Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Citric acid cycle wikipedia , lookup
Metabolomics wikipedia , lookup
Butyric acid wikipedia , lookup
Pharmacometabolomics wikipedia , lookup
Metabolic network modelling wikipedia , lookup
Basal metabolic rate wikipedia , lookup
Amino acid synthesis wikipedia , lookup
Fatty acid synthesis wikipedia , lookup
Clinical neurochemistry wikipedia , lookup
Biosynthesis wikipedia , lookup
Fatty acid metabolism wikipedia , lookup
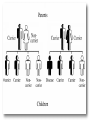
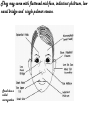
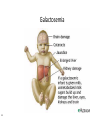
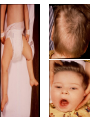
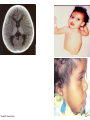
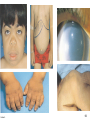
Metabolic Disorders Inborn Errors Of Metabolism DR. ABDULLAH ALOMAIR MB ChB, MRCP (Edin), FRCP (Edin.), DCH (Glas.) Associate Professor of Pediatrics Consultant Pediatrician Department of Pediatrics PRESIDENT SAUDI PEDIATRIC ASSOCIATION 1 Metabolic Disorders Inborn Errors Of Metabolism Inborn Errors Of Metabolism (IEM) -A large group of hereditary biochemical diseases. -In autosomal dominant disorders, the structural abnormality dominates over the chemical abnormality. -Specific gene mutation cause abnormal or missing proteins that lead to altered function. 2 Pathophysiology SINGLE GENE DEFECTS in synthesis or catabolism of proteins, carbohydrates, or fats. Defect in an ENZYME or TRANSPORT PROTEIN , which results in a block in a metabolic pathway. EFFECTS : - toxic ACCUMULATION of substrates before the block, - intermediates from ALTERNATIVE pathways - defects in ENERGY production and utilization caused by a deficiency of products beyond the BLOCK. Every metabolic disease has several forms that vary in AGE OF ONSET , clinical severity and, often, MODE OF INHERITANCE. Classification Transient Hyperammonemia of Newborn Inborn Errors of Metab: • • • • • Molybdenum Cofactor Deficiency • Organic Acidemias Fatty Acid Oxidation def Urea Cycle Defects Amino Acidurias Non-ketotic Hyperglycinemia Sulfite Oxidase Deficiency Metal Storage Disorders: Cholesterol Disorders: Leukodystrophies, other… • Krabbe disease Mitochondrial Disorders Glycogen Storage Disorders Hyperinsulinism Carbohydrate Disorders Lysosomal Disorders • • • Mucopolysaccharidoses (Xlinked Hunter’s, Hurler’s) Gaucher disease Tay-Sachs Disease Peroxisomal Disorders • • Zellwegger’s (CerebroHepato-renal) X-linked Adrenoleukodystrophy Metabolic Disorders • Due to inherited reduced activities of proteins involved in the synthesis, breakdown or transport of amino acids, organic acids, fats, carbohydrates and complex macromolecules. • Most are autosomal recessive due to mutations that result in reduced enzyme activity or reduced amount of enzyme. • Pathogenesis may include: accumulation of a toxic intermediate, reduced amount of a necessary end product or activation of an alternate pathway. 7 Metabolic Disorders Features suggestive of metabolic disorder : From history: Parental history : Consanguineous parents Previous unexplained neonatal deaths Particular ethnic group (in certain diseases) 8 Metabolic Disorders Features suggestive of metabolic disorder : Examination findings: Organomegaly (e.g. hepatomegaly) in the absence of viral infection. Cardiac disease Ocular involvement (e.g. cherry red spot) Skin manifestations e.g. pigmentations. Unusual odour. Due to change in the chemicals of the urine. Non-specific neurological findings. In a non-meningitis child you have to think of metabolic disorders. Neonatal and Post Neonatal Presentation Neonatal presentation • • • • • • • Normal-appearing child at birth (some conditions are associated with dysmorphic