Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
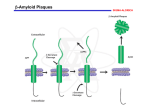
Current Alzheimer Research, 2007, 4, 473-478 473 Alzhemed: A Potential Treatment for Alzheimer's Disease Paul S. Aisen1,*, Serge Gauthier2, Bruno Vellas3, Richard Briand4, Daniel Saumier4, Julie Laurin4 and Denis Garceau4 1 Georgetown University, Washington, DC, US. 2McGill University, Montreal, Canada, 3Alzheimer Clinical and Research Center, Toulouse, France, 4 Neurochem Inc. Laval, Canada Abstract: As a potential disease-modifying treatment for AD, Alzhemed (tramiprosate) is a compound that binds to soluble amyloid-beta peptide (A) and inhibits the formation of neurotoxic aggregates that lead to amyloid plaque deposition in the brain. The safety, tolerability, and pharmacodynamic effects of Alzhemed were assessed in a double-blind study in which 58 individuals with mild-to-moderate AD (MMSE 13-25) were randomized to receive placebo or Alzhemed 50, 100 or 150 mg BID for 3 months. At the end of the double-blind phase, 42 of these subjects entered a 36-month openlabel (OL) phase in which they received Alzhemed 150 mg BID. Assessments included plasma and cerebrospinal fluid (CSF) Alzhemed concentrations, CSF levels of A, as well as cognitive (Alzheimer’s Disease Assessment Scale-cognitive subscale, Mini-Mental State Examination) and clinical performance (Clinical Dementia Rating scale, Sum-of-Boxes) measures. Alzhemed was safe and well tolerated, crossed the blood-brain barrier, and dose-dependently reduced CSF A42 levels after 3 months of treatment. Mild AD subjects (MMSE 19-25 at entry) displayed greater reduction of CSF A42 levels than moderate AD participants (MMSE 13-18 at entry). There was no effect of Alzhemed on the cognitive or clinical measures after 3 months of treatment. The OL follow-up suggested a stabilization of cognitive function especially in mild AD subjects over the 36-month study period. Alzhemed thus appears to be well tolerated with long-term exposure and reduces CSF A42 levels in mild-to-moderate AD subjects. These findings will be discussed in the context of two large-scale randomized, double-blind, placebo-controlled Phase III clinical trials that are currently being conducted to test the long-term safety and efficacy of Alzhemed. INTRODUCTION Alzheimer’s disease (AD), among the most important health care problems worldwide, is a progressive neurodegenerative disorder causing decline in memory, learning and other aspects of cognitive function associated with impaired activities of daily living, frequent behavioral complications, and eventually death. AD is now a treatable condition. Elucidation of the central role of cholinergic dysfunction in the symptoms of the disease led to the development of cholinergic therapies, specifically the acetylcholinesterase inhibitors, for AD treatment [1]. These drugs partially compensate for the cholinergic deficiency by slowing acetylcholine clearance from the synapse via inhibition of the primary clearance enzyme. Treatment with cholinesterase inhibitors donepezil, rivastigmine and galantamine results in modest improvement on cognitive symptoms evident in the first few months of treatment, as well as stabilization of daily function and behavior. Current AD treatment options also include memantine. Memantine acts on glutamate neurotransmission; it is a moderate affinity antagonist at the NMDA receptor. While it was hoped that NMDA inhibition might reduce excitotoxic neuronal damage and thus slow disease progression, clinical study results suggest symptomatic improvement without clear evidence of altered rate of decline. The apparent ab*Address correspondence to this author at the Department of Neurology, Georgetown University Medical Center, Building D, Room 202B, 4000 Reservoir Rd NW, Washington, DC 20057, USA; Tel: 202-784-6671; Fax: 202-784-4332; E-mail: [email protected] 1567-2050/07 $50.00+.00 sence of disease modification may be explained the high drug concentrations necessary to