phase. + – + + – – Waves interfere if they are out of phase and create a wave which is of lower amplitude. Complete interference results in the cancelling of one wave by another. + + – – + – Chem 61 Quantum Mechanics Chapter 1-15 1s, 2s, 2p orbitals 1s is spherical with the same phase throughout + 2s is spherical with a node node is where ψ2 = 0 node + - 2p... three orbitals of equal energy (degenerate) node 2px 2py 4s __ 3p __ __ __ 3s __ 2p __ __ __ order of filling of atomic orbitals 2s __ 1s __ 2pz Chem 61 Chapter 1-16 Molecular Orbitals MOLECULAR ORBITALS Molecular orbitals = Linear combination of atomic orbitals 2 Atomic Orbitals must produce 2 Molecular Orbitals (the number of molecular orbitals equals the number of atomic orbitals which were combined to form them) the hydrogen molecule σ* >∆E H1s antibonding Ψ1 − Ψ2 H2 . . H1s ∆E σ Ψ1 bonding Ψ2 .. Ψ1 + Ψ2 the energy of the hydrogen molecule with two electrons in the sigma orbital is 104 kcal/mole more stable than the separate hydrogen atoms; ∆E = 52 kcal/mole if one electron is in the sigma and one in the sigma*, the molecule is of higher energy than the two separate atoms because the s* is slightly >∆E higher than the s orbitals while the s is ∆E lower Bonding orbital...high electron density between nuclei Antibonding orbital..node between nuclei (zero electron density) σ bond is cylindrically symmetrical Aufbau principle...fill lowest energy orbitals first Hund's Rule...place one electron in each degenerate orbital first, then pair up Pauli Exclusion Principle...two electrons in the same orbital must have opposite spins Chem 61 Chapter 1-17 Molecular Orbitals of Carbon MOLECULAR ORBITALS ON CARBON Overlap between atomic orbitals in complex molecules often results in electron repulsions which destabilize the molecule. Hybrid orbitals allow for better overlap and a more accurate prediction of molecular structure. Methane CH4 has a central carbon with four equivalent bonds to hydrogen bond angles = 109.5° bond lengths = 1.09Å sp3 Hybridization carbon has electronic configuration 1s22s22p2 2p 2p 96 kcal 2s 1s hybridize 2s sp3 1s 1s + 2s + - + 3-3p's 109.5° apart 4-sp3's sp3 orbitals point toward the corners of a tetrahedron, 109.5° apart any carbon bonded to four other atoms is sp3 hybridized, e.g. CH4, H3CCH3, C—C bond length = 1.54Å C-H σ bonds require 104 kcal/mol to be broken H H C methane H H bond angles = 109.5°; tetrahedral Chem 61 sp2 Molecular Orbitals of Carbon Chapter 1-18 sp2 Hybridization sp2 hybridized carbons are trigonal planar with atoms 120° apart 2p 2p 2p 96 kcal 2s sp2 hybridize 2s 1s 1s 1s ethylene: trigonal planar H C H C H H 2p H H C C H H H C H 120° H H H pi orbital H sigma orbitalH sp2 H H C H C C C H H H H pi* orbital H C H sigma* orbital bond angles = 120°; C=C bond length = 1.34Å pi (π) bonds are formed by the side by side overlap of two p-orbitals (approx. 68 kcal/mol) pi bonds are above and below the plane where the sigma bond is located pi bonds make the molecule rigid between the two atoms preventing rotation E sigma σ* pi π* pi π sigma σ ethylene C=C double bond: one sigma, one pi Chem 61 sp2 Molecular Orbitals of Carbon Chapter 1-19 sp Hybridization sp hybridized carbons are linear with atoms 180° apart 2p 2p 2p 96 kcal 2s 1s sp hybridize 2s 1s 1s ethylene: trigonal planar + 180° apart; the remaining two 2p orbitals are 90° to the sp and each other + 1-2p 2s 2-sp's Acetylene: linear; bond angles 180° C=C bond length = 1.20Å acetylene has two perpendicular pi bonds and one sigma bond H H H 2-sp's H C H acetylene σ-orbital H 2-2p's on each carbon combine to form pi-orbitals E C C—C σ* C—C π* C—C π C—C σ H C C H acetylene π-orbitals Chem 61 Functional Groups Chapter 1-20 FUNCTIONAL GROUPs CH3CH3 Oxygen: sp3 CH2 CH2 HC CH alkanes alkenes alkynes CH3 OH CH3 OCH3 CH3 NH2 CH3 Br alcohols ethers amines alkyl halides O CH3 C H O CH3 C CH3 O CH3 C OH O CH3 C OCH3 aldehydes ketones Chem 61 2p 2s carboxylic acids esters 1s 1s O: 1s22s22p4 .. .. O H H Hybrid orbitals of oxygen and nitrogen water Nitrogen: sp3 2p sp3 hybridize 2s 1s 1s N: 1s22s22p3 one sp3 orbital already filled with an unshared pair of electrons bonding can occur to three other atoms .. N H .. H H ammonia N CH3 H methyl amine H sp3 hybridize two sp3 orbitals already filled with an unshared pair of electrons bonding can occur to two other atoms .. .. O H CH2CH3 ethanol sp3 .. .. O CH CH 2 3 CH2CH3 diethyl ether .. .. O sp2 CH3 C CH3 acetone Resonance Structures Chapter 1-21 RESONANCE STRUCTURES Chapter 6; Bruice, Pages 260 - 282: Some molecules cannot be accurately represented by one simple line-bond formula: they are "hybrids" of two or more structures – O O C O– O– – O C O– O– – O C O 2/3– O –2/3 O C O –2/3 1. Resonance structures exist only on paper...the actual structures are hybrids of all the resonance structures 2. Resonance structures differ only in the position of electron pairs....not atoms 3. All structures should be proper Lewis structures (exceptions) 4. All resonance structures should have the same number of unpaired electrons Nonequivalent Resonance Structures 1. Structures with a maximum number of octets is preferred. 2. Charges should be located on atoms with compatible electronegativity. 3. Minimize charge separation. 4. Charge separation may be enforced by the octet rule (atoms may be charged if they have an octet.) Chem 61 Hydrocarbons Chapter 2-1 CHAPTER 2 Hydrocarbons: compounds containing only hydrogen and carbon: alkanes, alkynes, alkenes alkanes contain only C—H and C—C single bonds CnH2n+2; alkenes CnH2n contain C— C double bonds and alkynes CnH2n-2 contain C—C triple bonds Alkanes do not react with hydrogen, but alkenes and alkynes can react with hydrogen under certain conditions H2, catalyst CH2 CH2 CH3 CH3 alkene alkane 2H2, catalyst CH3 C C CH3 H3C–H2C CH2–CH3 alkyne ISOMERISM Structural Isomers: compounds with the same molecular formula that differ in the order in which the atoms are bonded to one another CH3 CH3 C CH3 CH3 C5H12 2,2-dimethylpropane H CH3 CH3 C C CH3 H H H H H CH3 C C C CH3 H H H C5H12 2-methylbutane C5H12 n-pentane Chem 61 Nomenclature Chapter 2-2 Chem 61 NOMENCLATURE Alkanes are named by the following parent names CH4 CH3CH3 CH3CH2CH3 CH3(CH2)2CH3 CH3(CH2)3CH3 CH3(CH2)4CH3 CH3(CH2)5CH3 CH3(CH2)6CH3 CH3(CH2)7CH3 CH3(CH2)8CH3 methane ethane propane butane pentane hexane heptane octane nonane decane C1 C2 C3 C4 C5 C6 C7 C8 C9 C10 Cyclic alkanes are named the same but with cyclo as a prefix cyclohexane cyclobutane Branched hydrocarbons are named from the parent with a substituent as a prefix hydrocarbon branches are named by dropping ane from the parent and adding -yl thus methane: methyl ethane: ethyl propane: propyl , etc. special trivial names for branches CH3 C CH3 H CH3 C CH3 CH3 isopropyl tert-butyl H CH3 C C CH3 H H isobutyl H CH3 C C CH3 H H sec-butyl – 23 – Nomenclature Chapter 2-3 Chem 61 BASIC RULES OF NOMENCLATURE 1. Find the longest continuous chain (not necessarily drawn as a straight line) and name the parent 2. Number the parent chain starting at the end nearest the branch 3. Identify the branch and its position 4. Attach the number and the name of the branch to the parent name. H CH3 C 3 4 H the branch is CH3; therefore methane CH3 C CH3 2 1 H methyl butane is the parent 2-methylbutane H H CH3H C C C H 3C H CH3H CH3 C CH3 3,3,5-trimethylhexane Other Functional Substitutents —CO2H increasing priority -oic acid C H -al O C -one O —OH —NR2 —C=C— —C≡C— R—, C6H5—, Cl—, Br—, —NO2 -ol -amine -ene -yne prefix substituents – 24 – Nomenclature Chapter 2-4 Chem 61 alkenes H H C C C C CH3H H CH3 H H CH3 C C C HO H H CH3 CH3 2,5-dimethyl-2-hexene alcohols CH3 H 2-pentanol carboxylic acids H H H H C C C Br H H COOH 4-bromobutanoic acid aldehydes and ketones CH3 H H CH2CH3 C C C H CH3H CHO 2-ethyl-3-methylpentanal CH3 H O CH2CH2CH3 C C C H CH3 H 4-methyl-3-heptanone – 25 – Alkanes Chapter 2-5 Chem 61 ALKANES Physical Properties nonpolar compounds C1 to C4 are gases; C5 to C17 are liquids; >C17 are solids Boiling points increase about 30°C for each additional CH2 unit branching lowers the boiling point due to disruption of van der Waals attractions insoluble in water; soluble in organic solvents like diethyl ether, benzene Chemical Properties very unreactive compounds Halogenation CH3CH3 + Cl2 light CH3CH2Cl + HCl + other products Oxidation of Alkanes Combustion spark CH3CH2CH2CH2CH3 + 8O2 5CO2 + 6H2O oxidation: a reaction that either removes a hydrogen atom from a carbon or adds an electronegative element to the molecule (O, N, S, halogen). low oxidation level CH3CH3 CH2=CH2 CH3CH2OH CH3CH2NH2 CH3CH2Cl high oxidation level HC≡CH CH3CH=O CH3CH=NH CH3CHCl2 CH3CO2H CH3C≡N CH3CCl3 CO2 CCl4 – 26 – Oxidation and Reduction Chapter 2-6 "Complete"/"incomplete" oxidation (combustion) of propane 5O2 CH3CH2CH3 2 CH3CH2CH3 CH3CH2CH3 7O2 2O2 3 O=C=O + 4 H2O "complete" combustion 6 CO + 8 H2O "incomplete" combustion 3 C + 4 H2O "incomplete" combustion Heat of Combustion: energy released when a compound is completely oxidized to CO2 and water; depends mostly on number of CH2 units: approximately 157 kcal/ methylene unit Reduction of Alkenes, Alkynes reduction: a reaction that either adds H atoms or removes an electronegative atom from the molecule CH3CH3 Pd, Ni,or Pt; H2 CH2=CH2 Pd, Ni,or Pt; H2 HC≡CH Pd, Ni,or Pt; H2 Pd, Ni,or Pt; H2 heat no reaction (NR) CH3CH3 CH2=CH2 Pd, Ni, Pt H2 CH3CH3 Chem 61 Chapter 2-7 Conformations of Acyclic Hydrocarbons Chem 61 Butane CONFORMATIONS OF OPEN CHAIN COMPOUNDS Molecular Mechanics E eclipsed methyls Steric Energy: (isolated molecule in gas phase at 0° K): relative energy of a conformation or stereoisomer calculated using classical mechanics (atoms and bonds treated as balls and springs) eclipsed 4.5 kcal 3.8 kcal Stretch (bond length): energy associated with stretching or compressing bonds from their optimal length gauche Bend (bond angle): energy associated with deforming bond angles from their optimal angle 0 60 120 0.9 kcal 180 240 anti 300 360 Stretch-Bend: energy required to stretch two bonds involved in a severely compressed bond angle dipole-dipole: energy associated with interaction of bond dipoles H out of plane: energy required to distort a trigonal center out of planarity H torsional strain: destabilization from eclipsing of bonds on adjacent atoms Ethane Newman projection H C H H H H C H H H H H H H H H staggered HH staggered HH eclipsed E ∆H = 3 kcal/mole eclipsed Ethane 0 kcal staggered 0 60 120 180 240 300 360 H CH3 anti van der waals strain: destabilization from two atoms being too close together dimensional CH3 H HH H CH3 H HCH3 eclipsed1 H H H CH3 CH3 gauche HH H H H3CCH3 eclipsed2 H H CH3 H HCH3 H CH3 gauche H H HCH3 eclipsed3 Chapter 2-8 Conformations of Cyclic Hydrocarbons CYCLIC COMPOUNDS Strain energy -∆H (kcal/mole) cyclopropane cyclobutane cyclopentane cyclohexane 499.8 655.9 793.5 944.5 -∆H per CH2 166.6 164.0 158.7 157.4 strain energy per CH2 total strain energy 9.2 6.6 1.3 0 27.6 26.4 6.5 0 cyclopropane: bond angles 60°; tetrahedral 109.5° sp3 orbitals are 109.5° apart cyclopropane bond angles 60° maximum overlap cannot be achieved cyclobutane and cyclopentane H H H H H HH H H H H H H puckered cyclobutane H H H H H envelope cyclopentane puckering allows bond angles to be at or close to the tetrahedral angle and minimizes torsional strain (electron—electron repulsions in eclipsed bonds) between adjacent C—H bonds Chem 61 Chapter 2-9 Conformations of Cyclohexane cyclohexane H H H H H H H H H equatorial bonds H H H chair cyclohexane axial bonds half chair boat E twist boat 7.1 chair 0 60 H H H H H H H chair H H H H H 120 5.5 10.8 kcal 180 240 H H H H H H H H H H 300 360 H H H half chair H H H H H H H H H H H boat chair twist boat Chem 61 Chapter 2-10 Conformations of Cyclohexane Substituted Cyclohexanes substitutents on cyclohexanes preferntially occupy equatorial positions due to 1,3 diaxial interactions in axially substituted cyclohexanes H H H H C H H H H H 1,3-diaxial interactions H H H gauche H CH3 C H H E more stable by 1.8 kcal/mole CH3 H H H C H H anti H H H C H CH3 C H H H axial substitution similar to gauche butane equatorial substitution similar to anti butane H CH3 CH3 H