Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Size-exclusion chromatography wikipedia , lookup
Multi-state modeling of biomolecules wikipedia , lookup
Clinical neurochemistry wikipedia , lookup
Biosynthesis wikipedia , lookup
Amino acid synthesis wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
Oligonucleotide synthesis wikipedia , lookup
Biochemistry wikipedia , lookup
Peptide synthesis wikipedia , lookup
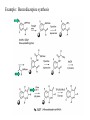
Molecular diversity and Drug Design Molecular diversity: differences in physical properties that exist in different molecules. (shape, size, polarity, charge, lipophilicity, polarizability, flexibility). Access to a diverse set of molecules increases the likelihood of finding an effective drug. Sources of Diversity: Natural products •Organisms, manipulation of biosynthetic machinery of microorganisms •Rate of discovery of truly novel natural product drugs has decreased. Synthetic compounds •Pharmaceutical companies optimizing their own drugs •Wide variety of starting materials and reaction types (unlike biosynthesis) All 10 largest selling drugs in 1994 were small organic molecules (<700g/mole) 8 were heterocycles and 1 was a natural product. Challenges for molecular diversity by organic synthesis: 1. Develop a variety of reactions with good yields for a broad set of starting materials 2. Efficiently identify the synthetic products Synthetic approaches: traditional (“target-oriented”) method is to make a desired compound (natural product or drug) using retrosynthetic analysis to plan the route: Synthetic approaches: A. Traditional- (solution phase) - linear or convergent. 25-50 compounds/person/year Linear: A A B + B D + C A D C B A B C D B Convergent: C + A D C D Traditional- (solid phase). Ex. - Peptide synthesis=optimized amide bond formation. Plan of synthetic route is simple…Linear polymer! amino acid R2 H2N R1 O C H dipeptide amino acid OH + H2N electrophile O R2 poor yields H2N C OH H Nucleophile O R1 H N C H O C H OH (and other combinations) How to solve problem of low yield of desired dipeptide? •Use protecting groups (“P” below) -so the correct groups react •Improve reactivity of CO2H (make a better electrophile) •Use solid phase resin beads which allow the use of excess reagents (high yield) improved purification because you can physically separate product R2 P H N C H R2 P H N O "activation" Resin bead (insoluble solid) OH electrophile O C H Better electrophile OR R1 + H2N R2 O C H Good Nucleophile "coupling" P H N C H O R1 H N C H O Traditional- (solid phase, continued) Solid phase peptide synthesis (SPPS) details 1. N-protected amino acid (Boc or Fmoc) is attached to bead by the C-terminus 2. Deprotect N-terminus, rinse with solvent. 3. Couple: Add solution of activated next amino acid. Let react, rinse with solvent. 4. Repeat deprotection and coupling for each subsequent monomer 5. Cleave peptide from resin Note: synthesize peptide from C to N terminus “Linker” attached to polystyrene resin bead R Synthetic approaches (continued): Parallel synthesis - simultaneous synthesis of multiple products, each in different reaction vessels with aid of automation. (Increased productivity 100-fold or more; more efficient). Each well has one compound, and can be identified by its position on the grid. Ex.: Parallel synthesis of a 96-member library of dipeptides in a microtiter plate with 8 rows and 12 columns. (partially shown) Step 1: each row starts with a different amino acid attached to a bead Step 2: each column adds a different second amino acid. (results in 96 different dipeptides) Step 3: Remove dipeptide from the bead Note: Each well contains one compound Step 4: Test each for biological activity. Note: pin and well grid technique also Synthetic approaches (continued): Example. Hydantoins can help control epilepsy. Parke-Davis made a library of 39 hydantoins using parallel synthesis BOC = protecting group for amines Synthetic approaches (continued): B. Combinatorial Synthesis: Multiple reactions in one reaction vessel, quickly generating a very large set of somewhat diverse products known as a combinatorial library. Usually the reaction is the same, but reactants are different. (more than one product in each reaction vessel/well) Number of compounds possible: bx (x = # steps) Design of combinatorial synthesis: sequential (a) or template (b): Combinatorial Synthesis Statistical: several reagents react at same time to get all possible combinations of reactants. (works if reaction rates are equal; hard to identify products). Mix and Split