provide neuroprotection [2]. Memantine treatment appears to be particularly useful in moderate to severe AD, either as monotherapy [3] or in combination with a cholinesterase inhibitor [4]. These symptomatic AD treatments, all developed within the past 15 years, provide modest but clinically meaningful benefits to people with AD and their caregivers. But there is no definitive evidence that any of these drugs alters the underlying disease process, or significantly slows the relentless progression of dementia. This remains the major goal of current drug development efforts in AD research. AMYLOID HYPOTHESIS As noted above, the current treatments target neurotransmission, and thus modulate cognitive function without addressing the initiating events in the neurodegenerative cascade. But steady progress in identifying the pivotal events in that cascade presents clear targets for disease-modifying therapy. The two principal pathological lesions in the AD brain are the extracellular amyloid plaque and the intracellular neurofibrillary tangle. The primary constituent of the amyloid plaque is the A peptide, a fragment derived by two sequential cleavages from a parent protein, the amyloid precursor protein (APP). The A peptides aggregate into oligomers and amyloid fibrils, and ultimately deposit as amyloid plaques. Neurofibrillary tangles consist primarily of hyperphosphorylated tau protein. Tau is an essential micro©2007 Bentham Science Publishers Ltd. 474 Current Alzheimer Research, 2007, Vol. 4, No. 4 tubule stabilizing protein; hyperphosphorylation is associated with conformational change, fibrillization and loss of function. Both amyloid and tau cascades have been viewed as plausible therapeutic target systems for disease modifying AD treatments [5]. However, compelling evidence suggests that it is the amyloid cascade that is pivotal in driving the neurodegenerative process [6]. Most convincing is the genetic evidence. Each of the three genes that, when mutated, can cause familial autosomal dominant AD (FAD), are directly involved in the cleavage of APP to release the A peptide; these genes code APP itself (the mutations increase susceptibility to secretase cleavage at the or sites at either end of the A sequence), and presenilins 1 and 2 (components of the secretase complex, that when mutated increase secretase cleavage). This means that the cause of FAD must be related to APP cleavage with generation of A. Since FAD and sporadic AD differ only in age of onset and inheritance, it follows that APP cleavage must be pivotal in sporadic AD as well (though in sporadic AD there are no pathogenic mutations; rather, insufficiently characterized processes presumably related to aging must increase A generation). The mechanism by which APP cleavage to release A initiates the neurodegenerative cascade is less clear. The amyloid plaques may contribute, as suggested by the characteristic neuritic changes surrounding plaques. But there is growing evidence that A is damaging in its diffusible monomeric and oligomeric forms (Klein, 2006). It has long been known that A is highly toxic to neurons in culture; recent studies using transgenic mouse models show that oligomeric A is implicated in synaptic dysfunction [7] and tau hyperphosphorylation [8]. It is thus clear that the amyloid pathway is pivotal in AD, and offers primary targets for disease modifying drug development [9]. But whether the specific goal of treatment should be reduction of plaque density, or reduction of diffusible forms of A, or even protection of neurons against damage caused by either or both, remains uncertain. ANTI-AMYLOID DRUG DEVELOPMENT In theory, the first step in the amyloid cascade, APP cleavage, is optimal for drug intervention. But there have been some practical obstacles to this approach. An inhibitor of -secretase would be a promising candidate, as this enzyme is required for A generation, though inessential for life [10, 11]. But development of a specific and drug-like secretase