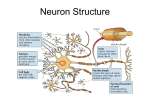
Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Nanofluidic circuitry wikipedia , lookup
Flux (metallurgy) wikipedia , lookup
Photoconductive atomic force microscopy wikipedia , lookup
Energy applications of nanotechnology wikipedia , lookup
Tunable metamaterial wikipedia , lookup
Sessile drop technique wikipedia , lookup
Surface tension wikipedia , lookup
Self-assembled monolayer wikipedia , lookup
Ultrahydrophobicity wikipedia , lookup
Low-energy electron diffraction wikipedia , lookup
3
In Situ STM Studies of Model Catalysts
Fan Yang and D. Wayne Goodman
3.1
Introduction
The surface science approach to studying heterogeneous catalysis dates back to the
pioneering work of Langmuir [1] in the 1910s that addressed the adsorption ofgases
on catalyst surfaces. Since then surface science studies of catalytic processes have
played a central role in our understanding of catalysis and have aided in the design
and improvement ofcatalysts for energy and environmental uses. The goal ofsurface
science investigations has been to provide structural and spectroscopic information
of catalyst surfaces at the spatial and temporal limit. Scanning tunneling microscopy
(STM), with the capacity to reach the spatial limit at the atomic level, has ignited
considerable interest since its discovery and has become a widely used tool in catalytic
science.
By following a selected area or a molecule at a model catalyst surface, in situ STM
can provide temporal measurements regarding the elementary steps of catalytic
transformations. The capabilities of in situ STM allow one to follow the dynamic
change of surface species and identify what Taylor described as the "active site" in
catalytic reactions. Such studies also provide kinetic measurements at the atomic
scale, enabling the most precise modeling of macroscopic reactions. With the
development of STM techniques, a large body of in situ STM work has emerged
in the past decade regarding catalytic processes such as adsorption and diffusion,
surface reaction, and catalyst deactivation. These experiments provide invaluable
insights into the fundamental issues of catalysis.
The timescale ofcatalytically important processes ranges from 10- 12 to 104 s, with
the chemisorptions and reactions taking place within picoseconds whereas catalyst
deactivation occurs in seconds or minutes. Although the timescale of the funda
mental process of adsorption and reaction is beyond the time resolution of STM,
information about the pathway and energetics ofadsorption/reaction can be acquired
with STM by monitoring the change in the spatial distribution ofsurface adsorbates.
Indeed, in situ STM can be combined with femtolasers to probe surface processes at
both the spatial and the temporal limits. The feasibility of combining these two
Scanning Tunneling Microscopy in Surface Science. Nanoscience and Catalysis
Edited by Michael Bowker and Philip R. Davies
Copyright © 2010 WILEY·VO! Verlag GmbH & Co. KGaA. Weinheim
ISBN: 978-3·527·31982·4
,
~. I
,1
I
561
3 In Situ STM Studies of Model Catalysts
techniques has been demonstrated in a recent study by Bartels et al. [2]. Such studies
are extremely promising with respect to our understanding of surface chemical
processes. In this chapter, we show how in situ STM has helped to visualize
elementary steps of chemical reaction and to elucidate mechanisms of catalyzed
processes.
Like most surface science techniques, conventional in situ STM studies have been
carried out in UHV on model catalysts consisting ofextended planar surfaces. When
extrapolating the information obtained in UHV surface science studies to real·world
catalysis, two issues have generally concerned the catalysis community, namely. the
pressure and material gaps.
The pressure gap refers to the fact that surface science studies are conducted under
UHV conditions (10- 10_10-- 14 bar). whereas industrial catalytic reactions typically are
carried out at high pressures (l-toOO bar). Over 10 orders ofmagnitude difference in
the pressure of reactant gases can drastically change the interaction of reactants on
the catalyst surface. a process essential to a catalytic reaction. The material gap refers
to the gap between the surface structure of metal single crystals often studied in
surface science and that oftechnical catalysts. "Real-world" catalysts usually consist of
small metal clusters ranging from 1 to 100 nm in size. finely dispersed onto a high·
surface-area oxide support. These metal dusters can have structures and properties
that are quite different from the bulk metal. In catalytic research, it is well
documented that the reactivity and selectivity of catalysts often depend on the size
and shape of supported metal clusters [3]. Furthermore, the presence of the oxide
support can modify the structure and properties of supported metal clusters.
The effect of cluster size and metal support interaction cannot be addressed in
surface science studies on well-defined Single crystal metal surfaces. As a local
structural probe. STM has the advantage ofaddressing these two issues. Although the
operational range ofSTM extends from UHV to high pressures, there are challenges
in maintaining the stability of STM at catalytically realistic operating temperatures
and pressures. Nevertheless. it is possible to apply STM to study catalytic reactions
under realistic conditions. To bridge the material gap, supported model catalysts.
consisting of small metal clusters supported on planar oxide surfaces, have been
introduced into surface science studies. STM is ideally suited to precisely charac
terize the structure of these supported model catalysts. In this chapter, we show the
recent progress in bridging the pressure and material gaps by applying in situ STM
to the study of model catalysts under realistic reaction conditions.