CH3 C H H H H C(CH3)3 H H H H 5.6 kcal/mole more stable ∆G = -RTlnK ∆G = -(1.98 kcal/mol°)(298)(2.94) = - (1.98 kcal/mol°)(298)(ln19) = -1.74 kcal/mole for methylcyclohexane Chem 61 Stereochemistry: Geometric Isomers Chapter 3-1 Chapter 3 STEREOCHEMISTRY Geometric isomerism in cyclic compounds OH OH OH H OH H OH OH cis trans-1,2-cyclohexanediol CH3 CH3 H H CH3 Br trans-1,3-dimethylcyclobutane Br Br cis cis Geometric Isomerism in alkenes 68 kcal/mole to cleave a carbon-carbon pi bond thus no "free" rotation CH3 H C CH3 C H CH3 H C C CH3 trans-2-butene CH3CH2 H H C CH3 cis-2-butene H H C C H trans-2-pentene Cl C Cl cis-1,2-dichloroethene Chem 61 Absolute Configuration Chapter 3-2 Stereoisomers: compounds with the same structures differing only in their arrangement of atoms in space. not cis or trans Cl CH3 C CH3CH2 F Cl C C H C Br E entgegen (across) I Z zusammen (together) Cahn-Ingold-Prelog Sequence rules 1. if the atoms are different, highest atomic number gets highest priority 2. if two isotopes of the same element, the one with the higher mass gets higher priority 3. if the atoms are the same, the atomic numbers of the next atoms are used to assign priority 4. atoms attached by double or triple bonds are given single bond equivalencies O R (O) C R' R = O C R' R (O) OH = R C O (C) (C) R C CR' = R C (C) R' C (C) (C) C OH O (C) (C) R C C—R' R C H H H H C R' (C) Chem 61 Stereochemistry: Chirality Chapter 3-3 CHIRALITY An object or molecule which cannot be superimposed on its mirror image is said to be chiral If an object or molecule can be superimposed on its mirror image, it is achiral. Enantiomers: isomers which are nonsuperimposable mirror images. I I H Br H 0000 0000 0000 0000 0000 0000 0000 0000 000 000 000 0000 000 0000 0000 0000 0000 0000 0000 0000 0000 0000 Br Cl 000 000 000 000 000 000 000 000 000 000 000 000 Cl 000 000 000 000 0000 0000 0000 0000 0000 0000 0000 mirror plane Cl Cl I Br H Br I H Stereogenic carbon atom: a carbon with four different groups bonded to it (designated *). H Cl H C* CH3 CH3 C* Cl CH2CH3 CH2CH3 enantiomers OH CH3 Br C* CH2CH3 H CH2CH2CH3 Fischer Projections by convention: horizontal bonds come out of the paper vertical bonds go back into the paper CH3 H C OH CH2CH3 CH3 H OH CH2CH3 H H C OH H C OH CH3 H H H OH OH CH3 C* CH3 Chem 61 Stereochemistry: Chirality Chapter 3-4 OPTICAL ROTATION enantiomers have almost all the same physical and chemical properties properties which differ are: 1. interaction with other chiral substances 2. interactions with polarized light Polarimeter ordinary light lamp polarized light polarizer rotated light solution of sample if plane polarized light is passed through a solution of a single enantiomer, the light is rotated either to the right or the left; the opposite enantiomer will rotate the light in the opposite direction optically active: a compound which rotates plane polarized light optical isomers: enantiomers dextrorotatory: rotates plane polarized light to the right, also (+) or d levorotatory: rotates plane polarized light to the left, also (–) or l racemic mixture: a 1:1 (50:50) mixture of two enantiomers does not rotate plane polarized light; therefore optically inactive Chem 61 Stereochemistry: Absolute Configuration Chapter 3-5 Absolute configuration: the order of arrangement of the four groups around a stereogenic center enantiomers have opposite configurations (R) and (S) Cahn-Ingold-Prelog System R: rectus or right S: sinister or left To assign R and S to an asymmetric atom 1. Rank the four attached groups from 1 (highest) to 4 (lowest) priority based on the Cahn-Ingold -Prelog Sequence rules 2. Project the molecule with the lowest priority group to the rear 3. Draw a semicircle from 1 to 2 to 3 4. If the direction of the semicircle is clockwise; configuration is R; if counterclockwise; configuration is S 1 Br 3 3 CH3 CH3 C Cl 2 2 CH3CH2 C OH 1 H H 4 4 R R If lowest priority group is out; assign the configuration as usual and then reverse it 1 Br 2 2 Cl Cl C CH3 3 H 4 lowest priority group out 3 CH3 C Br 1 H R S 4 S Chem 61 Stereochemistry: Diastereomers Chapter 3-6 Chem 61 Molecules with Two or more Asymmetric centers If a molecule has n asymmetric carbon atoms, it contains a maximum of 2n isomers; may not have that many CH2OH CH2OH CH2OH H C OH HO C H H C OH H C OH HO C H HO C H CH3 enantiomers CH3 CH2OH HO C H H C OH CH3 CH3 enantiomers two asymmetric carbons; 4 isomers diastereomers: stereoisomers which are not enantiomers; may have different physical and chemical properties Meso compounds meso compounds contain an internal plane of symmetry and are achiral: that is the mirror image is identical to the original diastereomers meso: identical CH2OH CH2OH CH2OH H C OH HO C H H C OH H C OH HO C H HO C H CH2OH CH2OH superimposable H CH3 diastereomers diastereomers meso CH2OH HO C H H C OH CH2OH enantiomers CH2OH H CH3 mirror plane chiral H CH3 H CH3 OH HO H meso H mirror plane H HO OH H chiral Stereochemistry: Asymmetric Synthesis, Resolution Chapter 3-7 Preparation of Enantiomerically Enriched Compounds Generating chiral compounds from achiral compounds Enzymes O O yeast H HO O OEt OEt >98% R Asymmetric Reagents H OH t-BuOOH O OH (+)-diethyl tartrate Ti(i-OPr)4 H >95% one enantiomer Resolution of a Racemic Mixture separated salt converted back to acid (R) R*COOH + (S) R*NH2 + (S) R*COOH racemic (R) R*COO–(S) R*NH3+ + (R) R*COOH (S) R*COO–(S) R*NH3+ diastereomeric salts (separable) (S) R*COOH Chem 61 Chapter 4-1 Acids and Bases ACIDS AND BASES Bronsted-Lowry: acid: a proton donor base: a proton acceptor Strong acid: completely ionized or dissociated in water e.g., HCl, H2SO4, HNO3, HBr Weak acid: only partially dissociated in water: carboxylic acids are weak acid CH3CO2H acid + CH3CO2- H2O + conjugate base base H3O+ conjugate acid amines are weak bases CH3NH2 base + HO – CH3NH3+ + H2O acid conjugate acid conjugate base Generally: strong acids have weak conjugate bases and weak acids have strong conjugate bases that is, as acid strength increases, the basicity of the conjugate base decreases Thus the ability of the conjugate base to stabilize a negative charge determines the strength of an acid Conjugate Acids H 2O pKa HCN 15.75 CH3CO2H 6.37 H3PO4 HCl 2.12 -7 4.75 increasing acid strength Conjugate Bases HO – NC – CH3CO2– H2PO4 – C l – decreasing base strength Chem 61 Chapter 4-2 Acids and Bases Factors affecting Acidity: the electronegativity and the size of the atom which carries the negative charge influence its ability to stabilize the negative charge size of the atom H—F H—Cl pKa 3.45 -7 H—Br -9 H—I -9.5 increasing size of halogen, increasing acid strength electronegativity pKa (CH3)3C—H 50 (CH3)2N—H 35 CH3O—H 15.5 F—H 3.45 increasing electronegativity of atom, increasing acid strength ACIDITY CONSTANTS, Ka's for acetic acid CH3CO2H + H2O Ka = CH3CO2– + H3O+ [CH3CO2–] [H+] [CH3CO2H] since stronger acids are more ionized, the larger Ka, the stronger the acid pKa = -logKa, the lower the pKa, the stronger the acid also, the higher the pKa, the weaker the acid or the stronger the base the stronger the acid, the more stable the anion produced by ionization of the acid. Chem 61 Chapter 4-3 Lewis Acids and Bases LEWIS ACIDS AND BASES Lewis acid: electron pair acceptor: any species with an electron deficient atom BBr3, AlCl3, H3C+ Lewis base: electron pair donor; any species with an unshared pair of electrons .. .. H3N:, CH3CH2..OH, H2C=O: Chem 61 Chapter 5-1 Alkenes: Structure and Isomerism ALKENES Alkene Structure sp2 hybridized carbons are trigonal planar with atoms 120° apart 2p 2p 2p 96 kcal 2s sp2 hybridize 2s 1s 1s 1s ethylene: trigonal planar H C H C H H 2p H H C C H H H C H 120° H H H pi orbital H sigma orbitalH sp2 H H C H C C C H H pi* orbital H H H C H sigma* orbital bond angles = 120°; C=C bond length = 1.34Å pi (π) bonds are formed by the side by side overlap of two p-orbitals (approx. 68 kcal/mol) pi bonds are above and below the plane where the sigma bond is located pi bonds make the molecule rigid between the two atoms preventing rotation E sigma σ* pi π* pi π sigma σ ethylene C=C double bond: one sigma, one pi Chem 61 Chapter 5-2 Alkenes: Structure and Isomerism Geometric Isomerism in alkenes 68 kcal/mole to cleave a carbon-carbon pi bond thus no "free" rotation CH3 H C H H C C CH3 H CH3 trans-2-butene C H H C C CH3CH2 CH3 cis-2-butene CH3 H C C Cl H Cl cis-1,2-dichloroethene trans-2-pentene Stereoisomers: compounds with the same structures differing only in their arrangement of atoms in space. not cis or trans Cl CH3 C CH3CH2 C H E entgegen (across) F Cl C Br C I Z zusammen (together) Cahn-Ingold-Prelog Sequence rules 1. if the atoms are different, highest atomic number gets highest priority 2. if two isotopes of the same element, the one with the higher mass gets higher priority 3. if the atoms are the same, the atomic numbers of the next atoms are used to assign priority 4. atoms attached by double or triple bonds are given single bond equivalencies Chem 61 Alkenes: Electrophilic Addition Reactions Chapter 5-3 Chem 61 Alkenes: Reactivity The reactivity of alkenes is due to their ability to donate a pair of electrons: their Lewis basicity. H H C Consider Ethylene: The site of reactivity is the pi-bond due to the exposed nature of the pi electrons H H C C H H 2p H H C C sp H pi orbital H sigma orbitalH H H H C C H H pi* orbital C H sigma* orbital The pi electrons act as a Lewis base (electron pair donor). These electrons react with electron deficient species (Lewis acids) Alkenes undergo electrophilic additions reactions. Electrophile: an electron deficient ion or molecule H+ Br+ BH3 +CH3 Nucleophile: an electron rich ion or molecule. I– CH3NH2 Br– H H Br– + CH2—CH2–H CH2=CH2 H–Br H C H H 2p C 2 H H H Br H H H H H H H C H 120° H H H H C H Br H H2O Reaction Mechanisms of Electrophilic Addition Reactions reaction mechanism: a detailed description of how a chemical reaction occurs. A roadmap of a reaction....curved arrows show which bonds are formed or broken. A mechanism includes the transition states involved in making and breaking bonds and reactive intermediates that are formed along the pathway from reactants to products. Br H H2C CH2 Thermodynamics Chapter 6-1 Thermodynamics describes the properties of a system at equilibrium ∆G = – RT ln Keq where Keq = [reactants] [products] R = 1.986 X 10-3 kcal K-1 mol-1 (gas costant) T = temperature in degrees Kelvin ∆Go = ∆Ho – T∆So free energy = enthalpy – T x entropy enthalpy = heat of reaction entropy = state of disorder transition state Energy Diagram ∆G ‡ E products reactants progress of reaction reactants – ∆G products exergonic + ∆G products reactants endergonic Chem 61 Kinetics Chapter 6-2 Kinetics describes the rate of progression of a reaction depends on the energy of activation (the stability of the transition state): the lower the transition state energy, the faster the reaction will be. transition state faster transition state slower ∆G ‡ E ∆G ‡ E products reactants progress of reaction reactants products progress of reaction first order reaction rate = k[A] rate is proportional to the concentration of one reactant second order reaction rate = k[A] [B] rate is proportional to the concentration of two reactants A two step reaction transition state E ∆G‡ reactants transition state ∆G‡ intermediate products Rate limiting step: is the slowest step (step with the highest energy of activation) Chem 61 Electrophilic Addition: Addition of H–X Chapter 6-3 Electrophilic Addition Reactions Nucleophilic addition does not occur with alkenes unless an electron-attracting group is attached to one of the carbon atoms to cause a polarity difference. C No Reaction Nu: + C However, in the electrophilic addition reaction, the reagent is E+A– (E+ = electrophile, A– = some anion) + E A + C C – E + C C A A – E C C Note the alkene acts as a Lewis base (or nucleophile) toward the Lewis acid (or electrophile) The intermediate, a carbocation, reacts with A– to yield a product in which E and A have added to the C=C double bond. Addition of HX (E+ = H– A– = F–, Cl–, Br–, I–) CH3 C CH2 + H—X CH3 CH3 + C X– CH3 CH3 X CH3 C CH3 CH3 Reactivity: H—I > H—Br > H—Cl > H—F The reaction is said to be regioselective