inhibitor has proven to be technically difficult [9]. Inhibitors of the -secretase complex have been identified, but -secretase activity is essential, and toxicity has been an issue with some drug candidates. A subset of non-steroidal anti-inflammatory drugs show -secretase modulating activity with reduction in amyloid accumulation in transgenic mice [12], though tolerable doses may be insufficient for brain amyloid reduction; R-flurbiprofen, an NSAID enantiomer that is tolerated at relatively high doses (because it lacks the cyclooxygenase inhibition of the L-form), is currently being tested in a Phase III AD trial [13]. It is also feasible to reduce amyloid accumulation by increasing clearance of amyloid by enhancing phagocytosis or Aisen et al. transport to the periphery. The interrupted active amyloid vaccination program, while unsuccessful (because of a serious adverse effect, encephalitis, in some subjects), demonstrated the promise of this approach [14]. The vaccine apparently induced clearance of amyloid plaques by augmenting phagocytosis, with cognitive benefit. At present, a number of companies and investigators are pursuing passive immunization programs aiming to augment phagocytosis, or alternatively promote clearance by sequestering A peptides in the periphery [15, 16]. Reducing aggregation of amyloid and fibrillogenesis may increase clearance of insoluble amyloid from brain. There are several approaches to reducing aggregation. Since heavy metals promote the aggregation process, chelating agents may increase clearance; this has been demonstrated with the zinc and copper binding compound clioquinol [17, 18]. Derivatives of -sheet binding dyes have also been considered candidate anti-aggregation compounds [19]. THE ALZHEMED PROGRAM The strategy that led to the development of Alzhemed (tramiprosate) focused on the role of proteoglycans in the amyloid aggregation and fibrillogenesis process [20, 21]. The polyanionic glycosaminoglycan (GAG) chains of tissue proteoglycans bind to amyloid peptides promoting aggregation in brain. Small sulfated or sulfonated molecules were developed to interfere with this binding and slow the process of fibrillogenesis. One such low molecular weight molecule, Alzhemed, proved to be a potent inhibitor of aggregation and fibrillogenesis in vitro, and also inhibited the neurotoxicity of amyloid peptides [20, 21]. A study in an APP transgenic mouse model confirmed the impact of systemic Alzhemed treatment on amyloid accumulation in brain [20]. Treatment of 9 week old TgCRND8 mice for 8 weeks reduced the area occupied by amyloid plaques in brain by 29%. There was a reduction of similar magnitude in brain levels of both soluble and insoluble amyloid peptides. While there continues to be debate regarding the relative contribution of fibrillar amyloid versus other forms of the peptide to neurodegeneration, increasing evidence points to the importance of diffusible oligomeric forms. For example, treatment of transgenic mice with anti-amyloid antibodies produces a rapid improvement in memory prior to any change in amyloid plaque burden [22]; this suggests that the interaction of the antibodies with diffusible rather than plaque-bound amyloid peptide improves function. It has been confirmed that oligomeric A peptides inhibit long term potentiation in brain slices, a model for synaptic information storage. The rapid cognitive improvement reported with treatment of AD with pooled human immunoglobulin (which contains natural anti-amyloid antibodies) [23] may be mediated by an immediate impact on diffusible A. The growing acceptance of the importance of oligomeric A in the pathophysiology of Alzheimer’s disease provides a strong rationale for targeting non-fibrillar A with antiamyloid interventions against AD. There is even concern that drugs or antibodies that interact with fibrillar deposits may increase levels of diffusible A, aggravating cognitive Alzhemed: A Potential Treatment for Alzheimer's Disease dysfunction. It