3.2
Instrumentation
To visualize the fundamental steps of chemisorptions and reactions that occur at
surfaces, in situ STM investigations typically monitor the diffusion or transformation
of adsorbed molecules. A series of snapshots of preselected surface regions,
compiled into a STM movie, can reveal the evolution of surfac~ phenomena.
On metal surfaces, the surface diffusion of adsorbates is usually so rapid that the
surface temperature mu
STM viewing. Low-ternl
potentially allows the det
enables the precise contI
the STM tip.
To measure reaction ki
at temperatures relevant
ature (VT) STM is requir'
typical scan rate of on
Considering the scannil
acquisition time ofSTM
the scanning component
few STM groups adrieve
model catalyst surfaces [
and using high perform
pushed the scan rate abc:
atomic resolution and 31
Recently, a few grmq
investigations to high ~
over a wide pressure
small changes at the
ambient gas. Efforts
temperatures and
For in situ STM
able to track a preirele~
junction instabilities
contact with a
approaches that
ofthe particular areas
developed where
with the collimated
monitor a pn$ellecte<lll
pressure over 12
In addition to
for in situ STM
extensively studied
tip is important for
and in situ tip
transition metals
gases, especially
ical and thermal
3.2 Instrumentation
surface temperature must be lowered below room temperature (RT) for successful
STM viewing. Low-temperature (LT) STM, developed in the mid-1990s, not only
potentially allows the determination ofreaction intermediates and pathways but also
enables the precise control and measurement ofthe bond activation processes using
the STM tip.
To measure reaction kinetics, STM should have the capability to resolve adsorbates
at temperatures relevant to catalytic reactions. For this purpose, a variable temper
ature (VT) STM is required, as well as capabilities for rapid scanning. VT STM with a
typical scan rate of one frame per minute was developed in the mid-1990s.
Considering the scanning probe is a mechanical probe driven by electronics, the
acquisition time of STM images is typically restricted by the mechanical behavior of
the scanning components and the performance ofthe electronics. In the mid-1990s, a
few STM groups achieved a fast scan rate of approximately 20 frames/s on extended
model catalyst surfaces [4-6]. Working on the compact design of the scanner probe
and using high performance electronics, Frenken and coworkers [7] have recently
pushed the scan rate above the video rate (",,50frame/s) on a graphite surface, with
atomic resolution and an image with 256 x 256 pixels.
Recently, a few groups have taken up the challenge to extend the in situ STM
investigations to high pressures. A major challenge in imaging surfaces with STM
over a wide pressure range is the sensitivity of the tunneling current to extremely
small changes at the tunneling junction resulting from induced instabilities by the
ambient gas. Efforts have emphasized the design of a STM that can work at high
temperatures and pressures with greater stabilities [8-11].
For in situ STM studies at high temperature and pressures, the inability of being
able to track a preselected surface area is often the limiting factor given the tunnel
junction instabilities and sample drifts. To overcome this challenge and to maintain
contact with a specific surface region, it is important to develop experimental
approaches that pattern the surface without influencing the kinetics and dynamics
of the particular areas under study. A "shadowing" technique (Figure 3.1a) has been
developed where metal atoms are dosed with the STM tip in the tunneling position
with the collimated metal flux creating a shadow of the tip on the substrate [12, 13].
For metal clusters supported on an oxide surface, tip manipulation is another method
ofchoice (Figure 3.1c). This technique removes clusters from a specific area through
aggressive scanning. Using the STM tip to pattern the surface, it is now possible to
monitor a preselected surface area at elevated temperatures while changing the gas
pressure over 12 orders of magnitude.
In addition to the instrumental performance, the STM tip is ofprimary importance
for in situ STM measurements. Methods for the preparation of STM tips have been
extensively studied with a goal ofpreparing an atomically sharp tip [14-19]. The STM
tip is important for high-pressure studies with respect to two aspects, tip selection
and in situ tip regeneration. To ensure a continuous DOS near the Fermi level,
transition metals are usually selected to prepare STM tips. In the presence ofreactant
gases, especially under high.pressure and high-temperatur;;: conditions, the chem
ical and thermal stability of the STM tip becomes the ultimate limit for reaction
studies and thus the major concern in tip selection. Tungsten tips are very stable in
1
I
57
58/ 3 In Situ STM Studies of Model Catalysts
(a)
Gas purification i
the surface is expo
completely contami
N2 can greatly imp '
studies and preven
backfilling the STM
large volume of gas
carbonyl contamina
i evaporator
i
o
0
sample
3.3 Visualizing the
(e)
•
f·
'
(cr): : ::_.: .:.