since an unsymmetrical alkene gives a predominance of one of two possible electrophilic addition products. The term regiospecific is used if one product is formed exclusively. In these reactions, the halogen (A–) is found attached to the most substituted carbon atom of the alkene (Markovnikoff's Rule): Chem 61 Electrophilic Addition: Addition of H–X Chapter 6-4 CH3 C CH2 + H—X CH3 CH3 R C C X CH3 CH3 C CH3 CHR + H—X R R R X– + C + C CH3 X– X CHR R R 3° carbocation more favored X– + R CH C—R CHR + H—X R CH3 C CHR R R R H X C CHR R 2° carbocation less favored Thus regioselectivity is explained by the lower Eact leading to the 3° carbocation intermediate. If a nucleophilic solvent is employed in the electrophilic addition reaction solvent may compete with A – for the intermediate carbocation. Cl CH CH2 OCOCH3 CH HCl CH3 CH CH3 + CH3COOH (solvent) Note the C=C's of the aromatic ring do not undergo this type of reaction. Since a carbocation intermediate is involved, rearrangement may sometimes occur. + HCl (CH3)2CH CH CH3 (CH3)2CH CH CH2 (CH3)2CH CH CH3 40% Cl + (CH3)2C Cl CH2 CH3 60% Chem 61 Carbocation Stability Chapter 6-5 Carbocation stability Carbocations are highly reactive intermediates ; they cannot be observed directly in the reaction mixture since they react as soon as they are formed. Any structural feature that will disperse a positive charge will stabilize the carbocation. This stabilization has been quantitated by so-called hydride H:– affinity measurements (gas phase). The lower the value of the hydride affinity, the more stable the carbocation. Name Structure methyl primary 1° secondary 2° tertiary 3° vinyl allyl secondary allyl tertiary allyl CH3+ CH3CH2+ (CH3)2CH+ (CH3)3C+ CH2=CH+ CH2=CH—CH2+ CH2=CH—CH+(CH3) CH2=CH—C+(CH3)2 Hydride Affinity (kcal/mol) 314 274 247 230 287 256 237 225 298 phenyl + 233 benzyl CH2+ secondary benzyl + CHCH3 226 tertiary benzyl + C(CH3)2 220 diphenylmethyl + CH 215 triphenylmethyl + C 210 Chem 61 Carbocation Stability Chapter 6-6 Thus the order of carbocation stability is triphenyl methyl > diphenylmethyl > 3° ≈ benzyl ≈ allyl > 2° > 1° >> methyl Carbocation centers can be stabilized by overlap with adjacent pi orbitals (resonance) or adjacent sigma orbitals (hyperconjugation). H + CH3 C CH3 + C H H p H σ .. C C C + CH2 adjacent sigma bond donates some electron density to the empty p orbital on the carbocation center more adjacent sigma orbitals result in a more stable carbocation thus 3° > 2° > 1° > methyl H H p sigma donation H H empty p orbital ovelaps with adjacent pi orbital to disperse the positive charge pi CH CH2 CH2 CH + CH2 equivalent resonance structures Since the pi orbital is closer in energy to the empty p than the sigma orbital, the pi overlap stabilizes the cation more efficiently. Chem 61 Electrophilic Addition: Addition of H2O Chapter 6-7 Addition of Water (E+ = H+; A– = HOH) use H2SO4, H2O H+ Step 1 R 2C + R 2C CHR .. + R 2C Step 2 Step 3 H O: R 2C H + :O H :OH2 CH2 R R 2C .. H :OH2 + CH2 R CH2 R CH2 R .. :O H R 2C CH2 R + H3O+ TS1 TS2 + R 2C Eact CH2 R TS3 H +O H R 2C CHR R 2C CH2 R O H R 2C CH2 R Addition of water to alkenes is a reversible reaction and whether the alkene or the alcohol predominates at equilibriuim depends on the reaction conditions. Low temperatures and high concentrations of water favor the alcohol. Higher temperatures and removal of water favor the alkene. Chem 61 Electrophilic Addition: Oxymercuration Chapter 6-8 If alcohol is used as the solvent in this process ethers are obtained as the products. Addition of Mercury acetate and Water ( E+ = +HgOCOCH3; A– = OH2) To avoid carbocation rearrangements under the conditions used in addition of H2O (H2SO4/H2O) a better system for the addition of water to a C=C has been devised. O CH3 C O O Hg O C O + CH3 Hg O E mercury acetate C CH3 CH3 CH3 C O CH3 + – O CH2 + +Hg O C or C CH2 Hg O C CH3 CH3 carbocation O + H2O: CH3 C CH2 Hg O C CH3 Hg O O C CH3 CH2 CH3 bridged carbocation +OH2 Since no rearrangement is observed in reactions with +HgOCOCH3 the bridged carbocation is believed to be the true intermediate. Attack by H2O on the more electron deficient carbon opens the bridged cation (backside attack). NaBH4 reduction of the C—Hg bond yields the alcohol in a separate step. CH3 CH3 C CH2HgOCOCH3 +OH2 H 2O CH3 CH3 C CH2 HgOCOCH3 HO CH3 CH3 HO NaBH4 C CH3 O + CH3 C CH3 C + O CH3 Chem 61 CH3 + Hg° CH3 C CH3 CH2 1. Hg(OCOCH3)2, ROH 2. NaBH4 CH3 CH3 RO C CH3 Electrophilic Addition: Hydroboration Chapter 6-9 Hydroboration (E+ = B, A– = H–) Discovered by H.C. Brown in the 1950's. The reagent BH3 (borane) is used in electrophilic addition reactions. Borane is generated by 2 B2H6 (diborane)+ 3 NaBF4 3 NaBH4 + 4 BF3 4 BH3 (borane) Addition of BH3 to C=C, C≡C is termed hydroboration. R—CH=CH2 + BH3 R H H B C C H H H R H H H B C C H H H H ‡ R H B C C H Note B is the electrophile and H acts as A-. The addition of H– and B occur from the same face of the alkene (syn addition). Further reaction of R—CH2–CH2—BH2 with more alkene yields first the dialkyl borane and then the trialkyl borane as the stable product. R—CH=CH2 + R—CH2—CH2—BH2 ( R—CH2—CH2)2BH R—CH=CH2 ( R—CH2—CH2)3B Brown discovered that trialkylboranes were easily oxidized by alkaline hydrogen peroxide (H2O2 in HO–). The oxidation reaction proceeds as shown. H H H H Chem 61 Electrophilic Addition: Hydroboration Chapter 6-10 – ( R—CH2)3B CH2R OOH RCH2 B–O OH RCH2 RCH2 RCH2 – RCH2CH2 B O CH2R + —OH OCH2R RCH2 CH2R OOH B O B–O O – OH OCH2R OOH CH2R CH2R RCH2O B O – OH CH2R 3 R—CH2—OH + B(OH)3 The overall reaction is R—CH=CH2 1. BH3 R—CH2—CH2—OH – 2. H2O2, OH Note the reaction is regioselective and yields an alcohol that is isomeric to that obtained by Hg(OCOCH3)2—NaBH4 or H2SO4—H2O. R—CH=CH2 H3O+ or Hg(OCOCH3)2 then NaBH4 R—CH—CH3 OH Treatment of a trialkylborane with CH3CO2D yields a monodeuterated alkane (CH3CH2)3B CH3CO2D CH3CH2D Chem 61 Electrophilic Addition: Hydroboration Chapter 6-11 Treatment of a trialkylborane with CH3CO2D yields a monodeuterated alkane (CH3CH2)3B CH3CO2D CH3CH2D Bromination of a trialkylborane yields the alkyl bromide. (C6H5CH2CH2)3B Br2 C6H5CH2CH2Br HO– Stereochemical Results of Hydroboration Hydroboration is regioselective and stereospecific CH3 H B BH3 H H CH3 cis-addition Thus H and OH add cis to the C=C. H2O2 HO– OH OH H H + H CH3 CH3 enantiomers H Chem 61 Electrophilic Addition: Stereochemistry of Halogen Addition Chapter 6-12 Stereochemistry of Halogenation Br + C C C C Br C C Br Br— Alkenes containing alkyl substituents react with Br2 via a bridged bromonium ion intermediate. Nucleophilic opening of the bridged ion by backside attack of Br – at carbon gives overall anti (trans) addition of E+(Br+) and A– (Br–) to the double bond. Chlorination reactions proceed by a similar pathway. Some examples: CH3 C H C X CH3 Br2 H or Cl2 CH3 C C CH3 X H CH3 meso C Br2 or Cl2 C (enantiomers) X H H + X H CH3CO2H (solvent) CH3 C H C CH3 C X H X Br2 or Cl2 X H H CCl4 X X CH3 CH3 H + HH X C H CH3 Chem 61 Electrophilic Addition: Mixed Addition Chapter 6-13 Mixed Addition Br C C Br Br C C Cl Br— C C Br+ Br+ C C Cl— Br2 or Cl2 with H2O, HO– present: C C X+ X+ C C HO– X C C OH Mixed Reagents R S+ RS—Cl + O=N—Cl + C C C C C RS C C Cl NO NO + C C C C C Cl I I—Cl + C C C + C I C C Cl Chem 61 Chapter 6-14 Hydrogenation of Alkenes Hydrogenation of Alkenes Alkene Heat of Hydrogenation, ∆Hh (kcal/mol) addition of H2 is cis (syn) CH3CH2CH=CH2 -30.3 CH3CH=CHCH3, Z -28.6 CH3CH=CHCH3, E -27.6 CH3 H H2/Pd Thus the stability of the three alkenes is H CH3 CH3 CH3 C C CH3 CH3CH=CHCH3, E > CH3CH=CHCH3, Z > CH3CH2CH=CH2 CH3 H2/Pd These comparisons indicate H H C C CH3 CH3 i) increasing alkyl substitution stabilizes an alkene ii) conjugated dienes are more stable than non-conjugated dienes, iii) trans alkenes are more stable than cis alkenes (steric repulsions in cis) ii) Heat of Hydrogenation Studies The heat of hydrogenation of an alkene is the energy difference between the starting alkene and the product alkane. It is calculated by measuring the amount of heat released in a hydrogenation reaction. H H C C CH3CH2 H H H C CH3CH2CH2CH3 + heat C CH3 CH3 CH3 H C CH3 C H CH3CH2CH=CH2 E ∆Hh progress Z-CH3CH=CHCH3 ∆Hh progress Chem 61 E-CH3CH=CHCH3 ∆Hh progress Chapter 6-15 Radical Addition Reactions Addition of HBr in the presence of a radical initiator such as peroxides effects so called anti Markovnikov addition of HBr CH3 C CH2 + H—X X– + C CH3 CH3 Reversal of regioselectivity (position of bromine and H addition) results from the inital addition of Br radical rather than H+. X CH3 CH3 CH3 C CH3 CH3 Stability of Carbon Free Radicals (Which H atom is abstracted?) Free Radical: any atom or group of atoms that contains one or more unpaired Stability of carbon free radicals follows the same pattern as carbocations: electrons. . A free radical is symbolized as a single dot representing the unpaired electron: Cl , . Chem 61 . Br , H3C , etc. . CH3 Free radicals are usually encountered as high energy short-lived, non-isolable . . . . CH2=CH—CH2 , C6H5CH2 > (CH3)3C > (CH3)2CH > CH3CH2 > . allyl benzyl tertiary secondary primary methyl intermediates in certain reactions. heat or light RO CH3 R initiation + H—Br C CH2 + Br RO–H + Br . . CH3 CH3 C . propagation Br H–Br CH2 CH3 CH3 H C Br CH2 + Br . CH3 propagation combination of any two radicals Br . + Br . . Br CH3 C CH2 Br2 termination + Br . CH3 CH3 2 RO . C R . Br Br C CH2 CH3 RO–OR . ROOR 2 RO . RO–OR peroxide CHR + H—Br R C Br CHR R 3° carbocation more favored H R C R Br CHR Radical Reactions of Alkanes Chapter 7-1 Free Radical Substitution Reactions Free Radical: any atom or group of atoms that contains one or more unpaired electrons A free radical is symbolized as a single dot representing the unpaired electron: . . . Cl , Br , H3C , etc. Free radicals are usually encountered as high energy short-lived, non-isolable intermediates in certain reactions. (Recall that carbocations were seen to be intermediates in certain reactions in Chapter 5). CH3 a) CH3 C CH3 Br – C+ Br CH3 carbocation intermediate CH3 CH3 double barbed arrows indicate the movement of two electrons CH3 b) CH 3 C CH3 Br CH3 CH3 C . Br. free radical intermediate CH3 single barbed arrows indicate the movement of one electron Note that the carbon in a) has lost an electron pair to the bromine atom, thus the carbon has an sp2 configuration and a single "+" charge resulting from the empty p orbital. In b) the carbon atom has lost one electron; the carbon is also in the sp2 configuration with the unpaired electron in the p orbital. CH3 CH3 C CH3 (CH3)3C+ CH3 CH3 . C CH3 . (CH3)3C equal probability of electron being in either lobe Chem 61 Radical Reactions of Alkanes Chapter 7-2 Free Radical Substitution Reactions Chem 61 Hence CH2Cl2, CHCl3, and CCl4 are formed Chlorination of methane in the presence of light is the classic example c) Termination of the Chain Reaction CH4 + Cl2 light CH3Cl + CH2Cl2 + CHCl3 + CCl4 + HCl . . . terminate the reaction. . . Cl a) Initiation of the radical chain reaction Cl Cl Cl light . . H Cl i) C . H—Cl + CH3 H requires 1 kcal/mole a methyl radical . ii) . CH3 + Cl—Cl CH3—Cl + Cl chlorine radical can now react with more CH4 and propogate the chain Note: Cl has substituted for a H and therefore free radical substitution . iii) As the concentration of CH3—Cl increases in the reaction mixture Cl starts to react with both CH4 and CH3Cl. . Cl . + CH3—Cl CH2—Cl + Cl—Cl H—Cl + . CH2—Cl (chloromethyl radical) Cl—CH2—Cl + Cl . CH3—Cl + . . CH2Cl . . CH2Cl + CH2Cl b) Propagation (self-perpetuating the chain reaction) H . + CH3 CH3 + CH3 2Cl requires 58 kcal/mole two chlorine radicals . . CH2Cl, CHCl2, CCl3) that disrupts the propagation of the chain will Reaction mechanism involves three general steps: Initiation, propagation, and termination heat or . Any reaction of Cl or of the carbon free radical intermediates (CH3 , CH2Cl2 CH3—CH3 Cl—CH2—CH2—Cl Reactivity of the Halogens Chapter 7-3 Reactivity of the Halogens F2 Bond Dissociation Energy Cl2 Br2 I2 37 58 46 36 kcal/mole . . . Thus the energy of activation for formation of F and I is lower than Cl and Br . however, the order of reactivity of X. with alkanes is: F2 >> Cl2 > Br2 >> I2 (I2 