Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Lymphopoiesis wikipedia , lookup
Adoptive cell transfer wikipedia , lookup
Multiple sclerosis research wikipedia , lookup
Pathophysiology of multiple sclerosis wikipedia , lookup
Sjögren syndrome wikipedia , lookup
Immunosuppressive drug wikipedia , lookup
X-linked severe combined immunodeficiency wikipedia , lookup
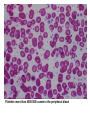
Myeloproliferative Disorders Myeloproliferative disorders make up a group of chronic conditions characterized by clonal proliferation of one or more marrow cell linage. It is important to determine if it is clonal or not because if it isn’t clonal that means it is a reactive. process such as increasing RBC in hypoxia, increasing WBC in infections, and increasing platelets in haemorrhage. Remember that the most physical sign in all myeloproliferative disorders is the splenomegaly. These disorders include: 1. Chronic myeloid leukaemia ‘CML’ 2. Polycythaemia Vera ‘PRV’ in RBC. 3. Essential Thrombocythemia for the platelets. 4. primary Myelofibrosis These disorders have possibility to progression from one to another e.g. PRV to Myelofibrosis, and may be terminated to AML. Polycythaemia Vera (PRV) A neoplastic (clonal) stem cell disorder, leads to excessive production of all myeloid cell lines, predominantly red cells. Clinical features The increase in whole blood viscosity causes vascular occlusion and ischemia, compounded by the increase in platelets. Headaches, Itch, Thrombosis, TIA, stroke, and Splenomegaly are also findings. Polycythaemia categories. POLYCYTHAEMIA VERA. SECONDARY POLYCYTHAEMIA. HYPOXAEMIA PO2 < 92% RENAL DISEASE TISSUE HYPOXIA - HIGH AFFINITY HB TUMOURS - HEPATOMAS, FIBROIDS, CEREBELLAR HAEMANGIOBLASTOMAS HIGH ERYTHROPOIET PRODUCTION IDIOPATHIC ERYTHROCYTOSIS. WHO criteria for PRV diagnosis. Major criteria 1- Hemoglobin more than 18.5/dL in men and more then 16.5/ in women. 2-presence of JAK2 mutation. Minor criteria 1-Bone marrow panmyelosis. 2-Low serum erythropoietin. 3-Endogenous colony formation in vitro. Diagnosis requires both major and one minor or first major and two minor. Bone marrow panmyelosis Investigations of Polycythaemia PULSE OXIMETRY RENAL - URINALYSIS + RENAL ULTRASOUND ABDOMINAL ULTRASOUND NEUTROPHIL COUNT PLATELET COUNT MARROW CYTOGENETICS MARROW EXAMINATION AND CULTURE SERUM ERYTHROPOIETIN ASSAYS. Essential Thrombocythemia (ET) The malignant proliferation of megakaryocytes ,Constitutive production of thrombopoietin by liver” results in a raised level of circulating platelets those are often dysfunctional. Prior to making a diagnosis of ET it is essential to exclude reactive causes of increase platelets. WHO Criteria for ET diagnosis. 1-Platelets count more than 450 000/cumm. 2-Megakaryocytic proliferation in the bone marrow. 3-Not meeting the criteria of other myeloproliferative disorders. 4-demostration of JAK2 mutation and in absence of JAK2 mutation, there is no evidence of reactive thrombocytosis. Diagnosis requires all these criteria. Platelets more than 4500 000/ cumm in the peripheral blood Megakaryocytic proliferation in the bone marrow Clinical Features Asymptomatic Haemorrhage – 25% Thrombosis – 20% Splenomegaly – 30% Recurrent Miscarriage Primary Myelofibrosis Background Primary Myelofibrosis , first described by Heuck in 1879, is a clonal disorder arising from the neoplastic transformation of early hematopoietic stem cells. primary Myelofibrosis is characterized by anaemia, bone marrow fibrosis, extramedullary hematopoiesis, leukoerythroblastosis, teardrop-shaped red blood cells (RBCs) in peripheral blood, and hepatosplenomegaly Diagnostic WHO criteria of primary myelofibrosis Major criteria 1-Megakaryocytic proliferation with marrow fibrosis. 2-Not meeting the WHO criteria of other myeloproliferative disorders 3-Demonstration of JAK2 mutation. Minor criteria 1-Leucoerythroblastosis. 2-Increased LDH level 3-Anaemia. 4-Splenomegaly. Diagnosis requires all 3 major and 2 minor. Peripheral blood shows teardrop cells and leukoerythroblastosis. Bone marrow fibrosis Extramedullary hematopoiesis in the spleen Pathophysiology The cause of the excessive marrow fibrosis observed in primary myelofibrosis remains unclear. Platelets, megakaryocytes, and monocytes are thought to secrete several cytokines, such as transforming growth factor beta (TGF-β), platelet-derived growth factor (PDGF), and basic fibroblast growth factor (bFGF), which may result in fibroblast formation and extracellular matrix proliferation. In addition, endothelial proliferation and growth of capillary blood vessels in the bone marrow are observed and may be a result of TGF-β and bFGF production. Clinical features One fourth of patients with primary myelofibrosis are asymptomatic, and the diagnosis is made as a result of detecting splenomegaly or checking blood cell counts for an unrelated cause. Symptoms may occur as a result of anemia, splenomegaly, hypermetabolic states, extramedullary hematopoiesis, bleeding, bone changes, portal hypertension, and immune abnormalities. Anemia may occur as a result of ineffective erythropoiesis, erythroid hypoplasia, and hypersplenism. Splenomegaly may result in early satiety and left upper quadrant discomfort. Splenic infarcts. Physical signs Splenomegaly is the most common finding in patients with primary myelofibrosis, and it is present in approximately 90% of patients. Hepatomegaly is also observed in 60-70% of patients with this disease. Pallor is observed in 60% of patients. Other physical findings include petechiae and ecchymosis (20%), lymphadenopathy (10-20%), signs of portal hypertension (10-18%), and gout (6%).