features) poor feeding lethargy vomiting seizures coma unusual odour Hypoglycaemia is very dangerous, acidosis (in some defects) 10 Neonatal and Post Neonatal Presentation Post neonatal presentation • • • • • • Encephalopathy without the presence of infection. Developmental regression Reye syndrome ( damage of the brain and liver eventually leading to encephalopathy). Motor deficits Seizures Intermittent episodes of vomiting, acidosis, hypoglycaemia and/or coma triggered by stress e.g. infections, surgery. Newborn Screening the earlier its detected the fewer the complications PKU - in NICU even if not advanced to full feeds Galactosemia Hypothyroidism Hemoglobinopathies Biotinidase defic, CAH (21-OH’ase def), Maple syrup urine disease ( MSUD ) - GUTHRIE TEST: it’s a cheap test that requires only one drop of blood to check for multiple metabolic disorders. PROCEDURES FOR DIAGNOSIC CONFIRMATION Non – Specific Tests: Specific Tests: • • Direct biochemical assays of metabolites or their metabolic byproducts, or of an enzyme’s function. • DNA studies • Neuro-radiology • • Blood glucose, ammonia, bicarbonate and pH Peripheral Blood smear – WBC or bone marrow vacuolization , foam cells or granules. C.S.F. glycine , other amino acids , lactate. Amino acids shouldn’t be present in the CSF if its there it indicates a metabolic disorder. Bone marrow transplantation is a treatment of both inborn errors of metabolism 13 INBORN ERRORS OF AMINO ACID METABOLISM ASSOSIATED WITH ABNORMAL ODOR Inborn Error of Metabolism Gultaric Acidemia Sweaty feet Maple Syrup urine disease Maple syrup Hypermethioninemia Boiled cabbage Phenylketonuria Mousy or musty Trimethylaminuria Rotten fish Urine Odor 14 They may come with flattened mid-face, indistinct philtrum, low nasal bridge and single palmar crease. Small chin is called micrognathia Low-set ears: >1/3rd of the ears lower than the line connecting the 2 pupils. Low nasal bridge: common sign, which is also seen in Down. MANAGEMENT OF IEM Genetic: Establish diagnosis. Carrier testing. Pedigree analysis, risk counseling. Consideration of Prenatal diagnosis for pregnancies at risk. 19 MANAGEMENT OF IEM PSYCHOSOCIAL , EDUCATIONAL , FAMILIAL Family counseling and support. Education to promote increased compliance with special form of therapy such as Protein – restricted diet. Assessment of community resources and support groups. 20 TREATMENT OF GENETIC DISEASES • Modify environment, e.g., diet, drugs • Avoid known environmental triggers • • BMT Surgical, correct or repair defect or organ transplantation • Modify or replace defective gene product, megadose vitamin therapy or enzyme replacement • Replace defective gene • Correct altered DNA in defective gene Galactosemia 22 : Carbohydrates Galactosemia Enzyme deficiency: Galactose-1-phosphate uridyl transferase deficiency. It is a rare autosomal recessive. ● Follows feeding with lactose containing (breast milk / formula) ● Patient feeds poorly , have vomiting, jaundice, hepatomegaly and hepatic failure ● Chronic liver disease ● Cataracts ● Developmental delay develop if condition is untreated., if they were given galactose free diet you will avoid the social and mental damage but they might complain of dyslexia. 23 CYSTIC FIBROSIS Cause : Loss of 3 DNA bases in a gene for the protein that transports Cl ions so salt balance is upset. Causes .a build up of thick mucus in lungs and digestive organs. It is diagnosed by sweat test: measuring the chloride concentration in the sweat AMINO ACID DISORDERS Phenyl Ketonuria (PKU) Phenylalanine Phenylalanine Hydroxylase Phenyl ethylamine Tyrosine Phenyl pyruvic acid Phenyl pyruvic acid is what gives the urine its smell because its ketonic and acidic. 