does not normally react; F2 reacts explosively) Thus the rate determining step is not: 2X. X—X Rather, hydrogen atom abstraction is the rate determining step. . X + CH4 . H—X + CH3 Since chlorination is faster than bromination, Eact for . Cl + CH4 H—Cl + . CH3 must be lower than the Eact for . . Br + CH4 H—Br + CH3 . [CH3 + HCl +Cl2] . [CH3 + HBr + Br2] E Eact . Cl + CH4 CH3Cl + Cl Progress . Eact . Br + CH4 CH3Br + Br . Chem 61 Reactivity of the Halogens Chapter 7-4 Stability of Carbon Free Radicals (Which H atom is abstracted?) Stability of carbon free radicals follows the same pattern as carbocations: . . . . . CH2=CH—CH2 , C6H5CH2 > (CH3)3C > (CH3)2CH. > CH3CH2 > CH3 allyl benzyl tertiary secondary primary methyl Unlike carbocations, free radicals do not rearrange to a more stable free radical. CH3 CH3 CH3 CH3 C CH CH3 + CH3 2° C + CH3 . C CH CH3 CH3 CH CH3 C CH CH3 OH CH3 CH3 3° CH3 CH3 H 2O CH3 Cl2 CH3 CH3 2° C CH CH3 CH3Cl Since both carbocations and free radicals involve planar sp2 carbon atoms, racemization will be observed in the products resulting from both intermediates. H CH3CH2 C CH2Cl CH3 R Cl2 Br CH3CH2 CH3 CH3OH C CH2Cl S Cl CH3CH2 + CH3 CH3CH2 C CH2Cl CH3 S (50:50) OCH3 CH3CH2 C CH2Cl CH3 S C Cl R CH3CH2 + CH3 (50:50) CH2Cl C CH2Cl OCH3 R Chem 61 Selectivity of Halogenation Chapter 7-5 Selectivity of Hydrogen Atom Abstraction Chem 61 Other examples of this are: The more stable free radical should determine the nature of the product CH3 Cl . CH2 Br. CH3 CH3 . a) 1° . CH3 . c) allyl b) 3° . CH3 CH2CH3 Cl2, light CH3 d) allyl e) 2° Br Br2, light Br and as products. Br Chlorination is much less specific than bromination Cl CH3CH2CH3 CH3CH2CH3 Cl2 Br2 CH3 CH CH3 55% Br CH3 CH CH3 100% + CH3CH2CH2Cl 45% . . The difference in selectivity of H atom abstraction by Cl and Br is explained by the Hammond postulate. The transition state in the chlorination reaction is less influenced by the stability of the intermediate free radical; thus both (CH3)2CH . . and CH3CH2CH2 result. In bromination the more stable free radical intermediate is highly favored; thus . (CH3)2CH is formed exclusively. CH2CH2Cl + (56:44) Thus intermediates c) and d) should be more stable and should lead to CH3 CHCH3 CHCH3 100% Halogenations Chapter 7-6 Bromination with N-Bromosuccinimide (NBS) O O CCl4 NH + NBr + light or peroxide O O Br NBS acts as a Br2 source; the reaction is initiated by either light or a peroxide (ROOR). NBS is used to introduce a Br atom at allylic or benzylic positions. Other Sources of Free Radicals Although light (hν) and heat (∆) are used for halogenation reactions other reagents are often useful for initiating free radical reactions. Some of these reagents include: a) dibenzoyl peroxide C6 H5 C O O O C 2 C6 H5 C 6H 5 C O . O O b) hydrogen peroxide H O O H 2 c) alkyl hypochlorites ∆ R—O—Cl H . O. RO + Cl . d) azobisisobutyrylnitrile CH3 CH3 C N C≡N. CH3 CH3 N C CH3 2 CH3 C≡N. Clues to Whether a Reaction Involves Free Radicals a) Reaction requires high temperatures (>200°) b) Reaction requires light energy (hν) c) Reaction requires an initiator (a-d above) or oxygen. . C C≡N. + N≡N Chem 61 Nucleophilic Substitution Reactions Chapter 8-1 Nucleophilic Substitution Reactions (SN1, SN2) alkyl halides: CH3—X methyl RCH2—X R2CH—X R3C—X primary, 1° secondary, 2° tertiary, 3° X aryl halide vinyl halide CH2=CH—X do not easily undergo nucleophilic substitution reactions Examples CH3OH CH3—I + Na+ – OCH3 CH3CH2CH2—Br + Na+ – OH CH3OH CH3—OCH3 + Na+ + Na SH SH CH3OH + NaCl Br + I CH3CH2CH2—OH + Na+ – Br Cl +– – OCH3 CH3OH + HBr CH3 CH3 Anatomy of a nucleophilic substitution reaction SH CH3 CH I CH3 + Na+ – SH CH3OH CH3 CH CH3 + Na+ I – Chem 61 Nucleophilic Substitution Reactions Chapter 8-2 Anatomy of a nucleophilic substitution reaction CH3 CH CH3 + Na+ – SH CH3OH I CH3 CH CH3 + Na+ – I SH (L) Leaving Group: any group that can be displaced from a carbon atom (in this case I—) (Nu) Nucleophile: the species that attacks the carbon atom bearing L and donates the electron pair to form the nucleophile—C bond (in this case —SH) (R—I) Substrate: the molecule, containing the leaving group that is acted on by the nucleophile (here: 2-iodopropane) Solvent: the medium used to dissolve the substrate and the nucleophile (in this case methanol, CH3OH); solvent can sometimes be the nucleophile (solvolysis) Curved arrows are used to indicate the movement of electrons during a nucleophilic substitution reaction. By convention electron movement is written from negative to positive. δ+ δ − Nu:– + R—CH2—L electron pair for new bond at C R—CH2—Nu + L:– electron pair that accompanies L Chem 61 Nucleophilic Substitution Reactions Chapter 8-3 Reaction Mechanisms of Nucleophilic Substitution Reactions Energetics of SN2 reaction mechanism: a detailed description of how a chemical reaction occurs. A roadmap of a reaction....curved arrows show which bonds are formed or broken. A mechanism includes the transition states involved in making and breaking bonds and reactive intermediates that are formed along the pathway from reactants to products. potential energy E CASE 1:SN2 Substitution Nucleophilic 2nd order (bimolecular): H C CH3O CH3 δ− CH3O Br H C H ∆H of reaction δ− Br CH3O C H + Br– products CH3 H H partial bonds in transition state Orbital Picture H H H H C Nu L H C—L sigma* Nu nonbonding orbital H H Nu Eact sp3 CH3 – energy of transition state reactants sp2 sp3 Chem 61 + C Nu—C sigma H L L nonbonding orbital Nu C H L progress of reaction Nucleophilic Substitution Reactions Chapter 8-4 Orientation of Nu: and L in SN2: Nucleophile approaches substrate from the backside of L Point of highest energy along the reaction coordinate is the transition state: the O—C bond is partially formed and the C—Br bond is partially broken The Nu is bonded to the carbon on the opposite side of that occupied by the leaving group L: Net inversion of the carbon atom is observed in all SN2 reactions Reaction Rate: the time required for all substrate molecules to be converted to product The rate of an SN2 reaction is proportional to the concentrations of the substrate and the nucleophile, thus the reaction is second order Rate = k[substrate][nucleophile] k is the proportionality constant called the rate constant Rate and Eact Under the same reaction conditions, the reaction with the lower Eact has a faster rate Reaction 2 below is faster Eact Eact E Rxn 1 Rxn 2 Increasing alkyl group substitution at the carbon atom bonded to the leaving group hinders approach of the nucleophile. The E act increases due to steric hindrance of nucleophile approach and thus the SN2 rate decreases Chem 61 Nucleophilic Substitution Reactions Chapter 8-5 Relationship between Substrate Structure and SN2 Rate Alkyl halide Relative rate of SN2 CH3—X 30 (CH3)2CH—X 0.025 CH3CH2—X 1 (CH3)3C—X ~0 CH3CH2CH2—X 0.4 Relative SN2 rate CH3—Cl + I – CH3—I + – Cl 93 CH3CH2—Cl + I– CH3CH2—I + Cl– 1 (CH3)2CH—Cl + I– (CH3)2CH—I + Cl– 0.0076 Increasing alkyl group substitution at the carbon atom bonded to the leaving group hinders approach of the nucleophile. The E act increases due to steric hindrance of nucleophile approach and thus the SN2 rate decreases SUMMARY SN2 reactions occur by the attack of a nucleophile on substrates containing the leaving group attached to a methyl, primary carbon, or secondary carbon. SN2 reactions do not occur with tertiary alkyl halides SN2 exhibit a single transition state (concerted reaction, i.e. not stepwise, no intermediates) Inversion of configuration at the carbon results from backside attack of the nucleophile The rate of SN2 reaction depends on the concentration of the nucleophile and the substrate The rate of SN2 follows the order: CH3—X > CH3CH2—X > CH3CH2CH2—X > (CH 3) 2CH—X >> (CH 3) 3C—X Chem 61 Nucleophilic Substitution Reactions Chapter 8-6 Chem 61 CASE 2: SN1: Substitution Nucleophilic First Order (Unimolecular) Substrates containing the leaving group attached to a tertiary carbon atom react with weakly basic nucleophiles by an alternative nucleophilic substitution reaction mechanism (CH3)3C—Br + CH3OH (CH3)3C—OCH3 + HBr The process involves three steps: Step 1: ionization (loss of halide ion) (CH3)3C—Br + CH3OH slow (CH3)3C+ Br – + The transition state is pictured as (CH3)3Cδ+ ------Br δ– . 1 Step 2: attack by the nucleophile .. CH3O—H .. + – (CH3)3C Br fast 2 (CH3)3C O+ CH3 H H O+ CH3 H H CH3 Step 3: deprotonation (loss of proton) (CH3)3C The transition state is pictured as (CH3)3Cδ+------δ+OCH3 (CH3)3C O CH3 + HBr fast 3 Br– Step 1 involves ionization of the alkyl halide to the halide ion and the tertiary carbocation. The step with the highest Eact is the slowest step (rate determining step) in the pathway. Ionization is aided by polar solvents (H2O, ROH) which solvate and thus stabilize the carbocation and the leaving group anion. Step 3 involves acid-base reaction between either Br– or more likely solvent CH3OH in a very rapid reaction The transition state is pictured as (CH3)3C O + H O CH3 Nucleophilic Substitution Reactions Chapter 8-7 Stereochemistry of SN1 reaction Relationship Between Substrate Structure and SN1 Rate the intermediate carbocation contains an sp2 hybridized carbon atom which is planar Alkyl halide + CH3 C Chem 61 CH3 CH3 Relative SN1 rate CH3—Br 1.0* CH3CH2—Br 1.0* (CH3)2CH—Br 11.6 1.2 X 106 Thus attack of CH3OH on the carbocation can occur equally from the top and bottom. If the carbon atom containing the leaving group were asymmetric, racemization of the product would be observed Br CH3 H 2O C CH2CH2CH3 CH3CH2 R OH C CH3 CH3CH2 R + CH2CH2CH3 CH3CH2 CH3 (50:50) (CH3)3C—Br *These observed reaction rates probably proceed through SN2 not SN1 SN1 rates reflect the relative Eact leading to the different carbocations (transition state 1) C CH2CH2CH3 OH S Reaction Rate of SN1 The rate of the SN1 reaction does not depend on the concentration of the nucleophile, but depends only on the concentration of the substrate SN1 Rate = k[substrate] The reaction is first order because the rate is proportional to the concentration of only one reactant Since the relative stability of carbocations is 3° > 2° > 1° > methyl; tertiary alkyl halides proceed at the faster rate Nucleophilic Substitution Reactions Chapter 8-8 Chem 61 SUMMARY Allyl and benzyl primary halides Allyl and benzyl primary halides are reactive substrates in both SN1 and SN2 type reactions. Substrate Realtive SN1 Rate 1.0 30 CH3CH2—X 1.0 1 33 40 380 120 CH2=CH—CH2—X C6H5—CH2—X SN1 reactions occur with substrates that yield stabilized carbocations (allyl, benzyl, 3°, 2°, not primary or methyl) Relative SN2 Rate CH3—X SN1 reactions are first order in rate. SN1 reactions lead to racemization. Reactions of Carbocations 1. Reaction with a nucleophile: SN1 reactions R+ + Nu:— or Nu: 2. Elimination of H+ on an adjacent carbon: E1 reactions CH3 + CH CH2 CH3—CH=CH2 + H+ H 3. Rearrangement to a more stable carbocation: occurs whenever a more stable carbocation can be formed CH3 + CH H C CH3 CH3 CH3 SN1 reactions involve an intermediate carbocation which is attacked by the nucleophile to yield the product of nucleophilic substitution. + CH2 C CH3 CH3 Chapter 8-9 Elimination Reactions Elimination Reactions (E1, E2) Case 1: E1: Elimination First order Formation of a carbocation intermediate in a reaction is a first order process. Once formed the carbocation may react with a nucleophile (SN1) or lose a proton from an adjacent carbon to form an alkene (E1) Thus SN1 and E1 reactions are competitive CH3 (CH3)3C—Br CH3 – C + Br (CH3)3C – Nu +Br (SN1) H C H H CH3 C CH3 CH2 + HBr (E1) E1 reactions predominate when the reaction contains only poor nucleophiles; otherwise SN1 reactions are the more likely pathway Since a carbocation is involved, rearangement may occur TS1‡ TS2‡ E Eact R+ R–X alkene Chem 61 Elimination Reactions Chapter 8-10 Chem 61 Thus CASE 2: E2: Elimination Second Order Bimolecular elimination results when an alkyl halide is treated with a strong base (HO –, RO –, etc) at elevated temperatures (80°-120°). CH3CH2CH CH3CH2CH2 CHCH3 RO– 69% + 80°, ROH Br The E2 reaction does not involve an intermediate as in the E1 reaction E2 reactions are concerted as in the SN2 reaction RO– H H H C C H ROδ− H Br ROH + H