is notable that, in contrast to the amyloid vaccine, Alzhemed specifically targets diffusible A; there is no interaction between the drug and fibrillar deposits [20]. Preclinical studies of Alzhemed support its potential as a drug candidate [20]. Genotoxicity studies including AMES testing, the mammalian cell cytogenetic test and the mouse micronucleus test have been negative. hERG testing did not suggest any adverse effect on cardiac conduction. Long term oral toxicity studies in rats and dogs demonstrate safety over a wide dosing range, with only mild gastrointestinal symptoms at high doses. Studies with systemically-administered 14 C labeled drug demonstrated brain penetration. The half life of the drug after oral administration was about 3 hours in plasma and at least 16 hours in brain. CLINICAL DEVELOPMENT OF ALZHEMED A series of Phase I studies in both young and elderly adults explored the safety and tolerability of Alzhemed in humans (unpublished data provided by Neurochem, Inc.). Single (up to 200mg orally) and repeated doses (up to 200mg, bid orally) were safe and generally well tolerated; treatment with higher doses was limited by gastrointestinal side effects, specifically nausea and vomiting. The time to reach peak plasma level was 5 hours, with estimated terminal plasma half life in the range of 1.5 to 5 hours. A randomized double-blind placebo-controlled Phase II study was conducted at six U.S. centers; the aims were to assess safety, tolerability, pharmacokinetics and pharmacodynamic effects. The mechanism of action of the drug indicates that the impact on disease will be slowed progression with long-term treatment rather than symptomatic improvement; since no measurable decline in the placebo group is expected in 12 weeks, no effect of treatment on cognitive or clinical measures at 12 weeks was anticipated. A total of 58 individuals with mild to moderate AD were randomly assigned to one of four treatment groups: placebo, or Alzhemed 50mg bid, 100mg bid or 150mg bid orally. Eligible subjects had MMSE scores ranging from 13 to 25 (stratified into two groups, mild [19-25] and moderate [1318]), and could be taking stable doses of cholinesterase inhibitors. Study medication was administered for 12 weeks. Outcome measures included clinical and laboratory examinations, and cognitive (Mini Mental State Examination, or MMSE [24] and the cognitive subscale of the Alzheimer’s Disease Assessment Scale, or ADAScog [25]) and clinical (Clinical Dementia Rating Sum of Boxes, or CDR-SB [26]) assessments. Lumbar punctures were performed before and after the 12 week course of treatment, for measurement of drug and biomarker levels. The results of this Phase II trial indicated that treatment with Alzhemed at these doses was safe. There was a dosedependent occurrence of gastrointestinal symptoms, particularly transient nausea and vomiting. There were no serious adverse events related to treatment, and no adverse effects on vital signs, laboratory studies or electrocardiograms. As expected, there was no decline in cognitive or clinical measures in the placebo group during the 12 week blinded phase, and no difference on these measures between groups. Current Alzheimer Research, 2007, Vol. 4, No. 4 475 Analysis of plasma drug levels indicated that the time to maximum level was between 4.5 and 6 hours after dosing; the elimination half-life was approximately 2-2.5 hours. There was no evidence of plasma accumulation with repeated dosing. Measurement of Alzhemed levels in CSF confirmed penetration into the central nervous system; CSF levels were measurable in 21/36 actively treated subjects, with concentrations ranging from 2.6 to 7.3 ng/ml. Of particular interest, a regression analysis performed on A CSF concentration changes as a function of Alzhemed dosage level revealed a significant linear relation (p = 0.041) between decrease in A42 concentration and dose of Alzhemed administered after three months of treatment. The magnitude of the change