.
~
.
•
•• •••••••
"
"
:
"
.
,:, ~ .... - ~ ;.::, -:" 1 j . .;
i
- ,'-'-,I::
.,r·
Tip manipulation
, ~" , ':1
.' . 1 / .
: : , I,
.
: . •~ I ·
"
t.. ,
: . -. ' .
." ~.,:I .I : _. __.
..a . ,
it
!
.. .
-.
--,;
, - _ - _ ...:
..
"
Of
. -.
' '. • I . ,
. (..
With the introductil
fun damental steps 01
and catalytic transfor!
Wintterlin in 2000 [2.
to the visualization 0
follOWing section, we
STM for studying fu
.._
,
.
• ," _. 2UII ;1!!' :Ie ~I 1i;"1 .: - . ":"':
Figure 3.1 Method s of patterning the surface
for in situ STM studies. (a) Schematics of
"shadow" technique. (Reprinted with
permiSSio n from Ref. [12]. Copyright 2002,
Wiley, Inc.) (b) STM image of the surface created
by "shadow" technique. The shadow area
uncovered by metal clusters is distinguished
from the area covered with metal clusters by the
Path~
.
wh ite dash line. (c) Schematics of the tip
manipulation. (d) STM image of the su rface
created by tip manipulation. The dash rectangle
in (d) shows the area where most cl usters are
picked up by the STM tip. This area with lower
cluster densities can be distinguished from the
rest of the surface .and serves as a nanomarker
for in situ STM studies.
CO but perform poorly in the presence of O 2 or mixtures of CO and O 2 , Platinum or
platinum alloy tips are stable in O 2 but suffer from adsorption of CO, especially when
the sample surface temperature is above 550 K [20]. Gold is stable in both CO and O 2
but unstable at high temperatures, especially in the presence of water. In addition to
tip selection, in situ tip regeneration or cleaning is also critical for STM studies in the
presence of high-pressure reactant gases because the STM tip is susceptible to
picking up poorly conducting components during extended measurements at
elevated pressures. Tn situ tip regeneration refers to the method of applying a large
voltage pulse (from a few to hundreds ofvolts) between tip and sample while the tip is
in tunneling range. !'his method induces field emission, which cleans and regen
erates the STM tip . Wintterlin and cowurkers [2l.] recently reported a high-pressure
STM study, in which tungsten tips were used to study ethylene oxidation on Ag(II1.);
the tip could be recovered by applying high voltages to the tip (e.g. , + 300 V).
3.3.1
Imaging of Adsorbate
STM can induce ad~
and with spatial con
motion of adsorbate
reaction intermediatt
one of the most' imp
Ho [25] have recend
oxidation on Ag(ll 0
the Langmuir-Hinsr
and a pair ofoxygen a
atom pair was forme l
the sample bias to
is broken and the til
Ag( 11 0) surface shol
STM tip was then I
repeatedly, causing t
molecule moved dos
form the o-co- o
bias over the CO molt
atom on the surface
A second reaction f
absorbed oxygen aton
3.3 Visualizing the Pathway of Catalytic Reactions
Gas purification is another important issue for high-pressure STM studies. When
the surface is exposed to high pressures, even highly diluted impurities may
completely contaminate the surface. Purification of all reactant gases using liquid
Nl can greatly improve the operating pressure range for high-pressure reaction
studies and prevent the electrical breakdown often induced by humidity when
backfilling the STM chamber [13J. For a flow-reactor system, where purification of
large volu me of gases is required, a heated zeolite filter is effective in removing
carbonyl contaminants from the gas flow [22, 23J.
3.3
Visuali~ing th e Pathway of Catalytic Reactions
With the introduction of LT and VT STM, it is now possible to monitor the
fu ndamental steps of chemical reactions, that is, reactant chemisorption, diffusion,
and catalytic transformation. A detailed review covering this subject was published by
Wintterlin in 2000 [24J. Since then, in. situ STM studies have flourished and expanded
to the visualization of the reaction pathway and kinetics of surface processes. In the
following section, we highlight selected examples of recent progress in using in situ
STM for studying fundamental catalytic processes.