C H C H C H Brδ− C CH3CH2 CHCH3 H H Br Brδ− transition state for E2 Elimination Reaction rate E2 rate = k[base][substrate] halides tertiary halides > secondary halides > primary Direction of Elimination The more substituted (and more stable) product is normally the predominant E2 product CH2=CH2 RCH=CH2 unsubstituted monoincreasing alkene stability RCH=CHR di- If RO– is bulky (CH3)3CO– vs. CH3O–; a higher percentage of the less substituted alkene results CH3O – H H H ROδ− H H R2C=CH2 di- CH2 31% + Br– C H becoming more sp2 CH3CH2CH2CH H C H CHCH3 R2CH=CHR tri- R2C=CR2 tetra- CH3CH CHCH3 + 80% (CH3)3CO – CH3CH CHCH3 50% CH3CH2CH CH2 20% + CH3CH2CH 50% CH2 Elimination Reactions Chapter 8-11 Chem 61 Stereochemistry of E2 SUMMARY Generally, in E2 reactions the proton and the leaving group must be in an anti orientation. This allows for the best overlap of the developing pi orbitals. E1 reactions result from carbocation intermediates and are competivive with SN1 reactions. E1 reactions may be accompanied by carbocation rearrangements. E1 reactions generally yield the most substituted (most stable) alkene. RO– H b b a C c – + ROH + L C Relative rates: 3° > 2°. d a d E1 reactions are not stereoespecific and yield a mixture of E,Z alkenes. c E2 reactions result from a concerted reaction and are competitive with SN2 reactions. L In order to predict the stereochemistry of the alkene resulting from an E2 reaction: a) rotate the C atoms so that the H to be removed by RO– and the leaving group are in the anti conformation b) the stereochemistry of the alkene resulting from the E2 reaction is as shown RO– Br H H C rotate C Ph CH3 Ph C Ph CH3 H C Ph H Ph Ph C CH3 Br C H Z In cyclohexane systems the H— and the leaving group must be trans and diaxial Cl RO– D H H D H E2 reactions generally produce the more stable alkene (except with bulky bases or leaving groups). E2 reactions are stereospecific and give anti elimination of H and L. Relative rates: 3° > 2° > 1°. Chapter 8-12 Substitution vs. Elimination Reactions Factors Governing Substitution and Elimination Reactions Structure of Alkyl Halide Leaving group attached to methyl or primary carbon: SN2 reaction observed unless Nu: is a strong base and elevated temperature employed; then observe E2. Leaving group attached to tertiary carbon: E2 reaction observed unless Nu: is weak base; then SN1 observed. Solvent nucleophilicity increases with increasing electron releasing capacity of molecule. If the solvent has low nucleophilic properties; i.e. H2SO4, H3PO4 then E1 reaction results CH3CH2OH > H2O > CH3CO2H > CF3CH2OH > CF3CO2H increasing solvent nucleophilicity Leaving group attached to secondary carbon: if the nucleophile is less basic than HO – (e.g. – CN, – N3, – SR) then the reaction will follow SN2 path If the Nu: is HO – or more basic (e.g. RO–, R2N–, RC≡C–), then the reaction will follow E2 path If the Nu: is a weak nucleophile (e.g. H2O, ROH, RCO2H) the reaction will follow SN1 path with some E1 The leaving group is attached to an allylic or benzylic carbon: SN1 Solvent Highly polar solvents (high dielectric constant) favor SN1, E1 reactions Concentration of Nucleophile or Base Increasing the concentration of the nucleophile has no effect on SN1, E1 but increases SN2,E2 rates. Chem 61 Temperature Increase in temperature increases the rates of all reaction types but has a larger effect on E2 reactions. Nucleophilicity Chapter 8-13 NUCLEOPHILICITY General comments Nucleophilic Constants of Various Nucleophiles Chem 61 Nucleophile n(CH3I)a pKa of conjugate acid 1. Note that nucleophilicity toward CH3I does not correlate directly with basicity. (N3–, PhO–, Br- are equivalent nucleophilicity but differ greatly in basicity. Also N3– and CH3CO2– are nearly identical in basicity but N3– is 30 times [1.5 log units] more nucleophilic). CH3OH NO3– F– CH3CO2– Cl–≠ (CH2)2S NH3 N3– PhO – Br – CH3O– HO– NH2OH NH2NH2 (CH3CH2)3N NC– I– HOO– (CH3CH2)3P PhS– PhSe– Ph3Sn– 0.0 1.5 2.7 4.3 4.4 5.3 5.5 5.8 5.8 5.8 6.3 6.5 6.6 6.6 6.7 6.7 7.4 7.8 8.7 9.9 10.7 11.5 -1.7 (CH3OH2) -1.3 3.45 4.8 5.7 2. Among neutral nucleophiles while (CH3CH2)3N is more basic than (CH3CH2)3P (pKa 10.7 vs. 8.69) the phosphine is 100 times (n = 8.7 vs 6.7) more nucleophilic. 9.25 4.74 9.9 -7.7 15.7 15.7 5.8 7.9 10.7 9.3 -10.7 8.69 8.5 an(CH3I) = log(knucleophile/kCH3OH) in CH3OH 25°C n = nucleophilic constant 3. Correlation of nucleophilicity with basicity is better if the attacking atom is the same. Thus CH3O– > PhO– > CH3CO2–> NO3– 4. Nucleophilicity usually decreases going across a row in the periodic table. (HO– > F– ; PhS–> Cl–). This order is determined by electronegativity. 5. Nucleophilicity usually increases going down the periodic table (I– > Br– > Cl– > F–; and PhSe– > PhS– > PhO–) Related to weaker solvation and greater polarizability of the heavier atoms. Nucleophilicity Chapter 8-14 Examples E1: 3° > 2° > 1° E2: 3° > 2° > 1° SN1: 3° > 2° > 1° SN2: 1° > 2° > 3° Nu:– 1° R CH CH2 L R H H hindered R strong base heat 3° CH CH2 Nu SN2 (inversion CH CH2 E2 (anti elimination of HL) NuH C C H L C C C + C H – Nu: acts as base C C –H C C E2 C C C H Nu+ H + – H+ C E1 C C H Nu S N1 H S N2 2° H C C H L – Nu: or base: E2 H H C + C C H Nu C H poor Nu or base C C – C SN1 or E1 C C C C C Chem 61 Alcohols Chapter 9-1 Alcohols are similar to water: contain sp3 hybridized oxygen, —OH functional group bonded to an sp3 carbon, with two lone pairs on oxygen H H .. O H CH3CH2 .. .. water O .. ethanol Alcohols can hydrogen bond thus their boiling points are higher than similar polar compounds which cannot hydrogen bond. Low molecular weight alcohols are soluble in water Alcohols contain a hydrophobic alkyl group and a hydrophilic hydroxyl group As the alkyl group increases in length and size, the solubility in water decreases Acidity and Basicity alcohols act as bases in the presence of strong acids CH3CH2 base ..O. . H CH3CH2 H+ ..O+ H H acid and as acids in the presence of strong bases (CH3)3C ..O. . acid H + K+H– .. .– + + H2 (CH3)3C O . .. K base CH3CH2O—H + Na0 CH3CH2O– Na+ + H2 Chem 61 Alcohols Chapter 9-2 IUPAC Names For alcohols drop —e from the alkane name and add —ol CH3CH2OH: ethan + ol = ethanol CH3CH2CHOHCH3: butan + ol; —OH on carbon 2: 2-butanol Classification Alcohols are classified as: methyl, primary, secondary, tertiary, allylic, benzylic CH3OH CH3CH2CH2CH2CH2CH2OH methyl primary 1° CH3CHOHCH3 secondary 2° CH2=CHCH2OH allylic CH2OH (CH3)3COH tertiary 3° benzylic REACTIONS OF ALCOHOLS Substitution Reactions: Reaction with Hydrogen Halides (HX) Alcohols do not undergo nucleophilic substitution by X– since HO– is a poor leaving group. Alcohols do undergo substitution by X – in acidic solution. Here the alcohol is protonated and H2O, a neutral species, is the leaving group. R .. OH .. H+ R .. + O H H X– R—X + H2O Chem 61 Alcohols Chapter 9-3 Chem 61 Reactivity of Hydrogen Halides The reactivity of hydrogen halides in alcohol substitution reactions is as follows: HF < HCl < HBr < HI pKa 3.45 -7 -9 -9.5 E The reactivity is explained since the acidity of HX increases and the nucleophilicity of X– increases going from HF to HCl to HBr to HI. + CH3CH2CH2 CH3CH2CH2 Reactivity of Alcohols Toward HX OH O H H progress of reaction methyl primary secondary tertiary allylic and benzylic increasing reactivity of ROH toward HX All alcohols react readily with HBr and HI; 3°, allylic and benzylic react rapidly with HCl 2° and 1° alcohols require the addition of ZnCl2 for rapid reaction with HCl Mechanism of Alcohol Substitutions Methyl and primary alcohols follow the SN2 mechanism Step 1 protonation of the alcohol CH3CH2CH2 OH H+ CH3CH2CH2 + O H H Step 2 SN2 dsiplacement of Water CH3CH2CH2 O+H H Br – S N2 CH3CH2CH2 Br + H2O CH3CH2CH2 Br Alcohols Chapter 9-4 Secondary and tertiary alcohols follow the SN1 mechanism Step 1 protonation of the alcohol CH3 CH3 C CH3 H+ CH2CH3 CH3 C + OH CH2CH3 OH2 Step 2 loss of water to for the carbocation CH3 CH3 C + CH2CH3 CH3 – H 2O CH3 OH2 C + CH2CH3 Step 3 attack of halide ion on the carbocation CH3 CH3 C + CH3 X– CH2CH3 CH3 S N1 C CH2CH3 I Rearrangement can occur R+ E R–O+H R–OH H R–X Chem 61 Conversion of Alcohols to Alkyl Halides Chapter 9-5 Other Reagents for the Conversion of Alcohols to Alkyl Halides R—OH + SOCl2 R—Cl + HCl + SO2 R—OH + PBr3 R—Br + HOPBr2 H H O CH3CH2 C OH Cl CH3 CH3CH2 C O+ S Cl CH3 Cl H S Cl Cl – H + CH3CH2 C OSOCl CH3 H R3N: O– H O CH3CH2 C O S Cl CH3 H Cl C CH3CH2 CH3 + SO2 Esters of Alcohols Reaction of alcohols with carboxylic acids in the presence of acid produces esters of carboxylic acids O CH3CH2CH2 C OH + CH3CH2CH2OH alcohol carboxylic acid H+ heat O CH3CH2CH2 ester C OCH2CH2CH3 Chem 61 Reactions of Alcohols Chapter 9-6 Inorganic esters mineral acids such as H2SO4, HNO3 and H3PO4 form esters with alcohols to produce important compounds H O H O H C ONO2 H C O P H C ONO2 H C H OH H C ONO2 H C H H nitroglycerin, a nitrate ester O P O OH CH3O OH S OCH3 O dimethyl sulfate, a sulfate ester H a diphosphate ester P-toluenesulfonates (tosylates) and methanesulfonates (mesylates) are excellent leaving groups in nucleophilic substitution reactions. They are readily prepared form an alcohol and the corresponding sulfonyl chloride. CH3OH + ClSO2CH3 O (CH3CH2)3N CH3O S CH3 + (CH3CH2)3N+HCl – O a methanesulfonate (mesylate) O OH + O CH3 S Cl O S O O p-toluenesulfonylchloride (tosyl chloride) cyclohexyl tosylate Reaction of sulfonates with nucleophiles OTs CH3CH2CH2O– Na+ OCH2CH2CH3 CH3 Chem 61 Dehydration of Alcohols Chapter 9-7 Chem 61 Examples: Elimination (Dehydration) of Alcohols CH2 CHCH2CH3 Alcohols undergo elimination much like alkyl halides CHCH2CH3 CH H+ OH Tertiary alcohols readily undergo dehydration by an E1 pathway Secondary alcohols also follow an E1 path, but primary alcohols probably eliminate by an E2 mechanism OH conc. H2SO4 (CH3)3COH ease of dehydration (CH3)2CHOH CH3CH2CH2OH 60° C conc. H2SO4 100° C conc. H2SO4 CH3CH=CH2 +H2O CH3 CH3 C CH3 CH3 C CH3 OH CH3 C C+ H CH3 C C CH3 CH3 H CH3 H+ – H+ O+H2 CH3 CH3 C C CH3 H CH3 CH3 C C CH3 H C C CH3 CH3 R+ ROH2+ ROH progress of reaction alkene CH3 CH3 CH3 C C CH3 H CH3 + C 1,2 shift CH3 CH3 CH3 The most stable alkene predominates in dehydration reactions. Rearrangements can occur. E OH + CH3 CH3 H+ CH3 – H 2O CH3 C CH3 CH3 CH3CH=CH2 +H2O Mechanism: CH3 heat CH3 180° C H CH3 H2SO4 CH CH3 (CH3)2C=CH2 +H2O C CH3 CH3 O+H2 – H 2O CH3 C H CH3 – H+ Dehydration of Alcohols Chapter 9-8 ETHERS, EPOXIDES AND SULFIDES Ethers are derivatives of water where both hydrogens have been replaced by an alkyl group. Ethers are less polar than alcohols and water and are not capable of hydrogen bonding to themselves because of the lack of an —OH group. The boiling points of ethers are also much lower than alcohols of comparable molecular weight. O CH3CH2OCH2CH3 O diethyl ether CH2 tetrahydrofuran CH2 ethylene oxide Preparation of Ethers Williamson Ether Synthesis (SN2 Reaction of an Alkoxide with an Alkyl Halide) R1O– + R2—X S N2 R1—O—R2 + X– Best results are obtained if the alkyl halide is methyl or primary (2° and 3° give mostly elimination) There are few limitations on the alkoxide CH3CH2CH2O – + (CH3)2CHCH2—Br O– CH3CH2CH2O—CH2CH(CH3)2 OCH2CH3 + CH3CH2—I (CH3)3CO – + CH3—I (CH3)3CO—CH3 Chem 61 Chapter 9-9 Reactions of Ethers and Epoxides Substitution Reactions of Ethers Ethers are relatively unreactive compounds, but they do undergo substitution reaction when heated with hydrogen halides, particularly HI and HBr. This is a very similar reaction to the reaction of alcohols with HX Br – .. .. H—Br CH3CH2CH2—O—CH2CH2CH3 + CH3CH2CH2—O—CH2CH2CH3 H HBr CH3CH2CH2—Br CH3CH2CH2—Br + CH3CH2CH2—OH OCH2CH(CH3)2 OH HI + I—CH2CH(CH3)2 unreactive with HI Reactions of Epoxides Epoxides are strained much like cyclopropanes because their bond angles (60°) are far removed from the normal tetrahedral angle (109.5°). The orbitals have poor overlap and the bonds are weakened. The C—O bond is also polarized and consequently, epoxides are highly reactive compounds. Base Catalyzed Cleavage of Epoxides Alkoxides: O CH3 C CH3 H CH3O – H CH3OH – O CH3 C C H 3C H C H OCH3 HO HOCH3 CH3 C H 3C CH3O – nucleophile attacks at the least hindered carbon in an SN2 reaction H C H OCH3 