Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Polysubstance dependence wikipedia , lookup
Orphan drug wikipedia , lookup
Neuropsychopharmacology wikipedia , lookup
Compounding wikipedia , lookup
Pharmacognosy wikipedia , lookup
Neuropharmacology wikipedia , lookup
Pharmaceutical industry wikipedia , lookup
Pharmacogenomics wikipedia , lookup
Prescription drug prices in the United States wikipedia , lookup
Drug design wikipedia , lookup
Drug discovery wikipedia , lookup
Prescription costs wikipedia , lookup
Drug interaction wikipedia , lookup
Plateau principle wikipedia , lookup
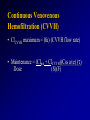
Basic Principles of Pharmacokinetics Betty Lee, Pharm.D. Lucile Packard Children’s Hospital February 8, 2008 Introduction of Pharmacokinetics • Basic Principles – – – – – Bioavailability (F) Volume of Distribution (V) Administration Rate (RA) Clearance (Cl) Elimination Rate Constant (K) and Half-Life (t1/2) – Creatinine Clearance (Clcr) Pharmacokinetics • Dialysis of Drugs – Continuous Venovenous Hemofiltration (CVVH) • Antimicrobial Agents – Aminoglycosides – Vancomycin Bioavailability (F) • The percentage or fraction of the administered dose that reaches the systemic circulation of the patient. • F = bioavailability factor • S = the fraction of the administered dose that is the active drug • Amount of Drug Absorbed = (S) (F) (Dose) Protein Binding • fu = Free Drug Concentration Total Drug Concentration • fu = C free C bound + C free • C free = (fu) (C total) • For example, gentamicin and vancomycin has fu value of 0.9. Volume of Distribution (V) • The apparent volume of distribution, does not necessarily refer to any physiologic compartment in the body. • V = (the total amt of drug in the body) / C • V is the major determinant of the loading dose • Loading dose = (V) (C) (S) (F) Administration Rate (RA) • The administration rate is the average rate at which absorbed drug reaches the systemic circulation. • This is usually calculated by dividing the amount of drug absorbed by the time over which the drug was administered (dosing interval). • RA = (S) (F) (Dose) Clearance (Cl) • The intrinsic ability of the body or its organs of elimination to remove drug from the blood or plasma. • Clearance is expressed as a volume per unit of time. • At steady state, the rate of drug administration (RA) and rate of drug elimination (RE) must be equal. Clearance (Cl) • Clearance (Cl) can best be thought of as the proportionality constant that makes the average steady-state plasma level equal to the rate of drug administration (RA ) • RA = (Cl) (Css ave) • and RA = (S) (F) (Dose) / • Cl = (S) (F) (Dose/ ) Css ave Clearance (Cl) • Factors that can alter clearance: – – – – – Body weight Body surface area Cardiac output Drug-drug interactions Extraction ratio – Genetics – Hepatic function – Plasma protein binding – Renal function Elimination Rate Constant (K) • The elimination rate constant (K) is the fraction or percentage of the total amount of drug in the body removed per unit of time and is a function of clearance and volume of distribution • K = Cl / V • First-order elimination– the amount or concentration of drug in the body diminishes logarithmically over time • C2 = (C1) (e-Kt1) Elimination Rate Constant (K) • • • • • C2 = (C1) (e-Kt) C2 / C1 = e-Kt ln (C2 / C1 )= -Kt ln (C1 / C2 )= Kt K = ln (C1 / C2 ) t Half-Life (t1/2) • Half-life is the time required to eliminate one-half of the drug • t1/2 = 0.693 / K • and K = Cl / V • t1/2 = 0.693 (V) Cl • K and t1/2 are dependent on clearance and the volume of distribution Clinical Application of K and t1/2 • Estimating the time to reach steady-state plasma concentration after initiation or change in the maintenance dose • Estimating the time required to eliminate all or a portion of the drug from the body once it is discontinued