synthesis: 1. Synthesize a “mix and split” library on beads. Each bead has one compound on it. 2. Identify active compound by Step 1 combination of analytical techniques of synthesis and reaction history (need encoding molecules or deconvolution). To create 10,000 compounds: for a 3-step synthesis, this technique requires 22 different reaction vessels (not 30,000 vessels for standard synthesis!). Step 2 of synthesis Mix and split: Each bead has one compound on it, but each vessel/well contains a mixture of different beads! Identification of structure of compound on a bead: Deconvolution 1. ID most active mixture. Recall: in each of the final mixtures (pots) of beads, the residue coupled last is the same. Here, the pot (B) with “Y” as the last residue is active 2. Take all saved resin beads from the prior synthetic step (beads attached to dimers) and couple the appropriate (active) last residue. (Here, Y). 3. ID most active mixture. Here it is the one with X as the second residue. 4. Resynthesize all compounds with variations in the first residue. ID active compound. Mix and split: Disadvantages of Deconvolution •Deconvolution takes a lot of time and reagents •Most active mixture identified in round 1 may be active due to the cumulative action of multiple compounds (not the very strong action of one very active compound). Identification of structure of compound on a bead: Chemical encoding or tagging For very large libraries in which deconvolution is too difficult… Building Coding Block compound If you isolate one bead with activity from that vessel/well, how do you know which compound it is? Chemical encoding! X Y Z A B C XYZ XYZ CBA ABC 1. Isolate one bead XYZ 2. Cleave test compound frombead XYZ = test compound ABC = encoding tag molecule CBA ABC 3. Assay 4. If active, analyze bead for encoding molecule Example: Chemical encoding or tagging Coding compound: must be determined easily in small amounts. (oligonucleotides). Identification of structure of compound on a bead: Chemical encoding or tagging To screen the beads for activity: The encoded tag is removed, amplified by PCR, and sequenced to determine the structure of the active compound. Other methods of encoding: Peptides (coding for non-peptide and peptide libraries) Haloaromatic tags, separated by CE Still’s Binary code; isotopic labeling Computerized tagging, radiofrequency tags (microchips) Identification of structure of compound on a bead: Chemical encoding or tagging Example of binary encoding: Library of dipeptides from three amino acids (9 possible) using 4 tags. The tag (or tags) that uniquely represent the library monomer are reacted with the resin just before the library monomer is attached. Ala Phe Leu Tag for positi on 1 1 2 1+2 Tag for positi on 2 3 4 3+4 Dipeptide Ala-Ala Phe-Phe Leu- Leu Tag 1,3 2,4 1,2,3,4 Dipeptide Ala-PHe Phe-Ala Leu-Ala Tag 1,4 2,3 1,2,3 Dipeptide Ala-Leu Phe-Leu Leu- Phe Tag 1,3,4 2,3,4 1,2,4 n tags will code for 2n-1 library compounds How does the synthetic approach fit in with drug design? The traditional (target-oriented) approach : trial and error. Chemists develop a hypothesis about the structure of a potential drug, synthesize this substance, and have biological tests conducted. The hypothesis is confirmed or falsified. In the latter case, the chemist then proposes a new structural hypothesis and synthesizes a new molecule. Statistically, this cycle has to be repeated an average of 10 000 times before a new drug is found. A characteristic feature of this approach is that the structure is known before the test. The combinatorial principle: trial and selection. By means of combination (permutation) of the individual components (scaffolds and building blocks), all possible molecules in a substance family (chemotypes) are synthesized simultaneously/in parallel. The active representatives are subsequently selected from this library of compounds by using an assay and their structure is determined. It follows the principle of evolution, in which the most active representative is selected from a number of compounds (survival of the fittest). Role of combinatorial chemistry in drug design: •Early combichem: peptides (HIV protease inhibitors, antimicrobial agents, opiate receptor ligands) •Now: want diverse structures to find a lead - start with scaffolds that are small and will allow a wide variety of substituents for determining favorable binding interactions. Features of orally active drugs: MW <500g/mole logP<5 <6 H-bond donating groups <11 H-bond accepting groups Potential scaffolds for drug design Example: Benzodiazepine synthesis Example: Protease inhibitor libraries: Target = thermolysin Pharmacophore: O P R OR OH (incorporated in a peptide) Trimer peptidyl phosphonates: 540 member library by split pool: Three side-chains: P1, P1’, P2’ Cbz-X(6)-Y(5)-Z(18)-NH-resin (“Prot” = protecting group) Library was assayed for thermolysin inhibition: Assays revealed: 1. An active compound that matched the most potent inhibitor found in literature (KI = 49nM) (P1= phe; P1’= Leu; P2’ = ala) 2. New active sequences P2’ = arg, his, and gln (polar, charged) - unlike any reported in literature (hydrophobic) Examining only related analogs may lead to biases in design. But, combinatorial chemistry can reveal new potent structures. How biologically relevant is the diversity created in chemistry? Limited “chemical structure space”… Even the most beautiful molecules synthesized using the most elegant methods are useless if they do not affect a biological target. Theoretically, one could make every possible drug structure, and you will find at least ONE that works. However, the universe does not contain enough atoms to synthesize even one copy of every conceivable molecule! Possible solution to better match of biological and chemical structure space: Natural Product-Guided Combichem. Analogs of known individual natural products. Example: active metabolite of vitamin D3 Variations: Synthetic approaches: C. Diversity-oriented synthesis (DOS) method is to make a large collection of structurally complex and diverse compounds using forward analysis to plan the route: Creates natural product-like molecules that are stereochemically and skeletally diverse. (Not available by conventional combichem). Synthetic approaches: Diversity-oriented synthesis (cont). Diversity-Oriented Synthesis-Based Combinatorial Chemistry: Libraries of Natural Product-like Compounds Evans asymmetric aldol Sharpless asymmetric dihydroxylation O Furan Note: furan can •Ring open (7.6) •Ring expand (7.7, 7.8) Note: addition of Br to furan allows more chemical additions to the molecule DOS libraries •10-100 compounds (not 10,000 as in combichem) •Mostly cyclic, complex, resemble natural products, lots of stereochem •few synthetic steps Potential role of diversity-oriented synthesis and combinatorial chemistry in more efficient drug discovery: Small molecules obtained by DOS may find use in “chemical genetics” (later) as well as drug design. Alternatives: Other ways to generate potent drugs… “Click chemistry” Biological receptor selects the best fitting partial ligands that don’t fill the binding site from a range of modules that can react with one another. When the modules bind, they can react and form a compound to block the entire binding site. Target selects its own ligand! Angew. Chem. Int. Ed. 2001, 40, 2004 p. 2021 Dynamic combinatorial chemistry Biological receptor is exposed to a library of potential ligands, each of which is formed by reversible combinations of small building block compounds in equilibrium. More of the more strongly bound ligands will form. Target selects its own ligand! Dozens of azides and acetylenes were added to the enzyme (acetylcholinesterase), but only one pair binds and reacts… Azide = blue Acetylene = yellow triangle Proc. Natl. Acad. Sci. USA, 101, 1449 (2004)]. References for Diversity in Drug Design. Patrick, G. L. An Introduction to Medicinal Chemistry; Oxford University Press: New York, NY, 2001 Silverman, R. B. The Organic Chemistry of Drug Design and Drug Action ; Academic Press: San Diego, CA, 1992. Thomas, G. Medicinal Chemistry An Introduction; John Wiley and Sons, Ltd.: New York, NY, 2000. Combinatorial Chemistry and Molecular Diversity in Drug Discovery; Gordon, E. M.; Kerwin, J. F., Eds.; Wiley-Liss: New York, NY, 1998 Terrett, N. K. Combinatorial Chemistry; Oxford Chemistry Masters; Oxford University Press: New York, NY, 1998. Arya, P.; Joseph R.; Gan,Z.; Rakic, B. “Exploring New Chemical Space by Stereocontrolled DiversityOriented Synthesis” Chemistry & Biology, 2005, 12, 163–180. Burke, M. D.; Schreiber, S. L. “A planning strategy for diversity-oriented synthesis” Angew. Chem. Int. Ed. 2004, 43, 46–58. Schreiber, S. L. “Target-oriented and diversity-oriented organic synthesis in drug discovery” Science, 2000, 287, 1964-1969. a) A. Kornb erg, Bioc hemistry 198 7, 26, 68 88 ± 689 1; comments on Korn berg s di scussi on of this top ic can be fo un d in further articles: b) A. Kornberg, C hem. Biol. 199 6, 3, 3 ± 5; c) L. H. Hurley, J. Med. Chem. 198 7, 30, 7A± 8A; d) G. deStevens, J. Med. Chem. 19 91, 34, 26 65 ± 267 0.