in CSF A42 in these two groups was greater for the mild subgroup (32% decline) than the moderate subgroup (14% decline). In general, CSF A42 levels reflect diffusible levels of the peptide in brain [27-29]; though amyloid plaque load influences CSF A42 levels, since plaque levels presumable remain stable over 12 weeks, change in CSF level should be an accurate indicator of change in diffusible A levels. Thus, the CSF A results suggest that Alzhemed has the anticipated pharmacodynamic impact in individuals with AD. Subjects in the Phase II study were invited to participate in an open-label extension trial; 42 individuals elected to participate. In the extension, subjects were treated with Alzhemed 150mg po bid, and were followed with cognitive and clinical assessments at 3 month intervals for up to 36 months. In the absence of a concurrent control group, the open-label extension assessment data must be interpreted with caution. However, the rate of decline, particularly in the mild group, appeared to be slow in comparison to expectations based on published data [30] (Figs. 1 and 2). The open label experience confirmed the long term tolerability of Alzhemed in individuals with AD. These encouraging results led to the launch of a large Phase III pivotal trial in the U.S. and Canada, followed by a similar trial in Europe. The Phase III trial design reflects the expected clinical response: slowing of decline in relatively mildly impaired individuals. Eligible subjects in the North Amercian trial are on stable symptomatic treatment (cholinesterase inhibitors, with or without memantine), and have an MMSE score between 16 and 26. To allow sufficient time for the cognitive and clinical trajectories for the active and placebo groups to separate, the treatment period is 18 months. Standard cognitive (ADAScog, MMSE), behavioral (Neuropsychaitric Inventory, or NPI [31]) and functional (Disability Assessment for Dementia, or DAD [32]) measures are used. To explore the effect of treatment on amyloid and the neurodegenerative process, lumbar punctures for CSF A and tau determination, and structural MRI scans for hippocampal, entorhinal cortex and whole brain volumes, are obtained in subsets of study participants. The U.S./Canada Phase III trial was launched in mid2004, and was fully enrolled with over 1000 subjects in less than one year. The European study started in the fall of 2005, with enrollment still in progress. The primary purpose of these pivotal trials is to establish the clinical efficacy of Alzhemed using standard cognitive 476 Current Alzheimer Research, 2007, Vol. 4, No. 4 Aisen et al. Fig. (1). Change in ADAScog scores after 24 months in open-label extension study. Fig. (2). Change in ADAScog scores after 36 months in open-label extension study. and clinical assessment tools, and to establish the safety and tolerability of the medication in a large study population. Disease-modification will be assessed by examining Alzhemed’s effect on hippocampal entorhinal cortex, and whole brain atrophy rate, as indicated by structural MRI scanning at Baseline and following 18 months of treatment. The analysis of the pattern of effect on cognitive and clinical assessment scores may also support disease modification. Specifically, absence of a short term treatment effect (eg, at 6 months), with increasing treatment group separation at 12 and 18 months (ie, a change in the slope of decline), would be suggestive of disease-modification. Ancillary data will be used to confirm the pharmacodynamic effect (reduction in soluble amyloid peptide in brain, as reflected by levels in cerebrospinal fluid). Alzhemed: A Potential Treatment for Alzheimer's Disease Current Alzheimer Research, 2007, Vol. 4, No. 4 ANTI-AMYLOID [7] The concept that systemic treatment with an anti-amyloid drug shown pre-clinically to prevent fibril formation and deposition in tissues leads to clinical benefits has now been tested in a multicenter trial. In amyloid A (AA) amyloidosis, the