Chem 61 Chapter 9-10 Reactions of Epoxides Grignard Reagents O H C H BrMgO CH3MgBr H C H H H HO H+ C C H H CH3 H C H CH3–MgBr+ Acid Catalyzed Cleavage + CH3 O H H C C H CH3OH CH3 CH3OH CH3 C C H CH3O + H – H+ H + O CH3 δ+ C CH3 H C H .. CH3O—H .. CH3OH CH3 C CH3O C H H H Nucleophile attacks at the most hindered carbon since the carbon is partially positive and the C—O bond is partially broken H + O OH H H H H H 2O OH trans diaxial opening in cyclohexanes H C H CH3 Chem 61 Chapter 9-11 Crown Ethers and Thiols Crown Ethers Crown ethers are macrocyclic ethers with repeating –OCH2CH2— units. Depending on the ring size, they effectively chelate alkali metal ions such as K+, Na+ or Li+ O O O O O Na+ O K+ O O O O O 12-crown-5 18-crown-6 Thiols and Sulfides The sulfur analog of an alcohol is a thiol, or mercaptan. Thiols are stronger acids than alcohols (pKa = 8, ROH = 16). Thiols form weaker hydrogen bonds than alcohols due to the lower electronegativity of sulfur. Thiols and thioethers (sulfides) are prepared by substitution reaction in the same way that alcohols and ethers are prepared. Br SH HS– CH3CH2CH2S– SCH2CH2CH3 I Disulfides are formed by the oxidation of thiols and are an important structural feature in some proteins. RSH I2 or K3Fe(CN)6 RS—SR a disulfide Chem 61 Chapter 9-12 Organometallic Compounds Grignard Reactions Grignard reagents are organometallic reagents derived from an alkyl halide and magnesium diethyl ether R—X + Mg Rδ−—Mgδ+X Grignard reagent Since the carbon carries a partial negative charge, the carbon is a strong base and a good nucleophile. CH3CH2—Br + Mg diethyl ether CH3CH2δ−—Mgδ+Br HOH CH3CH2—H Because carbonyl pi bonds are polarized, they can undergo a reaction called nucleophilic addition: the addition of a nucleophile to an electron deficient pi bond. R1 O– Oδ− O– C+ Cδ+ C R1 R1 R1 R1 Nu Nucleophilic Addition R1 Nu: O– Oδ− O– MgX+ C+ Cδ+ C R1 R1 R1 R2 R1 R1 R2 Mg-X A Grignard reaction with 1. formaldehyde produces a primary alcohol O 1. CH3 C H H OH Mg-X 2. H2O, H+ H C CH3 H R1 Nucleophilic Addition Chem 61 Chapter 9-13 Organometallic Compounds 2. an aldehyde produces a secondary alcohol O OH 1. (CH3)CHMgBr C CH3CH2 H 2. H2O, H+ H C CH2CH3 CH(CH3)2 3. a ketone produces a tertiary alcohol O C CH3CH2 1. (CH3)CHMgBr + CH3 2. H2O, H OH CH3 C CH2CH3 CH(CH3)2 4. an ester produces a tertiary alcohol (addition of two molecules of Grignard reagent) O OH 1. 2 CH3MgBr C OCH3 2. H2O, H+ CH3 C CH3 5. ethylene oxide produces a primary alcohol O 1. C6H5MgBr 2. H2O, H+ OH Chem 61 Alkynes: Structure and Bonding Chapter 10-1 sp Hybridization sp hybridized carbons are linear with atoms 180° apart 2p 2p 2p 96 kcal 2s 1s sp hybridize 2s 1s 1s ethylene: trigonal planar – + 180° apart; the remaining two 2p orbitals are 90° to the sp and each other + 1-2p 2s 2-sp's Acetylene: linear; bond angles 180° C=C bond length = 1.20Å acetylene has two perpendicular pi bonds and one sigma bond H H H 2-sp's H C H acetylene σ-orbital H 2-2p's on each carbon combine to form pi-orbitals E C C—C σ* C—C π* C—C π C—C σ H C C H acetylene π-orbitals Chem 61 Alkynes: Electrophilic Addition Chapter 10-2 Reaction of Alkynes with E+A– Reagents Reaction of alkynes with E+A– reagents proceed in the same manner as alkenes except different intermediates are possible R C C R E+ + C R C E vinyl carbocation R E+ R C bridged intermediate C R For H—X the vinyl carbocation is more stable than the bridged intermediate. Thus: R C C R E+ + C R C E vinyl carbocation R E+ R C bridged intermediate C R secondary vinyl carbocation is about the same stability as a primary carbocation Thus the order of carbocation stability is triphenyl methyl > diphenylmethyl > 3°≈ benzyl ≈ allyl > 2° > 1° ≈ 2° vinyl >> methyl Chem 61 Alkynes: Electrophilic Addition Chapter 10-3 For H–X CH3 C C H CH3 HCl C H C H Cl CH3 C CH3 CH3 C C + H Cl CH3 HCl C CH3 C Cl H C CH3 For Hg(OCOCH3)2: CH3CH2 Hg(OCOCH3)2 C2H5C≡CC2H5 HgOCOCH3 C C CH2CH3 CH3CO2 E For Cl2, Br2: X+ X2 R—C≡C—R C C6H5—C≡C—C6H5 X2 + C C6 H5 X C C R but, R R X R X X C C C 6H 5 C X C6 H5 C C 6H 5 + X C 6H 5 C C6 H5 C X Thus the choice of intermediate depends on structure; alkyl groups tend to favor the bridged ion; groups such as phenyl which stabilize the free carbocation tend to proceed via the vinyl carbocation. Chem 61 Alkynes: Electrophilic Addition Chapter 10-4 Addition of Water With alkynes this electrophilic addition reaction generates a vinyl alcohol (also called an enol). Hg(II) ion is often used. R—C H2SO4 C—R Hg(II) H 2O OH O R—C CH—R enol H +OH R—C C—R H2O: R—C R—C CH2—R keto C—R H2O: Hg2+ Hg2+ H+ R—C OH C—R H O R—C C—R Hg2+ Hg2+ H O R—C C—R OH C—R R—C H H Hydroboration H R—C C—H BH3 R—C BH2 CH H OH R—C CH enol H O R—C H C–H keto (aldehyde) Chem 61 Alkynes: Acidity Chapter 10-5 H C + base C C – .. C H pKa = 45 Chem 61 HF + base—H H pKa 3.2 H 2O 15.7 HC≡CH 25 NH3 36 H2C=CH2 H3C–CH3 44 50 increasing acid strength C C + base H C . C. – + base—H F– pKa = 25 The relatively high acidity of the alkyne —C≡C—H bond is associated with the large degree of s character in the sp C—H bond (50% compared with 33% in sp2 bonds). The carbon atom is more electronegative in the sp state; thus the C—H bond is more acidic. The acetylide ion may be formed by such strong bases as —:NH2 (pKa 33), RMgX or RLi (pKa 45-50). R C C H NaNH2 R NH3 R CH CH2 C C:– Na+ + NH3 acetylide ion NaNH2 No reaction NH3 Electronegativities Electronegativities acid strength pKa HC≡CH H2C=CH2 sp 2 > sp > N < O < F NH3 < H2O < HF 36 15.7 3.2 H3C–CH3 sp3 HO– HC≡C– NH2– H2C=CH– increasing base strength H3C–CH2– Alkynes: Acetylides Chapter 10-6 SN2 reaction with acetylide ion NH3 – + R'—C≡C: Na R' C + R—CH2—C≡C—R' R—CH2—L O C:– MgBr+ + R' C—CH2—CH2—O – MgBr+ C H+ R' C C—CH2—CH2—OH Nucleophilic addition reaction with acetylide ion. O – MgBr+ O R' C C:– MgBr+ R C H R' C R C C H CH3CH2MgBr R' C H+ OH C—H R' C C C H O R' C – C: MgBr + + O – MgBr+ C C R' H+ OH C C R' R Chem 61 Spectroscopy Chapter 12-1 Infrared and Nuclear Magnetic Resonance Spectroscopy electromagnetic radiation: energy that is transmitted through space in the form of waves wavelength: (λ): the distance from the crest of one wave to the crest of the next wave frequency: (ν): the number of complete cycles per second ν= c λ where c = speed of light Electromagnetic radiation is transmitted in particle-like packets called photons or quanta. The energy is inversely proportional to the wavelength and directly proportional to frequency. Ε= hc λ where c = speed of light; h = Planck's constant Ε = hν ultraviolet visible h = Planck's constant infrared radio decreasing energy Absorbtion of ultraviolet light results in the promotion of an electron to a higher energy orbital. Absorbtion of infrared results in increased amplitudes of vibration of bonded atoms. Intensity of radiation is proportional to the number of photons. Chem 61 Infrared Spectroscopy Chapter 12-2 Infrared Spectroscopy Infrared is recorded as %T versus wavelength or frequency When a sample absorbs at a particular wavelength or frequency, %T is reduced and a peak or band is displayed in the spectrum. Infrared is recorded as %T versus wavelength or frequency When a sample absorbs at a particular wavelength or frequency, %T is reduced and a peak or band is displayed in the spectrum. 100 %T 0 frequency Nuclei of bonded atoms undergo vibrations similar to two balls connected by a spring. Depending on the particular atoms bonded to each other (and their masses) the frequency of this vibration will vary. Infrared energy is absorbed by molecules resulting in an excited vibrational state. Vibrations occur in quantized energy levels and thus a particular type of bond will absorb only at certain frequencies. Both stretching and bending vibrations can be observed by infrared. O CH3 CH3 stretching O CH3 CH3 bending Chem 61 Infrared Spectroscopy Chapter 12-3 Interpretation of Infrared Spectra Chem 61 C—C and C—H Bonds Correlation tables Infrared spectra of thousands of compounds have been tabulated and general trends are known. Some common functional groups are shown below. C=O str OH and NH str CH str C—O str C=N str C≡N str C=C str NH bend C—N str C—C str sp3 C—C sp2 C=C sp2 C—C (aryl) sp C≡C sp3 C—H sp2 C—H sp C—H C(CH3)2 weak, not useful 1600-1700 cm–1 1450-1600 cm–1 2100-2250 cm–1 2800-3000 cm–1 3000-3300 cm–1 3300 cm–1 1360-1385 cm–1 (two peaks) CH bend OH bend 3500 3000 2500 2000 1500 1000 800 Alcohols and Amines Carbonyls One of the most useful absorbtions in infrared 1640-1820 cm-1 O—H or N—H C—O or C—N Ketones (saturated) C=O Ethers 1640-1820 cm–1 C—O Aldehydes C=O; C—H(O) 1640-1820 cm Carboxylic acids C=O; C(O)—OH 1640-1820 cm–1 Esters C=O ; C(O)—OR 1640-1820 cm–1 –1 2820-2900 and 2700-2780 cm–1 (weak but characteristic) 3330-2900 cm–1 1100-1300 cm–1 3000-3700 cm–1 900-1300 cm–1 1050-1260 strong Nuclear Magnetic Resonance Spectroscopy Chapter 13-1 Nuclear Magnetic Resonance (NMR) Spectroscopy Some atomic nuclei (1H, have a nuclear spin. 13 C, others) behave as if they are spinning...they Spinning of a charged particle creates a magnetic moment. If an external magnetic field is applied, these small magnetic moments (of the nuclei) either align with the field (α) or against the field (β), about 50% with and 50% against the field at any one time. β Ho ∆E hν α β Ho ∆E α Ho = the external magnetic field Resonance: the flip of the magnetic moment from parallel to antiparallel to the external magnetic field. Irradiation at the frequency equal to the energy difference, ∆E, causes resonance. ∆E depends on the external magnetic field. Protons (or other nuclei) in different magnetic environments resonate at different field strengths. A proton which resonates at a higher field is in a stronger magnetic environment or shielded. A proton which resonates at a lower magnetic field is said to be deshielded. Different magnetic environments are created by different electron densities in the vicinity of a proton. Chem 61 Nuclear Magnetic Resonance Spectroscopy Chapter 13-2 Chem 61 Adjacent electron withdrawing groups, highly electronegative atoms, or the hybridization of the carbon to which the proton is bonded can alter the magnetic environment. The pi system of benzene creates a magnetic field or ring current which deshields the protons attached to the ring. The local electrons create a small electric and magnetic field around a proton and shield it. Similarly, pi electrons in a C=O bond create a field which deshields the proton bonded to the C=O of an aldehyde. This is also affected by the inductive effect of the C=O. The more electron density present around the proton, the greater the field and the greater the shielding. Resonances are reported in chemical shifts (δ) downfield from tetramethylsilane (TMS) (CH3)4Si. δ= distance from TMS in Hz MHz of spectrum ppm In methyl halides, the more electronegative the halogen, the more deshielded the protons on the methyl. This is because F is inductively more electron withdrawing, causing the carbon to be more positive and thus pulling more electrons away from the hydrogen and causing it to be less shielded. In methyl halides, the more electronegative the halogen, the more deshielded the pr H3C—F δ H3C—Cl 3.0 4.3 H3C—Br H3C—I 2.7 2.1 Pi electron effects Magnetic fields created by pi electrons are directional and said to have an anisotropic effect. R Ho C H O H H deshielded H deshielded Nuclear Magnetic Resonance Spectroscopy Chapter 13-3 Equivalent and Nonequivalent Protons Protons that are in the same magnetic environment are equivalent and have the same chemical shifts. Protons in different magnetic fields are nonequivalent and have different chemical shifts. Magnetic equivalence is usually the same as chemical equivalence. Equivalence can be established by symmetry operations such as rotation, mirror planes and centers of symmetry Chemically equivalent protons have the same chemical shifts. To determine if protons are chemically equivalent, replace one by a different group, e.g. D or Br. Then replace a different one by the same group and compare the two compounds. If they are identical, the protons are equivalent. H H H C C H H OH H H H H C C C H Cl H H equivalent, but not to CH3 protons all six are equivalent equivalent Equivalent protons can be on different carbons. Protons which are homotopic or enantiotopic resonate at the same chemical shift in the NMR. If protons are interconverted by rotation about a single bond, they will average out on the NMR time scale and a single resonance will be observed. ClH2CCH2Cl anti and gauche forms rapidly interconvert and a single resonance is observed. Axial and equatorial hydrogens in cyclohexane average to a single peak because of rapid ring inversion. Diastereotopic hydrogens are chemically nonequivalent and thus give different chemical shifts in the NMR Chem 61 Nuclear Magnetic Resonance Spectroscopy Chapter 13-4 Intergration The spectrometer can integrate and determine the relative number of hydrogens associated with each resonance in the NMR spectrum by determining the area under the peaks. Spin-Spin Coupling for example... 