3.3.1
Imaging of Adsorbates and Reaction Intermediates
STM can induce adsorption-desorption and dissociation processes nonthermally
and with spatial control. At low temperatures, with limited surface diffusion, the
motion of adsorbates can be controlled and allows the determination of surface
reaction intermediates. The catalytic oxidation of CO on precious metal surfaces is
one of the most important model reactions in heterogeneous catalysis. Hahn and
Ho [25J have recently used in situ STM to visualize the reaction pathway of CO
oxidation on Ag(ll 0) at 4 K. Figure 3.2 depicts the pathway believed to be operative:
the Langmuir-Hinshelwood mechanism. Figure 3.2a and b shows a CO molecule
and a pair ofoxygen atoms adsorbed on the Ag(ll 0) surface, respectively. The oxygen
atom pair was formed by placing the tip over a molecularly adsorbed O 2 and raising
the sample bias to 0.47 V. Subsequently, the bond between two oxygen atoms
is broken and the two oxygen atoms adsorbed at the nearest fourfold sites of the
Ag(ll 0) surface show slight elongation along the [1 I 0) direction in Figure 3.2b. The
STM tip was then placed over the CO molecule with a + 0.24 V bias applied
repeatedly, causing the molecule to diffuse across the surface. Eventually, the CO
molecule moved close to the pair of 0 atoms (Figure 3.2c) and then joined them to
form the O- Co- o complex (Figure 3.2e). With an additional pulse of the sample
bias over the CO molecule, the O-Co-o complex is decomposed, leaving an oxygen
atom on the surface with the CO 2 des orbing from the Ag surface.
A second reaction pathway was also illustrated by moving a CO molecule toward the
absorbed oxygen atoms. In Figure 3.3a, an STM tip with a CO molecule adsorbed atthe
159
60'
3 In Situ STM Studies of Model Catalysts
(a)
Cd)
[110]
(f)
' - [001]
Figure 3.2 STM images obtain ed with a
CO-terminated tip. Vl = 70 mV and It = 1 nA.
(a) Iso lated CO molecule. (b) two 0 ato ms
(adsorbed o n the nearest fourfold hollow sites
alo ng the [1 I 0] di rectio n). (c) CO and two
o atoms se parated by 6.1 A along th e [0 0 1]
direction. and (e) O -CO -O complex. Grid
lines are d rawn through the silve r surface
atoms . Scan area of (a-c) and (e) is 25 Ax 25 A
Schematic diagrams for adsorption geometries
o f (c) and (e) are sh ow n in (d) and (f),
res pectively; a line ar atop and a ti lted off-site CO
are im pli cated. The black (red) circles represent
ca rbon (oxygen ) atoms and the lar ge gray circles
are sil ver atoms .·The sizes of the ci rcles are
scaled to the atomic covalent radii_ (Reprinted
with permission fro m Ref. [25] . Copyright 2001 .
The Ameri can Physica l Society.)
tip end was positioned over an oxygen atom, which lined up with another oxygen atom
fro m the O 2 disso(jation. T he two oxygen atom s were separated fr om each othe r by two
lattice spacings. With a pul se of + 0.47 V sample bias . the CO molecule is detached
from the STM tip and reacted with the oxygen below th e ti p to form CO 2 . Figure 3.3b
shows the two pul es of tunneling cunent. corresponding to the CO m olecule.
impin ging on the surface and reacting with the adsorbed oxygen, and to CO 2
desorption . res pectively. Figure 3.3c shows the oxyge n atom left on the Ag(l 1 0)
surface. The com bined im aging. manipulation. an d spectroscopic ca pabilities of the
STM provide direct visualization of reaction pathways at the single molecule leve l.
At low t mperatures. tip m anipulation has been regularly used tel promote smface
diffusion, to activate bon ds. an d to synthesize molecules. Rece nt progress along these
lines is desai bed by Hla an d Rieder [26 . 27J.
Figure 3.3 Reaction
fr om a CO-terminat.
ad so rbed on the sur!
with a CO-terminate
separated by two tat
alon g the [I 1 01dire
th rough the silver su
cu rren t during a 14)
with the CO-ter mlna
o atoms (denoted t
3.3.2
,Imaging Chemisol
Under catalytic re
diffusion of ads or
by in situ STM. C
adsorbates are ill
coverage is illCre,
attractive interacr
These adsorbate
the SllIface. Eve~
islands grow-intG
and by Wong et D
surfaces depend
coefficient measl
surface cove rag
ence on adsorba
The structure
relevant to earnl,
found for CO ot1
3.3 Visualizing the Pathway of Catalytic Reactions
(a)
(b)
6
<4
.s
2n__--'
0.0
0.2
0.4
Time (s)
(d)
(e)
Figure 3.3 Reaction of a CO molecule released
from a CO-terminated tip with an 0 atom
adsorbed on the surface. (a) STM image, taken
with a CO-terminated tip, of two 0 atoms
separated by two lattice spaci ngs (2 x 2.89 A)
along the [1 I 0] direction. Grid lines are drawn
through the silver surface atoms . (b) Tunneling
current during a 1470 mV sample bias pulse
with the CO-terminated tip over one of the two
o atoms (denoted by "'''). Two current rises
(f)
(at 250 and 310 ms) indicate the moments of
desorption and reaction of CO from the tip and
the moment of desorption of CO 2 into vacuum .
