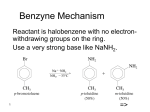
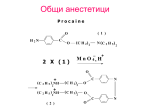
Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Symmetry in quantum mechanics wikipedia , lookup
Bohr–Einstein debates wikipedia , lookup
Wave–particle duality wikipedia , lookup
Ising model wikipedia , lookup
Particle in a box wikipedia , lookup
Rutherford backscattering spectrometry wikipedia , lookup
Rotational–vibrational spectroscopy wikipedia , lookup
Hydrogen atom wikipedia , lookup
Molecular Hamiltonian wikipedia , lookup
Rotational spectroscopy wikipedia , lookup
Theoretical and experimental justification for the Schrödinger equation wikipedia , lookup
Franck–Condon principle wikipedia , lookup