3 3 CH3CH2OCH3 2 TMS If a proton (Ha) is bonded to a carbon which is bonded to a carbon that has one proton (Hb), Ha will appear as a doublet Since in half the molecules, Hb will be in the α state and in half will be in the β state, Ha will experience two different magnetic fields and two peaks (a doublet) will appear for Ha. Ha without an adjacent hydrogen For one adjacent hydrogen α or β Ha with Hb adjacent in theβ state Ha with Hb adjacent in the α state Chem 61 Chapter 13-5 Nuclear Magnetic Resonance Spectroscopy For two adjacent hydrogens: Hb, Hc At any one time Hb or Hc could be in the α or β state (50:50) thus 4 combinations for Hb, Hc exist: αbαc αbβc βbαc βbβc gives 1:2:1 triplet When both Hb and Hc are α, a different field is observed than if both are β or one is α and one is β. When one is α and one is β, the field is the same. That is, βbαc and αbβc produce the same field and a single signal for Ha is observed with twice the intensity. Thus three signals are observed in a 1:2:1 ratio: a so-called triplet For three adjacent protons: ααα ααβ αββ βββ 1:3:3:1 quartet αβα βαβ βαα ββα Thus the splitting pattern of a particular proton or equivalent protons will be a pattern with n+1 lines where n is the number of adjacent equivalent protons. singlet 0 neighboring protons doublet 1 neighboring protons triplet 2 neighboring protons quartet 3 neighboring protons quintet 4 neighboring protons sextet 5 neighboring protons septet 6 neighboring protons The separation of the peaks in a splitting pattern is called the coupling constant, J. Chem 61 Nuclear Magnetic Resonance Spectroscopy Chapter 13-6 Splitting Diagrams Splitting patterns for protons can be constructed in diagram form by starting with one line to represent the unsplit proton resonance. If an adjacent proton Hb affects Ha it is split into a doublet; if another equivalent proton to Hb is present, each line of the double will be split into a doublet, since the coupling constant J is the same, the two center lines overlap and a only three lines are observed with the center line twice the height. This can be repeated for additional adjacent protons. Ha without an adjacent hydrogen 1 1 1 splitting diagram Ha split by one adjacent hydrogen 1 2 Ha split by a second adjacent hydrogen Ha split by a third adjacent hydrogen 1 3 3 1 Chemical Exchange and Hydrogen Bonding CH3OH, methanol would be expected to give an NMR spectrum of a doublet for the CH3 and a quartet for the OH. For a dilute sample at -40° in CCl4 this is the case. If the NMR spectrum is run at 25° as a more concentrated sample only two singlets are observed. This is because the intermolecular hydrogen bonding in methanol allows the rapid exchange of the OH proton from one CH3OH molecule to another, effectively averaging the spin states of the OH proton and resulting in no change in the magnetic field due to the OH. Amines and other compounds which can undergo hydrogen bonding can also show this effect. Thus the NMR spectra of alcohols, amines and carboxylic acids are temperature, concentration and solvent dependent. Chem 61 Nuclear Magnetic Resonance Spectroscopy Chapter 13-7 CHEMICAL SHIFTS Functional Group Shift,δ Primary alkyl, RCH3 Secondary alkyl, RCH2R Tertiary alkyl, R3CH 0.8-1.0 1.2-1.4 1.4-1.7 Allylic, R2C=C—CH2R 1.6-1.9 Benzylic, ArCH2R Iodoalkane, RCH2I Bromoalkane, RCH2Br Chloroalkane, RCH2Cl Ether, RCH2OR Alcohol, RCH2OH Ketone, RCH2C(=O)R 2.2-2.5 3.1-3.3 3.4-3.6 3.6-3.8 3.3-3.9 3.3-4.0 2.1-2.6 Aldehyde, RCH(O) Terminal alkene, R2C=CH2 Internal alkene, R2C=CHR Aromatic, Ar—H Alkyne, RC≡C—H Alcoholic hydroxy, ROH Amine, RNH2 9.5-9.6 4.6-5.0 5.2-5.7 6.0-9.5 1.7-3.1 0.5-5.0 (variable) 0.5-5.0 (variable) Chem 61 CHEMISTRY 61 Exam 1 Dr. M.T. Crimmins September 15, 1998 Name___________________________________________ Pledge: I have neither given nor received aid on this exam. Signature________________________________________ I. Nomeclature (12 points) Give the IPUAC name for the following compounds: Indicate R, S, cis, trans, E, or Z where appropriate. 1. CH 3 CH 2CH 2CH 2CH 3 CH 2 C H C H CH 2 CH 2 C H CH 3 CH 3 CH 3 ______________________________ 2. H 3C H C C CH 3 CH 2CH 3 ______________________________ 3. CH 3 H ______________________________ 4. CH(CH3)2 ______________________________ II. A. Write valid Lewis structures for the following species. Show all nonbonding (unshared) electrons and indicate any formal charges . (6 points). 5. [H2COH]+ 6. IO4 – 7. Give the hybridization of the indicated atoms in the species below (6 points) H 3C C N: H 3C N .. CH 2 1 O H 3C C O CH 3 8. Draw three structural isomers for C3H6O. Indicate what type of functional group is represented by each compound (e.g. carboxylic acid) (6 points). 9. Draw all the possible stereoisomers of 3-bromo-2-butanol. Indicate if they are chiral, meso or achiral and indicate their relationship to each other. (i.e. enantiomers, diastereomers) (8 points) 10. Draw both chair conformations of trans-1,2-dimethylcyclohexane. If one is more stable than the other, circle it. (6 points) 11. Draw an energy diagram for one 360° rotation about the C3-C4 C–C bond of hexane. Also draw a Newman Projection of the most stable conformation. (8 points). 12. Circle the molecule(s) which have a permanent dipole. In those which have a permanent dipole, show the direction of the overall dipole. (6 points) O H C H Cl Cl B Cl 2 H 3C O H 13. In the space to the right, indicate if each of the pairs of molecules below are identical compounds, enantiomers, diastereomers, structural isomers, or conformational isomers. (9 points). CH 3 CH 3 H 3C H 3C CH 3 a. Br H H OH H CH 3 C C H 3C H ________________________ Br H H OH b. c. CH 3 ________________________ H 3C CH 3 C C H H ________________________ 14. What two effects cause cyclobutane and cyclopropane to be higher in energy than cyclohexane? (3 pts) 15. Label the species below as Lewis Acids or Lewis Bases (4 points) Br + H3CO– _____________ _____________ 16. Circle the following which has the highest heat of combustion per CH2 unit. (4 points) a. cyclopentane b. cyclopropane c. cyclohexane d. cyclobutane 17. Indicate the geometry of carbon in the molecules below (e.g. trigonal bipyramidal). (6 points) a. CH3CH3 b. H2C=O c. HC≡CH ____________ ______________ ______________ 3 18. Circle the statement(s) which are true of enantiomers. (4 points) a. They have a non-superimposable mirror image. b. They have no asymmetric carbon atoms. c. They are chiral. d. They do not rotate the plane of polarized light. 19. Draw an energy diagram of the molecular orbitals of the C=C bond of ethylene (H2C=CH2) and label them (e.g. σ) and indicate their relative energies. Indicate the ground state electronic configuration of the C=C electrons. (6 points) 20. What kind of molecular orbital results (σ, σ*, π, π*) results when the pairs of orbitals show below are combined in the indicated manner? (6 pts) + a. + + c. b. 4 CHEMISTRY 61 Exam 1 Dr. M.T. Crimmins September 21, 1999 Name___________________________________________ Pledge: I have neither given nor received aid on this exam. Signature________________________________________ I. Nomeclature (12 points) Give the IPUAC name for the following compounds: Indicate R, S, cis, trans, E, or Z where appropriate. 1. CH 3CH 2 CH 2CH 2CH 2CH 3 CH 2 C H C H C H CH 2 C H CH 3 CH 3 CH 2CH 3 CH 3 ____________________________ 2. C(CH3)3 CH 3 _____________________________ 3. H 3C CH 2CH 3 C CH 2CH 2CH 3 H _____________________________ 4. OH _____________________________ 5. A. Write valid Lewis structures for the following species. Show all nonbonding (unshared) electrons and indicate any formal charges . (6 points). CH3– CH2N2 6. Give the hybridization of the indicated atoms in the species below (6 points) H 2C C C(CH3)2 CH 3 H 3C N .. CH 3 1 O H 3C C O CH 3 7. Write structures for the each of the following having a molecular formula of C4H8O (6 points). a. a n aldehyde b. a cyclic alcohol c. an ether 8. What intermolecular forces exist between molecules of each of the following. (6 points). a. CH3CH2CH2CH2CH3 .. CH 3 S CH 3 _________________________ O _________________________ c. CH3CH2OH _________________________ b. 9. Draw all the possible stereoisomers of 3,4-dibromohexane. Indicate if they are chiral, meso or achiral and indicate their relationship to each other. (i.e. enantiomers, diastereomers) (8 points) 10. Label the following molecules as chiral or achiral. OH HO OH H H _____________ H CH3 CH2OH ______________ C CH2CH3 CH3 ______________ 11. Draw both chair conformations of trans-1,2-dimethylcyclohexane. If one is more stable than the other, circle it. (6 points) 2 12. Draw an energy diagram for one 360° rotation about the C2-C3 C–C bond of butane. Also draw a Newman Projection of the most stable conformation. (8 points). 13. Circle the molecule(s) which have a permanent dipole. In those which have a permanent dipole, show the direction of the overall dipole. (6 points) 14. In the space to the right, indicate if each of the pairs of molecules below are identical compounds, enantiomers, diastereomers, structural isomers, or conformational isomers. (9 points). CH 3 CH 3 H 3C CH 3 H 3C H 3C CH 3 a. CH 3 CH 3 H 3C CH 3 CH 3 ________________________ Br H H OH H OH b. c. Br H H CH 3 C C H 3C H ________________________ H 3C CH 3 C C H H ________________________ 15. What effect(s) cause cyclobutane and cyclopentane to be non-planar? (3 pts) 3 16. Label the species below as Lewis Acids or Lewis Bases (4 points) H3C+ H2C=CH2 _____________ _____________ 17. Indicate the geometry of carbon in the molecules below (e.g. trigonal bipyramidal). (6 points) CH 4 ____________ CH 3 H 3C C CH 3 H 2C C CH 2 ______________ ______________ 18. Draw and label the atomic orbitals which combine to form the molecular orbitals of formaldehyde, H2C=O. . (6 points) 19. What kind of molecular orbital results (σ, σ*, π, π*) results when the pairs of orbitals show below are combined (mathematically) in the indicated manner? (6 pts) – a. + + c. b. 4 CHEMISTRY 61 Exam 2 Dr. M.T. Crimmins October 22, 1998 Name___________________________________________ Pledge: I have neither given nor received aid on this exam. Signature________________________________________ I. REACTIONS: Predict the major organic products of the following reactions. INDICATE STEREOCHEMISTRY AS NEEDED. (4 points each) 1. CH 3 HI 2. HBr CH 2 peroxide 3. H C H 1. BH3 CH 2CH 3 2. H2O 2, NaOH C H 4. CH 3 H2, Pd/C CH 3 5. H 3C H 3C C C CH 2 H 3C + CH 2 H C C H heat CO 2 CH 3 6. H 3C C C CH 3 Cl2 1 equivalent 7. H 2C H C C H HBr, -80°C CH 2 8. CH 3 1. Hg(OAc)2, H2O 2. NaBH4 1 II. Multiple Choice: Place the letter in the blank and Circle the best answer (only one). (4 points each) ___9. The rate limiting step for hydration of an alkene with water and acid is a. b. c. d. protonation of the alkene by a strong acid addition of water to a carbocation to form the protonated alcohol loss of a proton from the protonated alcohol to form the alcohol. simultaneous addition of H+ and HO – to the alkene. ___10. Which of the following free radicals is the most stable? a. c. c. CH 3 . H 3C C d. CH 3 H H 3C . C CH 3 CH 3 CH 3 H . C H 3C H . C ___11. Which of the following indicated hydrogens is the most acidic? a. CH3CH=CH2 c. CH3CH2CH3 b. CH3C≡C–H d. H–CH2CH2OCH3 ___12. In the addition of HBr to 1,3-butadiene the 1,2 product