(c) STM image of the same area rescanned after
the pulse, showing CO on the tip has reacted
away. Scan area of (a) and (c) is 25 A x 25A.
(d-f) are the schematic diagrams for (a-c).
respecti vely. (Reprinted with permission from
Ref. [25J. Copyright 2001 , The American Physi cal
Society.)
3.3.2
Imaging Chemisorption on Metals
Under catalytic reaction conditions, adsorbates are usually mobile on the surface. The
diffusion ofadsorbates has been studied both on metal surfaces and on oxide surfaces
by in situ STM. On metal surfaces, it has been shown that at low surface coverage,
adsorba tes are mobile and are distributed randomly on the surface. As the surface
coverage is increas d, the interaction betwcl'n adsorbates also changes such that an
attractive interaction begins to appear, leading to the formation of adsorbate islands.
These adsorbate islands are in equilibrium with the diffusing adsorbates (2D gas) at
the surface. Eventually, with an increase in the adsorbate coverage, the adsorbate
islands grow into an adsorbate overlay r. In situ STM studies by Wintterlin et al. [4J
and by Wong et al. [281 both illustrat that the diffusion rate of adsorbates on metal
surfaces depend on their coverage and/ or nearest neighbors. The surfac diffusion
coeffici.entmeasured for adsorbates on metal surfaces is meaningful only at very low
surface coverages where the adsorbate-adsorbate interaction has a negligible inA.u·
ence on adsorbate diffusion.
The structure of the adsorbate layer formed at high s urface coverage is more
relevant to cat lytic reactions at high pressures. Besenbacher and coworkers [29-33]
found for CO on I't(ll 0) and Pt(lll) and NO on Pd(111) that the structme ofhigh
161
621
3 In Situ STM Studies oj Model Catalysts
adsorbate coverages, formed at low-temperature and low-pressure conditions, is
identical to the structure formed at room temperature and high pressures . Th is
finding suggests that on metal surfaces, reaction studies at high surface coverage
conducted at low temperature likely connect with real catalytic processes at high
pressures.
By studying the diffusion of surface adsorbates (or adsorbate vacancies), in situ
STM can be used to determine the active site for chemisorptions. Mitsui et al. [34-36]
studied the process of hydrogen dissociation on Pd(l 1 1) using in situ STM. Pd is a
catalyst widely used in hydrogenation and dehydrogenation reactions. xposure ofpd
to H2 leads to dissociative adsorption. At approximately 65 K and in the presence of
2 x 1O- 7 1orr of H 2 , Pd(l 1 1) is nearly saturated with H atoms leaving only a few
vacancies as sites for the dissociation of adsorbed Hz molecules (Figure 3.4). Due to
the inversion of the image contrast caused by the adsorption ofH atoms on the ST M
tip, the empty ~urface sites are imaged as protrusions. This surface can be used to
model the Pd(l 1 1) surface under high -pressure Hz at room temperature.
Figure 3.5 shows a sequence of STM images acquired for the same surface region.
Figure 3.5a depicts a number of isolated vacancies (bright spots) separated by more
than one Pd lattice, as well as two aggregat s of vacancies (marked by dashed circles).
Each vacancy aggregate consists of a pair of vacancies (dimer) occupying neighboring
fcc sites. The dimers in Figure 3.5 are always imaged as a three-lobed object because
of the fast diffusion of a neighbor H atom, which can hop over bridging sites to
occupy the vacancy pairs without getting close to other H atoms. The vacancy pairs or
dimers are most frequ ently encountered in the STM study. Isolated vacancies can hop
Figu re 3.5 STM im ag~
the forma tion, separa l
H-vaca ncy clu sters. n
(3 nm " 2.S nm) are r<
ann otations. (a) Five \
are labeled (A-E) ana
pairs (2V) are lTlarh d
reference. (b) Vacanci,
2V cluster indica ted b
the number "2". Vaca .
Figure 3,4 The 6.5 nm x 6.5 nm STM image of Pd(l 1 1) with a H
cove ra ge near one monolayer. Numerous H vacancies, visible as
bri ght protrusions, are present. V, = 45 mV and I, = 2.7 nA.
Stre aks and fractional protrusions are due to vacancies moving
while the tip is scanni ng over them. (Reprinted with permission
from Ref. [35]. Copyri gh t 2005, Springer.)
randomly on the Pc
Figure 3.Sb. VacJnc
coaJesce to form a I
several minutes an
three-vaca ncy aggl
(Figure 3.Sc). The
aggrega te are OCelli
3.3 Visualizing the Pathway of Catalytic Reactions
Figure 3.5 STM images from a movie showing
the fo rmation, separation, and annihilation of
H·vacancy cl usters. The image s on the left
(3 nm x 2.5 nm) are repeated on the right with
annotations. (a) Five vacancies near the cen ter
are labeled (A-E) and two triangular vacancy
pai rs (2V) are marked with dashed triangles for
reference. (b) Vacancies A and 8 have formed a
2V cl uster indicated by the triangle containing
the number "2". Vacancies C-E have formed a
three-vacancy (3V) cluster, indicated by the
larger triangle containing the number "3".