amyloidogenic peptide is derived from serum amyloid associated protein, or SAA. SAA is an acute phase protein, elevated in many chronic inflammatory conditions including rheumatoid arthritis, familial Mediterranean fever and tuberculosis. As in any amyloid related disease, the pathogenic protein adopts a highly aggregable -sheet conformation, with resultant deposition in tissue. In AA amyloidosis, the kidney is a primary site of the deposition, leading to proteinuria and renal insufficiency. [8] FIBRILLEX: STRATEGY VALIDATION OF Fibrillex is an anti-amyloid drug, like Alzhemed, that has been shown to inhibit amyloid deposition in a mouse model of AA amyloidosis. To study its clinical efficacy, the trial enrolled 183 individuals with AA amyloidosis and renal involvement, and followed renal function and other clinical endpoints during 24 months of treatment. Treatment with Fibrillex resulted in preservation of renal function as measured by creatinine clearance and need for dialysis. The Fibrillex trial provides some support to this strategy, and suggests that the brain-penetrating, anti-amyloid drug Alzhemed may reduce amyloid-mediated damage in the AD brain. CONCLUSION The leading hypothesis regarding the pathophysiology of AD suggests that the amyloid peptide is the most promising target for disease-modifying interventions. Alzhemed is a small molecule that binds soluble A peptide and prevents amyloid formation and deposition, and inhibits A-induced neurotoxicity. The drug penetrates the blood-brain barrier, and is safe and tolerable in humans. Phase II testing has confirmed an anti-amyloid pharmacodynamic effect, as demonstrated by a reduction of CSF A42 in Alzhemed treated patients. Two pivotal phase III trials will now determine the efficacy of Alzhemed in slowing progression of AD. If the anti-amyloid activity of Alzhemed does indeed significantly slow disease progression, it will represent a major advance in AD therapeutics. REFERENCES [1] [2] [3] [4] [5] [6] Giacobini E, editor. Cholinesterases and Cholinesterase Inhibitors. London: Martin Dunitz Ltd, (2000). Creeley C, Wozniak DF, Labruyere J, Taylor GT, Olney JW. Low Doses of Memantine Disrupt Memory in Adult Rats. J Neurosci 26:3923-32 (2006). Reisberg B, Doody R, Stoffler A, Schmitt F, Ferris S, Mobius HJ. Memantine in moderate-to-severe Alzheimer's disease. N Engl J Med 348:1333-41 (2003). Tariot PN, Farlow MR, Grossberg GT, Graham SM, McDonald S, Gergel I. Memantine treatment in patients with moderate to severe Alzheimer disease already receiving donepezil: a randomized controlled trial. Jama 291:317-24 (2004). Cummings JL. Alzheimer's disease. N Engl J Med 351:56-67 (2004). Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297:353-6 (2002). [9] [10] [11] [12] [13] [14] [15] [16] [17] [18] [19] [20] [21] [22] [23] [24] [25] [26] [27] [28] 477 Lacor PN, Buniel MC, Chang L, Fernandez SJ, Gong Y, Viola KL, et al. Synaptic targeting by Alzheimer's-related amyloid beta oligomers. J Neurosci 24:10191-200 (2004). Oddo S, Caccamo A, Tran L, Lambert MP, Glabe CG, Klein WL, et al. Temporal profile of amyloid-beta (Abeta) oligomerization in an in vivo model of Alzheimer disease. A link between Abeta and tau pathology. J Biol Chem 281:1599-604 (2006). Aisen PS. The development of anti-amyloid therapy for Alzheimer's disease : from secretase modulators to polymerisation inhibitors. CNS Drugs 19:989-96 (2005). Roberds SL, Anderson J, Basi G, Bienkowski MJ, Branstetter DG, Chen KS, et al. BACE knockout mice are healthy despite lacking the primary beta- secretase activity in brain: implications for Alzheimer's disease therapeutics. Hum Mol Genet 10:1317-24 (2001). Luo Y, Bolon B, Kahn S, Bennett BD, Babu-Khan S, Denis P, et al. Mice deficient in BACE1, the Alzheimer's beta-secretase, have normal phenotype and abolished beta-amyloid generation. Nat Neurosci 4:231-2 (2001). Weggen S, Eriksen JL, Das P, Sagi SA, Wang R, Pietrzik CU, et al. A subset of NSAIDs lower amyloidogenic Abeta42 independently