predominates at -80°C while the 1,4 product predominates at 40°C. The 1,4 product is said to result from a. b. c. d. kinetic control. thermodynamic control. a Diels Alder reaction. the s-cis diene. ___13. Which of the following alkenes would have the lowest heat of hydrogenation? a. b. c. d. H H 3C CH2 CH3 C H 3C C H3C CH2 CH3 H 3C C CH H2C CH3 H C CH2 H 3C C CH 3 CH C CH CH H3C CH3 CH3 H H3C CH 2 ___14. What is the stereochemical relationship of the products of the following reaction? H H 3C a. b. c. d. e. C C H Br2 CH 2CH 3 diastereomers enantiomers identical (only one stereosiomer of the product is formed). cis-trans isomers conformational isomers ___15. Which of the carbocations below is the most stable? a. b. H c. H H C+ CH 3 C+ CH 3 2 d. + CH 2 C CH3 C+ CH 3 ___16. What is the hybridization of the positively charged carbon in the carbocation below? + CH 2 C a. sp3 V. b. sp2 c. sp d. s Mechanisms. Give a stepwise, detailed mechanism with arrows and intermediates for the following reactions. Be sure to account for stereochemistry as needed. (6 points each) 17. H3 C H H C H3 C Br2 H C CH3 C Br CH3 C H Br 18. CH3 C CH2 CH3 X+ Y– Y X CH3 C CH2 CH3 Syntheses: Give reagents to carry out the transformations below. (5 points each) 19. H H C H ClCH 2–CH 2Cl C H 20. O H 3C C C H H 3C 3 C CH 2CH 3 21. Consider the energy diagram below and answer the questions using the letters on the diagram. (10 points). B D C E N E R G Y F E M J H K N A L G PROGRESS OF REACTION a. What point(s) in the diagram represent transition states? _______________ b. What point(s) in the diagram represent intermediates? _______________ c. What is the energy of activation for the reaction? _______________ d. What is the rate-limiting step in the reaction? _______________ e. Is the reaction endergonic or exergonic? _______________ f. What is the free energy change of the reaction? _______________ g. Does G or C form faster from E? _______________ 22. Draw the HOMO (highest occupied molecular obrital) and the LUMO (lowest unoccupied molecular obrital) of 1,3-butadiene and label them. Circle the one which would interact favorably with the ethylene orbital below in a Diels-Alder reaction. (4 points) 4 CHEMISTRY 61 Exam 2: October 21, 1999 Dr. M.T. Crimmins Name___________________________________________ Pledge: I have neither given nor received aid on this exam. Signature________________________________________ I.REACTIONS: Predict the major organic products of the following reactions. INDICATE STEREOCHEMISTRY AS NEEDED. 1. CH3 1. Hg(OAc)2, H 2O 2. NaBH4 2. CH3 CH2CH3 1. BH3 CH3 2. H2O2, HO – C C H 3. HCl (1 equiv) CH3CH2 C C H 4. CH3 Br2 H 5. CH3CH2 H2 C C CH2CH3 poisoned catalyst (Pd/BaSO4) 6. H3 C Br2, H2 O C CH2 H3 C 7. H C C CH3 HBr, peroxide H 8. H3 C H2O, H+ C CH2 H3 C 9. CH3CH2 C C H NaNH2 CH3CH2Br 1 (4 points each) II. Multiple Choice: Circle the best answer (only one). (3 points each) 10. Which of the following alkenes is the most stable? CH3 CH3 b. a. c. CH3 11. CH3 d. CH3 CH3 CH3 Which of the following is the least stable carbocation? a. H2C=CHCH2+ b. (CH3)3C+ 12. CH2 c. C6H5(CH3)2C+ d. CH3CH2+ In the following reaction what is the relationship of the products formed? Br2 H CH3 a. enantiomers b. meso compound 13. The carbon -carbon triple bond of an alkyne is composed of_________ a. b. c. d. 14. two σ bonds and one π bond three σ bonds one σ bond and two π bonds three π bonds The free energy of reaction is a. b. c. d. e. 15. the difference in energy between the reactants and an intermediate in the reaction the difference in energy between the reactants and the transition state the difference in energy between the reactants and the products the difference in energy between the transition state and the products the difference in energy between the intermediate and the products What is the hybridization of the positively charged carbon in H3C+ a. p b. sp2 16. e. d2sp3 f. s c. sp d. sp3 A secondary cation is more stable than a primary carbocation because of a. b. c. d. 17. c. structural isomers d. diastereomers overlap of a filled p orbital with an adjecent σ* antibonding orbital overlap of an empty p orbital with adjacent σ bonding orbitals resonance deduction Which of the following free radicals is the most stable? a) H3 C . C H CH3 b) . c) CH3 H3 C . C CH3 CH3 2 d) H . C H CH3 18. Which of the following is not true of H2C=CH2? a) b) c) d) It contains 5 σ bonds. It has bond angles of 120°. All the atoms are in the same plane. It has free rotation about the C=C bond. Syntheses: Give reagents to carry out the transformations below. (4 points each) 19. CH 3 C CH2 OCH3 CH 3 CH 3 C CH3 CH3 20. H3 CC C–H CH3CH2CH2CH2CH3 21. O HC 22. CH3CH2 C CH3 Draw an energy diagram for the hypothetical exergonic reaction below where B is an unstable intermediate. Label the positions for A, B, and C on the diagram and indicate the energy of activation on the diagram. (5 points). A 23. CCH 2CH3 fast slow B C Draw resonance structures (4 pts each) for a) benzyl cation b) acetate ion (CH3CO2–) 3 V. Mechanisms. Give a stepwise, detailed mechanism with arrows and intermediates for the following reactions. (4 points each) 24. CH3 H2 O, H+ CH3 H3 C C C CH2 H CH3 CH3 OH CH3 25. H3 C H H C C Br2 H3 C H3 C CH3 H Br Br H 26. H3 C CH3 HBr C C CH CH3 CH2 H3 C H3 C C Br 4 CH3 CHEMISTRY 61 Exam 3 Dr. M.T. Crimmins November 24, 1998 Name___________________________________________ Pledge: I have neither given nor received aid on this exam. Signature________________________________________ I. Multiple Choice: Place the letter in the blank and Circle the best answer (only one). (4 points each) ___1. The rate limiting step for free radical halogenation is a. b. c. d. initiation hydrogen atom abstraction from carbon by the halogen radical attack of carbon radical on molecular halogen termination ___2. Which of the following are "concerted" reactions? a. SN1 b. SN2 c. E1 d. E2 e. SN1 and E1 f. SN2 and E2 ___3. Reaction of a strong base with a tertiary alkyl halide is most likely to result in: a. no reaction b. E2 elimination c. SN1 substitution d. E1 elimination ___4. Which of the following statements is correct? a. Free radical bromination is more selective than chlorination because the transition state is more reactant-like. a. Free radical bromination is more selective than chlorination because the transition state is more product-like. c. Free radical chlorination is more selective than bromination because the transition state is more product-like. d. Free radical chlorination is more selective than bromination because the transition state is more reactant-like. ___5. Which of the folowing species is the most nucleophilic? a. NH3 b. H3P c. H2S d. H2O ___6. SN1 reactions lead to a. formation of free radicals b. retention of stereochemistry c. racemization d. inversion of stereochemistry ___7. Which of the following would undergo the fastest dehydration reaction in the presence of acid? a. b. CH 3 c. CH 2 O H d. CH 3 OH OH CH 3 OH 1 ___8. Which of the following reactions will proceed the fastest? a. b. CH 2Cl CH 2 Br NaOH c. NaOH d. CH 3 CH 2 Br CH 2 Br NaOH NaOH . II. REACTIONS: Predict the major organic products of the following reactions. INDICATE STEREOCHEMISTRY AS NEEDED. (4 points each) 9. CH 2CH 3 Br2, light 10. O C H CH 2 CH 3 CH 2 CH 2 OH, H+ CH 3 1. NaH OH 2. CH3I 11. 12. H H CH(CH3)2 (CH3)3CO–K+ Br 13. H O H H NaOH, H2 O C(CH3)3 2 14. H CH3Se– Na+ Br H CH 3 15. H H 3C C C CH 3 C CH 3 CH 3 O H Br CH 3 16. 1. Mg CH 3CH 2CH 2Br 2. H2C=O 3. H2SO4 V. Mechanisms. Give a stepwise, detailed mechanism with arrows and intermediates for the following reactions. Be sure to account for stereochemistry as needed. (6 points each) 17. CH 3 CH 3 CH 3 H3PO4, heat OH CH 3 18. CH 4 + Br2 heat or light CH 3 Br + HBr 3 19. CH 3 CH 3CH 2 O C CH 3 CH 3 CH 3 HI CH 3CH 2 excess I + I C CH 3 CH 3 Syntheses: Give reagents to carry out the transformations below. (5 points each) 20. I 21. O O CH 3 CH 2CH 2CH 3 22. Only one monochlorination product is obtained from an alkane with the molecular formula C5H12. What is the structure of the alkane? (4 points) 23. Draw the transition state for the reaction of CH3Br with HO–. (4 points) 4 CHEMISTRY 61 Exam 3 Dr. M.T. Crimmins November 23, 1999 Name___________________________________________ Pledge: I have neither given nor received aid on this exam. Signature________________________________________ II. Reactions: Predict the major organic product of the following reactions. If more than one product is formed give both and indicate the major product. Indicate stereochemistry where necessary. (4 points each) 1. H CO2CH3 H CO2CH3 heat + 2. HBr -80 °C 3. CH3 H3C C CH2 CH2CH3 Br2, light H 4. Br H NaI H CH3 5. H 3C H H 2O low temperature Br CH3 6. Br Ph CH3 C C Ph H – (CH3)3CO K + H 7. CH3 Ph C CH3OH CH CH2 Br –1– 8. Indicate if the following compounds are aromatic, non-aromatic or anti-aromatic. (2 points each) + + H H a. b. c. d. e. 9. List three criteria for aromaticity. (6 points) 1.______________________________________ 2.______________________________________ 3.______________________________________ II. Multiple Choice: Place the letter in the blank and Circle the best answer (only one). (3 points each) ___10. In the following solvolysis reaction what is the relationship of the products formed? H 3C CH2CH3 C CH2CH2CH3 Br a. enantiomers b. meso compound CH3OH c. structural isomers d. diastereomers ___11. Which of the following reactions would proceed the fastest? a. CH3CH2CH3 + Br2 + light → CH3CHBrCH3 b. CH3CH2CH3 + F2 + light → CH3CHFCH3 c. CH3CH2CH3 + I2 + light → CH3CHICH3 d. CH3CH2CH3 + Cl2 + light → CH3CHClCH3 ___12. Which of the following is the strongest nucleophile? c. CH3Od. CH3Se- a. CH3NH– b. Cl___13. Which of the following alkyl halides would undergo the fastest SN2 reaction? a. CH2I b. b. CH3CH2CH2CH2—I CH2Cl d. (CH3CH2)2CH—I ___14. Which of the following are "concerted" reactions? a. SN1 b. Diels-Alder reaction c. E1 d. electrophilic addition e. SN1 and E1 –2– ___15. Reaction of a hydroxide ion (HO–) with a primary alkyl halide is most likely to result in: a. SN2 substitution b. E2 elimination c. SN1 substitution d. E1 elimination ___16. Which of the following statements is correct? a. Free radical bromination is more selective than chlorination because the transition state for bromination is more reactant-like. a. Free radical bromination is more selective than chlorination because the rate limiting step for bromination is more endothermic than for chlorination. c. Free radical bromination is more selective than chlorination because the rate limiting step for bromination is more exothermic than for chlorination. d. Free radical chlorination is more selective than bromination because the transition state for chlorination is more reactant-like. ___17. Which of the following would be the rate limiting step in a free radical halogenation? a. b. c. d. CH3CH2. + Br2 → CH3CH2Br + Br. Br2 → 2 Br. 2 CH3CH2. → CH3CH2CH2CH3 CH3CH3 + Br. → CH3CH2. + HBr ___18. Ionization to give a carbocation and a leaving group is the rate determining step for a. SN1 b. SN2 c. E1 and E2 d. E1 e. SN1 and SN2 f. SN1 and E1 Syntheses. Give reagents to show how to synthesize the compounds on the right from the compounds on the left. They may require more than one step. (4 pts each) 19. CH3 CH2OH 20. CH3 CH3 CH3 CH3 H 3C C H3 C CH CH3 C CH3 CH3 –3– C CH2 V. Mechanisms. Give a stepwise, detailed mechanism with arrows and intermediates for the following reactions. (5 points each) 21. Br2, light CH 3CH2CH3 CH 3CHBrCH3 22. Br OCH3 CH3 CH3OH CH3 CH3 + 23. H2C=CH CH=CH2 HBr H3 C–CH CH=CH2 40 °C Br –4– + H3 C–CH CH–CH2 Br 24. The energy diagram for the hypothetical reaction A + B → D → G + H is shown below. (6pts). C E F E D G A +B H reaction coordinate a. What is the rate determining step?_____________ b. What happens to the rate if the concentration of A is doubled? _____________ c. What is the rate expression? rate = _______________ d. If D is a charged species and A and B are neutral, what effect will a polar protic solvent have on the rate of reaction?___________________ e. What is the kinetically favored product?__________ f. What is the thermodynamically favored product?_________ –5–