(c) The 2V pair has separated into isolated
vaca ncies A and 8, while the 3V cluster has been
annihilated by dissociative adsorp tion of a Hz
molecule , leaving a si ngle remaining vacancy C.
The other 2V clusters separated a few frame s
later. (Reprinted with permission from Ref. [3 4J.
Copyright 2003, Nature Publishing Group.)
randomly on the Pd surface and occasionally coalesce to form aggregates, as shown in
Figure 3.Sb. Vacancies A and B aggregate to form a dimer while vacancies C, D, and E
coalesce to form a tnree-vacancy aggr gate. The vacancy dimer remains together for
several minutes and eventually disintegrates back to isolated vacancies. However, the
three-vacan cy aggr gate disappears and leaves only one vacancy on the surface
(Figure 3.Sc). The authors conclud d then that two vacancies in the three-vacancy
~ ggregate are occupied by H atoms from the clissociation ofadsorbed H 2 . The uthors
163
641
3 In Situ STM Studies oj Model Catalysts
also found , in the presence of2 x 10- 7 Torr H2 at 65 K, vacancy aggregates with four
or more vacancies are also filled by H atoms from the dissociation of H 2 within the
aggregates and transformed into a single vacancy or totally annihilated. Through a
series of ",TM movies on the diffu sion of surface hydrogen vacancies, the authors
found that on ly an aggregate with three or more vacancies could be annihilated by H2
dissociation . rne dimers or isolated vacancies are never occupied by H atoms.
Instead, the dimers always dissociate creating isolated vacancies with an average
lifetime of 10 min at 65 K. From these data, the authors concluded that three or more
empty palladium sites are necessary for the dissociation of H 2 molecules. This
finding is rather surprising since it has traditionally been assumed that two
neighboring empty sites are sufficient for the dissociation of a diatomic molecule.
The discovery of the active sites for H 2 dissociation on Pd(lll) illustrates the power
of in situ STM in addre ssing the elementary steps of surface reactions and in testing
the conventional assumptions in catalysis.
Yo
[llOl~
[110]
Figure 3.6 The b. 11 IT
3.3 .3
Determining the Sites for Chemisorption on Oxide Surfaces
On reduced oxide surfaces , the diffusion of an adsorbate is often limited by the
localized bonding, either ionic or covalent, between the adsorbate and the oxid e
substrate. The relatively slow diffusion of adsorbates allows chemisorption and
diffusion on oxides to be studied by STM at elevated temperatures. For example, Ti0 2
is an exc lIent photocatalyst for dissociation of wa ter and decomposition of organic
molecules, critical to pollution control and the hydrogen economy. Studies of the
adsorption and diffusion of wa ter, oxygen, and organic molecules on Ti0 2 are of
pri mary impor tance to our understanding of photocatalysis by Ti0 2.
Being th e m ost stable phase ofTi0 2, the rutile 1'i0 2(11 0) cryst<LI has been studied
most extensiv ly by STM and other surface science techniques [37]. Figure 3.6 shows
a structural model of the rutile Ti0 2(1 1 0)-(1 x 1) surface. 'The surface contains two
types of tita nium atoms that form rows along the [0 0 1J direction. Rows of six
coordinated Ti atoms alterna te with five-coordinated terminal Ti at.oms , which miss a
single 0 atom perpendi cular to the surface. The surface also con tains two kinds of
oxygen atoms, that is, three-coordin ated oxygen atoms, sitting in the surface plane,
and bridging oxygen atoms, sitting above the surface plane a nd bonded to two six
coordinated Ti atoms. Un dersaturated bridging oxygen atoms can be easily removed
from the surface by annealing, electron bombardment, or ion sputte ri ng to form
bridging xygen vacancies. Brid ging oxygen vacancies are tIl e most common and
well-defined defects on the 1'i0 2 (1 1 0) surface.
STM studies on the adsorption and diffu ion of small molecules on the 1'i0 2(11 0)
surface began in the late 1990s [37-3 91 and have provided a g neral u nderstan ding of
the impOltant role ofbri dgi ng oxygen vacancies in the dissociation of water, oxygen,
and small organic molecul ' 5 . However, due to the difficulties in distinguishing active
s urface siles and disso ·iation products, only recently have the ft.l ~da mental steps
of ad sorption and di ssociation processes been understood with the help of
in situ S1'M.