of cyclooxygenase activity. Nature 414:212-6 (2001). Molecule of the month. MPC-7869 (Flurizan). Drug News Perspect 18:141 (2005). Gilman S, Koller M, Black RS, Jenkins L, Griffith SG, Fox NC, et al. Clinical effects of Abeta immunization (AN1792) in patients with AD in an interrupted trial. Neurology 64:1553-62 (2005). DeMattos RB, Bales KR, Cummins DJ, Dodart JC, Paul SM, Holtzman DM. Peripheral anti-Abeta antibody alters CNS and plasma Abeta clearance and decreases brain Abeta burden in a mouse model of Alzheimer's disease. Proc Natl Acad Sci USA 3:3 (2001). Matsuoka Y, Saito M, LaFrancois J, Gaynor K, Olm V, Wang L, et al. Novel therapeutic approach for the treatment of Alzheimer's disease by peripheral administration of agents with an affinity to beta-amyloid. J Neurosci 23:29-33 (2003). Bush AI. The metallobiology of Alzheimer's disease. Trends Neurosci 26:207-14 (2003). Raman B, Ban T, Yamaguchi K, Sakai M, Kawai T, Naiki H, et al. Metal ion-dependent effects of clioquinol on the fibril growth of an amyloid {beta} peptide. J Biol Chem 280:16157-62 (2005). Klunk WE, Debnath ML, Koros AC, Pettegrew JW. ChrysamineG, a lipophilic analogue of Congo red, inhibits Abeta-induced toxicity in PC12 cells. Life Sci 63:1807-14 (1998). Gervais F, Paquette J, Morissette C, Krzwkowski P, Yu M, Azzi M, et al. Targeting soluble A peptide with 3APS for the treatment of brain amyloidosis. Neurobiol Aging in press (2006). Gervais F, Chalifour R, Garceau D, Kong X, Laurin J, McLaughlin R, et al. Glycosaminoglycan mimetics: a therapeutic approach to cerebral amyloid angiopathy. Amyloid 8 Suppl 1:28-35 (2001). Lee EB, Leng LZ, Zhang B, Kwong L, Trojanowski JQ, Abel T, et al. Targeting amyloid-beta peptide (Abeta) oligomers by passive immunization with a conformation-selective monoclonal antibody improves learning and memory in Abeta precursor protein (APP) transgenic mice. J Biol Chem 281:4292-9 (2006). Dodel RC, Du Y, Depboylu C, Hampel H, Frolich L, Haag A, et al. Intravenous immunoglobulins containing antibodies against betaamyloid for the treatment of Alzheimer's disease. J Neurol Neurosurg Psychiatry 75:1472-4 (2004). Folstein M, Folstein S, Mchugh P. The Mini-Mental State Examination. J Psychiatric Res 1975;12:189-98. Rosen WG, Mohs RC, Davis KL. A new rating scale for Alzheimer's disease. Am J Psych 141:1356-64 (1984). Morris JC. The Clinical Dementia Rating (CDR): Current version and scoring rules. Neurology 43:2412-4 (1993). Barten DM, Guss VL, Corsa JA, Loo AT, Hansel SB, Zheng M, et al. Dynamics of {beta}-Amyloid Reductions in Brain, Cerebrospinal Fluid and Plasma of {beta}-Amyloid Precursor Protein Transgenic Mice Treated with a {gamma}-Secretase Inhibitor. J Pharmacol Exp Ther 312:635-43 (2004). Best JD, Jay MT, Otu F, Churcher I, Reilly M, Morentin-Gutierrez P, et al. In vivo characterisation of A{beta}(40) changes in brain and CSF using the novel {gamma}-secretase inhibitor MRK-560 (N-[cis-4-[(4-chlorophenyl)sulfonyl]-4- (2,5-difluorophenyl)cyclohexyl]-1,1,1- trifluoromethanesulfonamide) in the rat. J Pharmacol Exp Ther 317:786-90 (2006). 478 Current Alzheimer Research, 2007, Vol. 4, No. 4 [29] [30] Lemere CA, Beierschmitt A, Iglesias M, Spooner ET, Bloom JK, Leverone JF, et al. Alzheimer's disease abeta vaccine reduces central nervous system abeta levels in a non-human primate, the Caribbean vervet. Am J Pathol 2004;165:283-97. Stern RG, Mohs RC, Davidson M, Schmeidler J, Silverman J, Kramer-Ginsberg E, et al. A longitudinal study of Alzheimer's disease: measurement, rate, and predictors of cognitive deterioration. Amer J Psychiatry 151:390-6 (1994). Received: May 30, 2006 Accepted: September 28, 2006 Aisen et al. [31] [32] Cummings J. The neuropsychiatric inventory: Assessing psychopathology in dementia patients. Neurology 48 (Suppl 6):S10-S6 (1997). Gelinas I, Gauthier L, McIntyre M, Gauthier S. Development of a functional measure for persons with Alzheimer's disease: the disability assessment for dementia. Am J Occup Ther 53:471-81 (1999).