25 Phenylketonuria PKU 26 PKU DIAGNOSIS CLINICAL FEATURES • Screening : Guthrie Test. • High Phenylalanine > 20 mg/dl. • High Phenyl pyruvic acid. 1. Hyperactivity, athetosis, vomiting. 2. Blond. 3. Seborric dermatitis or eczema skin. 4. Hypertonia. 5. Seizures. 6. Severe mental retardation. 7. Unpleasant odor of phenyl acetic acid. TREATMENT • DIET. • BH4 (Tetrahydrobiopterin). • L – dopa and 5hydroxytryptophan. 27 PKU 28 Albinism 29 Iris had fibrous tissue, and it’s colourless and is red due to vessels. Homocystinuria 31 Elevated homocystine levels affect collagen , result in a Marfanoid habitus, ectopia lentis but lens dislocation in homocystinemia is downward unlike in marfan its upward, mental retardation and strokes, its harmful to the bones and body. Araachnodyctly. Homocystinuria METHIONINE Cysathionine CYSTATHIONINE Synthatase DIAGNOSIS: High methionine and homocystine. TREATMENT: •High dose of B6 and Folic Acid. •Low methionine and high cystine diet, •Betain (trimethylglycine) 32 Homocystinuria 33 Amino acid disorders : Urea cycle defects and hyperammonemia All present with lethargy, seizures, ketoacidosis, neutropenia, and hyperammonemia Ornithine carbamyl transferase (OTC) deficiency Carbamyl phosphate synthetase deficiency Citrullinemia Arginosuccinic Aciduria Argininemia Transient tyrosinemia of prematurity First Steps in Metabolic Therapy for IEM • Reduce precursor substrate load • Provide caloric support • Provide fluid support • Remove metabolites via dialysis • Divert metabolites • Supplement with cofactor(s) Therapeutic Measures for IEM • D/C oral intake temporarily • Usually IVF’s with glucose to give 12-15 mg/kg/min glu and at least 60 kcal/kg to prevent catabolism (may worsen pyruvate dehydrogenase deficiency) • Bicarb/citrate Carnitine/glycine • Na Benzoate/arginine/citrulline • Dialysis--not exchange transfusion • Vitamins--often given in cocktails after labs drawn before dx is known • Biotin, B6, B12, riboflavin, thiamine, folate Important IEM Treatment supplements: • Carnitine for elimination of Organic Acid through creation of carnitine esters. • Sodium Benzoate, phenylacetate and phenylbutyrate for Hyperammonemia elimination. CARNITINE METABOLISM • An essential nutrient found in highest concentration in red meat. • Primary function : Transport long-chain fatty acids into mitochondria for oxidation. • Carnitine supplementation in fatty acid oxidation disorders and organic acidosis may augment excretion of accumulated metabolites , but may not prevent metabolic crises in such patients . • Carnitine is an endogenous metabolite but can be given as supplementations. CARNITINE METABOLISM • Primary defects of carnitine transport manifest as Reye syndrome , cardiomyopathy or skeletal myopathy with hypotonia • Secondary carnitine deficiency is due to diet ( esp. I.V alimentation or ketogenic diet ) , renal losses , drug therapy ( esp. valproic acid) and other metabolic disorders ( esp. disorders of fatty acid oxidation and organic acidemias ) • Prognosis depends on the cause of the carnitine abnormality. • Free and esterified carnitine can be measured in blood. • Oral or I.V. L-carnitine is used in carnitine deficiency or lnsufficiency in doses of 25-100mg/kgm/day or higher. 