-
- - - -
-
-
-
-
-
- - - -
(red) balls represent'
coordinated surface '
coord ina ted Ti atoms
single oxygen vacanCl f
5fTi row (Oat) are in
Ref. [401 Copyright 2
Wendt el al. [40]
water and O2 011
methods, aUow su
atoms, smface h~
illu strates the din
and adsorbed \Val,
ST 1 resolves the
bridging ox)'gen r
oxygen vacancies
oxygen vacancies
found that appl}i
atoms from the h;
situ STM , Wendt
bridging oxygen
hydroxyl groups, \
at the neare t bric
hut can be initiat;
five-coo rdinated "
cwes, hydrogen
bridging oxygen
Dohnalek ;md
hydroxyl
images (Figure
3.3 Visualizing the Pathway of Catalytic Reactions
[110]
~
o atoms
Ti atoms
[110]
Figure 3.6 The ball mode l of the Ti0 2 (1 1 0) surface . Large gray
(red) ba lls represent 0 atom s, small light gray (gray) balls fi ve
coordina ted s urfa ce Ti atoms (sf-Ti), and small black balls six
coordinated Ti atoms (6f-Ti). The bfjdging oxygen ato ms (Ob,),
si ngle oxygen vacan cie s (Vo) , and 0 atoms adsorbed o n top of the
sf-Ti row (Oo,) are indicated. (Reprin ted with permission from
Ref. [4 0J Copyright 2005 , Elsevier.)
Wendt et al. [40] and Bikondoa et al. [41 J studied the ad sorption and dissociation of
water and OL on TiO z(l 10). In situ STM studies, in parallel with the use of Drl
method ,allow smface features such as bridging oxygen vacancies, adsorbed oxygen
atoms, surface hydroxyls , and adsorbed water to be distinguished. Figure 3.7a
illustrates th e difference between a bridging oxygen vacancy, a surface hydroxyl,
and adsorbed water in their appearance in S M images. Due to electronic effects,
STM resolves the five-coordinated terminal Ti rows as bright rows whereas the
bridging oxygen rows are imag d as dark rows. The nuances in the appearance of
oxygen vacancies and hydroxyls in STM images are distinguished by visualizing
oxygen vacancies being transformed into OH species in situ. The authors have also
found that applying voltage pulse ( ~ 3 V) over the hydroxyls desorbs individual H
atoms from the hydroxyl groups while leaving the oxygen vacancies intact. Using in
situ STM, Wendt et al. [421 demonstrated that water dissociation takes place at
bridging oxygen vacancies of the TiOz(l 1 0) surface at 187 K and form pa ired
hydroxyl groups, wilh one positioned at the oxygen vacancy site and the other ormed
at the nearesl bri dging oll:ygen sit . The diffusion of these pairs is inhibited at 187 K
but can be initiated in the presence of neighboring water m olecules adsorbed in the
five-coordinated Ti trough. TIuough the interaction with neighboring wa ter mole
cules , hydrogen atoms h om the paired hydroxyl grou ps are transferred to adjacent
bridging oxygen rows, cau~ing the ross-row diffusion of hydroxyl groups.
Dolmalek and coworkers [43, 44 ] hav' further m easured the diffu sion kinetics of
hydroxyl groups (or H atom) at room tempera hlre an d above. In situ S M
images (Figure 3.8) have confi rmed thal I fl O dissociates at the bridging oxygen
165
661 3
In Situ STM Studies of Model Catalysts
.,
-
(a) •
.' •
.1
111 ,__ '
II
,
(
.
(bl
.t
---- II
II
•
•
•
[0011
. '-- 1
-
\ , . - Iliol
1. 5
.~ 1.0
~,
,(c)
I
!
.
•
1110 ,\ . 16() t\
•
!
.~
~ -.
~05 ~_'
0.0 '
.'-..-- .'
•
II I
Ti
0
o
__
10
0
n
0
TI
15
0
20
TI
TI
25
1)
0
5
0
TI
0
10
ci
15
20
Lcnglh along J iOI (A)
TI
25
r
o
•
•
•
10
15
20
25
Fi gure 3.7 STM images (16 nm x 16 nm) of clean , reduced
Ti0 2 (1 1 0) samples showing the difference between bridging
oxygen vacancies, surface hydroxyls , and adsorbed water. The
sample in (a) was less reduced than the sample in (b) . (c) STM
height profiles along the 1 i 0] direction of species indicated in (b). (Reprinted with permission from Ref [40] . Copyright 2005 , Elsevier.)
--I '"
,,;.
,
(a )
1'#
I
(b)
f
.... '~I
::.
•"
,
...
,(e)
..
ft ~
III
I(d)
,.
....
