Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Schrödinger equation wikipedia , lookup
Particle in a box wikipedia , lookup
Atomic orbital wikipedia , lookup
X-ray photoelectron spectroscopy wikipedia , lookup
X-ray fluorescence wikipedia , lookup
Relativistic quantum mechanics wikipedia , lookup
Theoretical and experimental justification for the Schrödinger equation wikipedia , lookup
Electron configuration wikipedia , lookup
Franck–Condon principle wikipedia , lookup
Rutherford backscattering spectrometry wikipedia , lookup
Hydrogen atom wikipedia , lookup
Chemical bond wikipedia , lookup
Molecular Hamiltonian wikipedia , lookup
Molecular orbital wikipedia , lookup
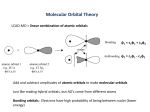
Covalent Bonding • We seek to solve the Schrödinger equation for H2 h2 2 − 2m ∇ + VAB (r)ψ AB (r) = Eψ AB (r) – where VAB(r) is the intermolecular potential, which we can approximate by writing VAB(r) = VA(r) +VB(r) • We look for a Linear Combination of Atomic Orbitals (LCAO) solution ... ψ AB = cAψ A + cBψ B – exact solution requires an infinite number of vectors Aim of this brief treatment of covalent bonding is to illustrate what happens to the energy levels of isolated atoms when a molecule is formed. In the coming lectures we will see that energy levels in molecules and solids are closely related. For simplicity we will consider the hydrogen molecule H2, for which the energy levels, potentials and atomic wavefunctions are the same. Consider two atoms A and B brought together from infinity. Each free atomic orbital satisfies its own effective one-electron Schrödinger equation - in other words, HAψA=EA and HBψA=EB h2 2 − 2m ∇ + VA (r)ψ A (r) = EAψ A (r) h2 2 − 2m ∇ + VB (r)ψ B (r) = EBψ B (r) In practice the intermolecular potential VAB(r) depends also on electronic wavefunction ψAB, and so the equation must be solved self-consistently. Here we will approximate the intermolecular potential as the sum of the individual free atomic potentials. For covalently bonded systems this is often quite a reasonable starting approximation. 1 Hamiltonian Matrix Elements • Our molecular Schrödinger equation looks like ( ) h2 2 √ √ H − E (cAψ A + cBψ B ) = 0 where H = − ∇ + VAB 2m • Premultiplying by ψA and integrating over all space yields (hAA − E )cA + (hAB − ES)cB = 0 – where hαβ = ∫ ψ α H√ψ β dr S = ∫ ψ Aψ B dr The slightly unusual looking eigenvalue equation is simply a rearrangement of the more familiar H√ψ = Eψ Manually do the premultiply by ψA. Since s-orbitals are real we can forget about using the complex conjugate. Since we are considering the hydrogen molecule we can definitively assert that the overlap S is a positive number, as the wavefunctions are real and do not change sign For higher angular momenta combinations (and higher principal quantum numbers) the integrals depend on the atomic separation due to the change in sign of the wavefunction with radius and direction. The same procedure using ψB yields (hBA − ES)cA + (hBB − E )cB = 0 2 Matrix Equation • A similar premultiplication by ψB and the approximation that the overlap S is zero yields the matrix equation hAB cA hAA − E =0 h hBB − E cB BA • Diagonal elements ( ) hAA = ∫ ψ A H√ A + VB ψ A dr = EA + ∫ ρ AVB dr ≈ EA • Off-diagonal elements ( ) hAB = ∫ ψ A H√B + VA ψ B dr = ∫ ψ AVAψ B dr = − β The Matrix Make the approximation that the overlap S is equal to zero. Simplifies the mathematics, without affecting the qualitative description. The case of finite S is a relatively simple algebraic exercise. Diagonal Elements Write HAB= HA + VB The integral over ρA reflects a small lowering in the energy of the electron on atom A due to the attractive tail of the potential from atom B. Again, for simplicity of argument we will ignore this term. The same approach applies to the other diagonal element hBB. Off-diagonal Elements: This time we express HAB as HB + VA. Get integral ψAHBψB=ψAEBψB=ES With the neglect of overlap this term vanishes. β is unambiguously +’ve By symmetry, the two off-diagonal elements are equal (for complex wavefunctions, the off-diagonal elements are complex conjugates). 3 Eigenvalues and Eigenvectors • Determinant must be zero for non-trivial solutions E0 − E −β −β =0 E0 − E • The eigenvalues and eigenvectors are E0 − β E= E0 + β (c A = cB ) (c A = − cB ) • To first order, the inclusion of a finite overlap S yields E = E0 + βS ± β Setting the determinant equal to zero yields ( E0 − E )2 − β 2 = 0 E0 − E = ± β E = E0 ± β For E=E0–β the values of cA and cB are determined by solving β − β − β c A =0 β cB ⇒ c A = cB ⇒ c A = − cB Similarly, for E=E0+β, we have − β − β − β c A =0 − β cB The quantity β is referred to as the bond integral Overlap repulsion is reflected in the term βS 4 Energy Levels and Molecular Orbitals • The free atom orbitals combine to form bonding and antibonding states separated in energy by an amount 2β E0 2β • The upper molecular state (with the anti-symmetric wavefunction) does not lead to bonding as it’s energy is higher than the energy of the free atoms. It is known as the anti-bonding state. • The lower molecular state (with the symmetric wavefunction) does lead to bonding as it’s energy is lower than the separated atoms. It is called the bonding state. Note that the lowering of the energy is given by β which explains why we called this quantity the bond integral. • From the previous slide we see that the molecular eigenvalues are shifted upwards by an amount βS due to overlap repulsion which reflects the Pauli exclusion principle. These are the two key ingredients of the covalent bond: an attractive bond energy pulling the atoms together, balanced at equilibrium by a repulsive overlap potential keeping the atoms apart. • We compute charge density for these two states by taking the square of the respective wavefunctions which shows that the anti-bonding state has zero electron density at the midpoint of the two points, whereas the bonding state has a heaping up of charge which is attracted to both nuclei and thus by electrostatics pulls the nuclei together. 5 Beyond the Hydrogen Molecule • He-He molecule (unstable) – populate bonding and anti-bonding levels, and the average energy is unchanged (in fact, slightly higher) • Li-Li molecule (stable) – 1s orbitals much close to nucleus than 2s orbitals – very small splitting for 1s levels – bonding arises from the 2s energy split • Be-Be molecule (unstable) – populated bonding and anti-bonding for 2s energy split, and so the average energy is unchanged We can study this numerically using a pair of square-well-potentials. It is very instructive to compute the energy difference numerically and compare it to the bond integral β. A very attractive aspect of this approach is that we can understand the bond integral b using a one-dimensional integral. Note that the energy split does not reflect simply the overlap between the free-atom wavefunctions - rather, it is integral involving the product of the two wavefunctions and the free-atom potential. 6










![Chem 400 Chem 340 Inorg Review [AR].S17](http://s1.studyres.com/store/data/000220292_1-82084c4723d43bb722b21295b237196f-150x150.png)