39 ORGANIC ACIDEMIA Disorder Enzyme • Methyl malonic Acidemia. • Methyl malonyl COA mutase. • Propionic Acidemia. • Propionyl COA Carboxylase. • Multiple carboxylase deficiency. • Malfunction of all carboxylase. • Ketothiolase deficiency . • 2 methylacetyl COA thiolase def. 40 ORGANIC ACIDEMIA Clinical Features Treatment Vomiting, ketosis. Hydration / alkali. Thrombocytopenia , neutropenia. Calories to catabolic state. Osteoporosis. Mental retardation. Exchange transfusion. Low protein diet. 41 ORGANIC ACIDEMIA 42 LYSOSOMAL STORAGE DISORDERS • Glycogen Storage Diseases • Sphingolipidoses common in eastern jews (Lipidoses And Mucolipidoses) • Mucopolysaccharidoses 43 Lysosomal Storage Disease Disease Enzyme Defiency Major Accumulating Metabolite Glucosidase Glycogen β-galactosidase GM1 gangliosides, galactose-containing oligosaccharides Hexosaminidase A Glucocerebrosidase Sphingomyelinase GM2 ganglioside Glucocerebroside Sphingomyelin MPS I H (Hurler) α-L-Iduronidase Heparan sulfate Dermatan sulfate MPS II (Hunter) (X-linked recessive) L-Iduronosulfate sulfatase Heparan sulfate Dermatan sulfate Glycogenosis Type II (Pompe disease) Sphingolipidoses GM1 gangliosidoses GM2 gangliosidoses Tay-Sachs disease Gaucher disease Niemann-Pick disease Mucopolysaccharidoses 44 Glycogen Storage Diseases Name Type O von Gierke (Type IA) Type IB Pompe (Type II) Forbe (Cori) (Type III) Andersen (Type IV) McArdle's (Type V) Her (Type VI) Tarui (Type VII) Type VIII Type IX Type X Type XI Enzyme Glycogen synthetase Glucose-6-phosphatase Symptoms Enlarged, fatty liver; hypoglycemia when fasting Hepatomegaly; slowed growth; hypoglycema; hyperlipidemia G-6-P translocase Acid maltase Same as in von Gierke's disease but may be less severe; neutropenia Enlarged liver and heart, muscle weakness Glycogen debrancher Enlarged liver or cirrhosis; low blood sugar levels; muscle damage and heart damage in some people Cirrhosis in juvenile type; muscle damage and CHF Glycogen branching enzyme Muscle glycogen phosphorylase Liver glycogen phosphorlyase Muscle cramps or weakness during physical activity Muscle phosphofructokinase Muscle cramps during physical activity; hemolysis Unknown Liver phosphorylase kinase Cyclic 3-5 dependent kinase Unknown Hepatomegaly; ataxia, nystagmus Hepatomegaly; Often no symptoms Hepatomegaly, muscle pain (1 patient) Hepatomegaly. Stunted growth, acidosis, Rickets Enlarged liver; often no symptoms Principle Groups of Glycogen Storage Diseases 47 Von Gierke Disease 48 LYSOSOMAL STORAGE DISORDERS Lipidoses And Mucolipidoses 49 In gaucher liver is enlarged but the rest of the body is very thin Gauch. cell 50 In gaucher you see the cherry red spot appearance in the macula Sandhoff - Dense thalam 52 Cerebral palsy -- scissoring of the legs Lipid accumalation around the retinal arteries and veins Lipid-retina 53 LYSOSOMAL STORAGE DISORDERS Mucopolysaccharidoses 54 Clinical And Pathological Ultra structure Of Mucopolysaccharidoses Disease Clinical Manifestation Ultrastructure of Stored Material MPS type I Earliest, most severe developmental regression coarse facial features Hepatosplenomegaly dystosis of bone cardiac involvement corneal clouding present in hurler but absent in hunter Fibrillogranular mucopolysaccharides in cells of viscera and brain MPS type II Later developmental regression Hunter coarse facial features Fibrillogranular mucopolysaccharides in cells of viscera and brain X-linked dystosis of bone Hurler hepatosplenomegaly cardiac involvement 55 minimal corneal clouding Hurler’s 56 In hurler : Nasal bridge is depressed , increase distance of philthrum , epicanthal folds, bossing of the head , thick eyebrows , upturn nostrils Hurler’s 57 58 Mcopolysacch. Morquio 59 PEROXISOMAL DISORDERS Peroxisomes = Subcellular organelles involved in various essential anabolic or catabolic processes, biosynthesis of Plasmalogens and bile acids. Due to dysfunction of a single or multiple peroxisomal enzymes, or to failure to form or maintain a normal number of functional peroxisomes. 