Clinical Application of K and t1/2 • Predicting nonsteady-state plasma levels following the initiation of an infusion • Fraction of Steady State Achieved at time t1 = 1- e-Kt1 • C1 = (Css ave) (Fraction of Steady State Achieved at time t ) 1 • C1 = (S) (F) (Dose/ ) (1- e-Kt1 ) Cl Clinical Application of K and t1/2 • Predicting a steady-state level from a nonsteady-state plasma level obtained at a specific time following the initiation of an infusion • Fraction of Drug Remaining at t2 = e-Kt2 • C2 = (C1) (e-Kt2) • C2 =(S) (F) (Dose/ ) (1- e-Kt1 ) (e-Kt2) Cl Clinical Application of K and t1/2 • Given the degree of fluctuation in plasma concentration desired within a dosing interval, determine that interval; given the interval, determine the fluctuation in the plasma concentration Dosing Interval () • If the goal of therapy is to minimize plasma fluctuations to no more than 50% between doses, the dosing interval should be less than or equal to the half-life. • Maintenance Dose = (Cl)(Css ave) () (S) (F) Creatinine Clearance (Clcr) • Clcr for Children = (0.48) (Height in cm) (BSA) (ml/min) SCrss (1.73m2) • Clcr in ml/min= (U) (V) P • Clcr in ml/min; U is the urine creatinine concentration in mg/dL, V is the volume of urine per time collection in mL/min, and P the plasma creatinine concentration in mg/dL. Dialysis of Drugs • Clpat = Clm + Clr • Clpat is the patient’s drug clearance during nondialysis periods and is the sum of the patient’s metabolic clearance (Clm) and residual renal clearance (Clr). • Postdialysis Replacement Dose = [Amt of Drug in the Body Prior to Dialysis] [ Fraction of Drug Lost during Dialysis] Dialysis of Drugs • Postdialysis Replacement Dose = (V) (Css ave) [(1-e –(Clpat + Cldial)(Td) ] V • Postdialysis Replacement Dose = (V) (Css ave) (1-e-Kdial(Td) ) • Kdial is the elimination rate constant during the dialysis; Td is the duration of dialysis. Estimating Drug Dialyzability • Divide the volume of distribution by fu or the usual free fraction to calculate the apparent unbound volume of distribution. If the unbound volume of distribution exceeds 3.5 L/kg, it is unlikely that the drug will be dialyzable. • Unbound Volume of Distribution = V / fu Estimating Drug Dialyzability • If patient’s clearance is > 10 ml/min/kg, it is unlikely that hemodialysis will add significantly to the patient’s intrinsic drug elimination process. This is because most drugs have a hemodialysis clearance less than 150 ml/min. Estimating Drug Dialyzability • If the usual dosing interval is much less than the drug’s t1/2, it is unlikely that hemodialysis will significantly alter the dosing regimen. The key is to schedule the drug administration shortly after rather than shortly before dialysis, so that even if the drug is dialyzable, very little is remaining to be removed by dialysis. Estimating Drug Dialyzability • Drugs with a low molecular weight are more likely to be removed significantly by dialysis. However, high-flux hemodialysis can remove molecules with molecular weight > 1000 Daltons. Continuous Venovenous Hemofiltration (CVVH) • Drug removal by means of CVVH is independent from drug molecule size • ClCVVH is clinically relevant for – drugs with dominant renal clearance, especially when presenting a limited Vd and poor plasma protein binding – most hydrophilic antimicrobial agents. (e.g. betalactams, aminoglycosides, glycopeptides) • The larger the Vd, the less likely will be removed by CVVH Continuous Venovenous Hemofiltration (CVVH) • Extent of drug removal is expected to be directly proportional to the device’s surface area and to be dependent on the mode of replacement fluid administration (predilution or postdilution) and on the ultrafiltration and/or dialysate flow rates applied. Continuous Venovenous Hemofiltration (CVVH) • ClCVVH maximum = (fu) (CVVH flow rate) • Maintenance = (Clpat + ClCVVH)(Css ave) () Dose (S)(F) Continuous Venovenous Hemofiltration (CVVH) • For time-dependent antimicrobials – Maintain the frequency of drug administration while modifying the amount of each single dose • For concentration-dependent antimicrobials – Maybe more useful to change the dosing interval while maintaining a fixed dosage Aminoglycosides • Volume of distribution is ~0.25 L/kg; pediatric patients younger than 5 years tend to have a volume of distribution of 0.5 L/kg • Aminoglycosides are eliminated almost entirely by the renal route (Cl = Clcr). • t1/2 = 2-3 hr • Peak level should be drawn 30 min. after a 30-min. infusion; trough level should be drawn within 30 min. before the next dose Aminoglycosides—during CVVH • • • • • Hydrophilic Low Vd Absence of plasma protein binding Almost complete renal clearance Rapid and consistent extracorporeal removal during CVVH Aminoglycosides • Bactericidal activity is concentration-dependent • Also has postantibiotic effect that results in depressed bacterial growth after plasma concentrations have fallen below the MIC • Saturable uptake mechanisms within the renal cortex and inner ear indicate that extended interval dosing may also minimize the likelihood of developing nephrotoxicity and ototoxicity. Aminoglycosides • Conventional dosing: – Gentamicin, Tobramycin: • Peak 5-8 mg/L, Trough <2 mg/L – Amikacin: • Peak 20-30 mg/L, Trough <10 mg/L • Once-daily dosing: – Gentamicin, Tobramycin: • Peak ~20 mg/L, Trough--Undetectable – Amikacin: • Peak ~60 mg/L, Trough--Undetectable Once-daily Aminoglycosides • Less intensive monitoring of serum concentrations • Nomogram developed by Nicolau D et al. Antimicrob Agents Chemother 1995; 39:650-655. – Recommends a single level be drawn 6 to 14 hours after the dose • With extended interval dosing there should be no significant accumulation with multiple dosing, therefore, measurements can be obtained after any dose Vancomycin • Volume of distribution: an average value of 0.7 L/kg or for patient older than 18 years: V (L) = 0.17 (age in yr) + 0.22 (TBW in kg) + 15 • Eliminated primarily by the renal route; approximately 5% of the dose is metabolized (Cl ~ Clcr). • t1/2 = 6 to 7 hours Vancomycin—during CVVH • • • • • • Low Vd Hydrophilic Moderately protein-bound Short t1/2 Mainly renal clearance Shown to be removed significantly removed during CVVH Vancomycin • Bactericidal for most gram-positive organisms, except against enterococci • Synergistic with gentamicin against most strains of S. aureus and enterococci • Therapeutic serum concentrations: – Peak 30-40 mg/L – Trough 5-15 mg/L Vancomycin • Some controversy about necessity for routinely monitoring plasma vancomycin concentrations: – Vancomycin exhibits concentration-independent killing, and specific peak plasma concentrations have not been correlated with efficacy. – Monitor those at highest risk for therapeutic failure or potential drug toxicity, which includes pediatric patients who have high clearances and short half-lives Vancomycin • As a general rule, vancomycin is dosed with an interval of approximately one half-life • Peak level should be drawn 1 hour after a 1-hour infusion; trough level should be drawn within 1 hour before the next dose • Css max = [Css min] [(S)(F)(Dose)] V Vancomycin • Vancomycin-induced ototoxicity has been primarily reported in patients with vancomycin concentrations > 80 mg/L. • As a single agent, vancomycin is associated with a low incidence of nephrotoxicity; however, when it is combined with aminoglycoside, the incidence may be as high as 30%. References • Winter, Michael. Basic Clinical Pharmacokinetics, 4th Ed. Baltimore: LWW, 2004. • Pea F, Viale P et al. Pharmacokinetic Considerations for Antimicrobial Therapy in Patients Receiving Renal Replacement Therapy. Clin Pharmacokinet 2007; 46 (12): 997-1038.