60 PEROXISOMAL DISORDERS Clinical Manifestations: Hypotonia. Dysmorphia. Psychomotor delay and seizures. Hepatomegaly. Abnormal eye findings such as retinitis pigmentosa or cataract. Hearing impairment. 61 Peroxisomal Disorders • Zellweger Syndrome is autosomal recessive disorder. (Cerebro-hepato-renal syndrome) • Typical and easily recognized dysmorphic facies. • Progressive degeneration of Brain/Liver/Kidney, with death ~6 mo after onset. • When screening for PDs. obtain serum Very Long Chain Fatty AcidsVLCFAs Zellweger 63 PEROXISOMAL DISORDERS Diagnosis: Immunochemical studies for Peroxisomes. V. Long Chain FA ( VLCFA ) level. Chor. Vill. Samp. or/ amniocytes culture Plasmalogens synthesis. Treatment: Supportive, multidisciplinary interventions. Diet: VLCFA, phytanic acid. Organ transplantation. 64 Peroxisomal Disorders GROUP I : BIOGENSIS OF PEROXISOME Zellweger syndrome (cerebrohepatorenal syndrome). Neonatal adrenoleukodystrophy. Infantile Refsum disease. Hyperpipecolic acidemia. GROUP II : PERSOXISOMAL ENZYME DEFECTS GROUP III : POSITIVE PEROXISOMES BUT MULTIPLE DEFECTIVE ENZYME Zellweger – Like. Pseudo – infantile Refsum disease. Rhizomelic chondro-dysplasia punctata Refsum disease. X - linked Adreno-Leuko-Dystrophy. Pseudo – Zellweger syndrome. Hyperoxaluria….etc. 65 Mitochondrial Disorders Classically involve mutations in mitochondrial DNA Follow a maternal pattern of inheritance Highly variable with regard to penetrance and expressivity based on the variability in tissue distribution of abnormal mitochondria 66 Mitochondrial Syndromes Presenting in Childhood to Adult 67 Syndrome Most Common Clinical Presentation Other Clinical Features Mt DNA Defect MELAS: myopathy, encephalopathy, lactic acidosis and stroke-like episodes Stroke-like episodes in the first and second decade of life often associated with migraine headache, blood lactate Deafness, myopathy, diabetes mellitus mtDNA mutations at 3243, 3271 tRNA mutations MERRF: Myoclonic epilepsy with ragged red fibers Progressive myoclonic epilepsy Ataxia, myopathy deafness, short stature MtDNA A8344G tRNA mutation NARP: Neurogenic weakness, ataxia and retinitis pigmentosa Peripheral neuropathy, myopathy, seizures Leigh syndrome MtDNA 8993 Complex V deficiency 68 Transient Hyperammonemia of Newborn: Markedly high NH4 in an infant less than 24 HOL (hours of life), or first 1-2 DOL (day of life) before protein intake occurs. Often in context of large, premature infant with symptomatic pulmonary disease. Very sick infant. Unknown precipitant, unknown etiology (possible slow delayed urea cycle initiation), with potential for severe sequelae (20-30% death, 30-40% abnl dev.) if not treated. Does not recur after being treated. Clinical Presentation of Amino Acid Disorders Clinical Abnormality Abnormal Amino Acid Presumptive Diagnosis Acute neonatal presentation with ketoacidosis Leucine, isoleucine, valine Organic Acid Disorders Maple syrup urine disease Methylmalonic acidemia Propionic acidemia Isovaleric acidemia Acute neonatal presentation with hyperammonemia Arginine, Citrulline Marfanoid, strokes, ectopia lentis, mental retardation Homocystine & methionine Ornithine transcarbamylase deficiency Argininosuccinate synthase deficiency Argininosuccinate lyase deficiency Severe Phenylalanine developmental delay 70 Urea cycle disorders Homocystinuria Phenylketonuria Metabolic Profiles Organic and Amino Acid Disorders Predominanat Biochemical Clinical Findings KetoAcidosis Lethargy Odor Acidosis Lethargy Odor Lactic Acidosis Lethargy Hypoglycemia Lethargy Hyperammonemia Lethargy Other Most Common Diagnosis Ammonia: Normal or slightly elevated Ketones: Elevated Glucose: Normal Maple syrup urine disease Ammonia: Elevated Glucose: Normal or decreased Ketones: May be elevated Lactate: Slightly elevated Methylmalonic acidemia Propionic acidemia Isolvaleric acidemia Acidosis: Usually present Ammonia: Normal or slightly elevated Ketones: May be elevated Pyruvate dehydrogenase Pyruvate carboxylase deficiency Respiratory chain disorder Ammonia: Lactate Acidosis Ketones: Absent or inappropriately low Fatty acid oxidation defects Acidosis: Absent Respiratory Alkalosis Urea cycle disorders Newborn screening is available dependent on population frequency for some Expanded newborn screening for fatty acid defects recently offered 71 CHILDREN AFTER THE NEONATAL PERIOD Clinical Manifestation Mental retardation, Macro/Microcephaly. Coarse facial features/dysmorphia. Developmental regression. Convulsion. Myopathy / cardiomyopathy. Recurrent emesis with coma and hepatic dysfunction. Hypertonia / hypotonia. Failure to thrive. Ophthalmic – related problems : e.g. cataract, corneal cloudiness, cherry red spot, optic atrophy. Renal failure or renal tubular acidosis. 72 Clinical Symptomatology of Inborn Errors of Metabolism (IEM) in the Neonate or Infant Symptoms indicating possibility of an IEM (one or all) Infant becomes acutely ill after period of normal behavior and feeding; this may occur within hours or weeks Neonate or infant with seizures and/or hypotonia, especially if seizures are intractable Neonate or infant with an unusual odor Symptoms indicating strong possibility of an IEM, particularly when coupled with the above symptoms Persistent or r ecurrent vomiting Failure to thrive (failure to gain weight or weight loss) Apnea or respiratory distress (tachypnea) Jaundice or hepatomegaly Lethargy Coma (particularly intermittent) Unexplained hemorrhage Family history of neonatal deaths, or of simila r illness, especially in siblings Parental consanguinity Sepsis (particularly Escherichia coli) Laboratory Assessment of Neonates Suspected of Having an Inborn Error of Metabolism Routine Studies Blood lactate and pyruvate Complete blood count and differential Plasma ammonia Plasma glucose Plasma electrolytes and blood pH Urine ketones Urine-reducing substances Special Studies Plasma amino acids Plasma carnitine Urine amino acids Urine organic acids Inborn Errors of Metabolism of Acute Onset: Nonacidotic, Nonhyperammonemic Features Neurologic Features Predominant (Seizures, Hypotonia, Optic Abnormality) Glycine encephalopathy (nonketotic hyperglycinemia) Pyridoxine -responsive seizures Sulfite oxidase/santhine oxidase deficiency Peroxisomal disorders (Zellweger syndrome, neonatal adrenoleuko dystrophy, infantile refsum disease) - Jaundice Prominent Galactosemia Hereditary fructose intolerance Menkes kinky hair syndrome a1-antitrypsin deficiency Hypoglycemia (Nonketotic) : Fatty acid oxidation defects (MCAD, LCAD, carnitine palmityl transferase, infantile form) Cardiomegaly Glycogen storage disease (type II phosphorylase kinase b deficiency Fatty acid oxidation def ects (LCAD) Hepatomegaly (Fatty): Fatty acid oxidation defects (MCAD, LCAD) Skeletal Muscle Weakness : Fatty acid oxidation defects (LCAD, SCAD, multiple acyl -CoA dehydrogenase 18 ) Management of IEM - NICU • Stop nutrient triggering disorder e.g. protein, galactose • Give high-energy intake • NICU care to correct tissue perfusion, dehydration, acidosis • Hyperammonemia Rx with Na benzoate, Na phenylbutyrate, arginine • Dialysis • Insulin to control hyperglycemia and reduce catabolism • Vitamins e.g Biotin, B6, B12 • Specific therapy e.g. carnitine, glycine MEDICAL Dependent on diagnosis and severity: Dietary or vitamin therapy Drug therapy BMT Avoid known environmental triggers Surgery 77