Figure 3.8 STM images of the same area on
Ti0 2 (1 10) at 357K (Vt = 1.5 V, It =O. 1 nA)
as a function of time (LH = 60 s): (a) clean
Ti0 2 (1 1 0) with bridging oxygen (880)
vacancies; (b) Ti0 2 (1 1 0) with a geminate
hydroxyl pair formed by adsorption and dis
socia tion of a water molecule. Hv marks the
~.
"',
.
'I
•
.,
vacancies, producing I
room temperature alor
in the paired hydroxyl
positioned at the heall
one adsorbed at neight
Hv and HB were meas
estimated to be apprc
these hydrogen atoms i
inga repulsiveOH - OI
state is responsible for
measured kinetic para
duced by OFf calculati
The pathwayofdissc
experiment aJso applie
been used to study the
Figure 3.9 shows a Sl
exposure to
bridging oxygen
methanol led to the
vacancies ofTiOl( l 1
group at the bridging
hydroxyl group at the
to the bridging
higher than the
entia ted from the
.I
OH hydrogen and He the hydrogen that s plit off
from the OH ; (c) same area after a single hop
of He; and (d) after su bsequent hop of Hv.
Insets exhibi t the ball models illustrating the
corresponding processes . (Reprinted with
permission from Ref [44] . Copyright 2008,
The America n Chemical Society.)
The adsorption
Wendt et al. [40]
dissoci ate at the b
and the other 0
previous vaca ncy
3. 3 Visualizing the Pathway of Catalytic Reactions
vacancies, producing paired hydroxyl groups. Hydrogen atoms readily diffuse at
room temperature along the bridging oxygen row. However, the two hydrogen atoms
in the paired hydroxyl groups exhibit inequivalent diffusivity. The hydrogen atom
positioned at the healed oxygen vacancy site (Hy) diffuses much slower than the
one adsorbed at neighbor bridging oxygen sites (Hs). The different diffusion rates of
Hyand HB were measured between 300 and 410K and the activation barrier ofHB
estimated to be approximately 0.22 eV lower than Hy. The diffusion barrier of
these hydrogen atoms increases with the separation between hydroxyl groups, suggest
ing a repulsive 0 H -OH interaction. The authors speculated that a long-lived polaronic
state is responsible for the inequivalent diffusion rates of Hy and H B . However, the
measured kinetic parameters (prefactors and diffusion barriers) could not be repro
duced by OFT calculations, suggesting a rather complex diffusion mechanism.
The pathway ofdissociation and diffusion discovered in the above water adsorption
experiment also applies to the adsorption of alcohols on Ti0 2 (1 10). In situ STM has
been used to study the adsorption of methanol [45] and butanol on Ti0 2 (1 1 0) [46] .
Figure 3.9 shows a series of STM images obtained on the same area following
exposure to methanol. Figure 3.9a depicts the clean surface before exposure, with
bridging oxygen vacancies marked as yellow circles. The exposure of 0.06 ML
methanol led to the dissociative adsorption of methanol at the bridging oxygen
vacancies ofTi0 2 (11 0) (Figure 3.%). The dissociation of methanol forms a methoxy
group at the bridging oxygen vacancy, resolved as bright features in Figure 3. 9b, and a
hydroxyl group at the nearest neighbor. The methoxy group has a similar appearance
to the bridging oxygen vacancy in STM images, except that the methoxy group is 0.8 A
higher than the bridging oxygen vacancy. The hydroxyl group could not be differ
entiated from the bright features of neighboring methoxy groups in Figure 3.9b.
However, with time, the hydrogen from the hydroxyl group (red dots in Figure 3.9c
and d) diffuses along the bridging oxygen row and across the bridging oxygen rows
through interactions with methanol molecules weakly bounded to the Ti trough. The
diffusing hydrogen atoms were identified by their apparent height in STM images
and by a tip desorption experiment (Figure 3.ge) proposed by Bikondoa et al. [41].
Figure 3.9f gives a graphic illustration of the dissociation and diffusion pathway of
methanol on Ti0 2 (1 1 0), which was also observed in the adsorption experiment of
2-butanol (CH 3 CH 2 CH(OH)CH 3) on Ti0 2 (1 1 0) at room temperature [45J.
The adsorption and dissociation of O 2 on Ti0 2 (1 1 0) have been investigated by
Wendt et al. [40] and Du et al. [47]. Both observed that O2 molecules adsorb and
dissociate at the bridging oxygen vacancies, with one 0 adatom healing the vacancy
and the other 0 ada tom bounded to the neighboring five-coordinated Ti site. Du et aL.
also analyzed the lateral distribution of the 0 adatoms upon dissociation and
discovered a transient mobility of 0 adatoms along the Ti trough in the [0 0 1]
direction. Unlike the dissociative adsorption of O 2 on metal surfaces where both
adatoms have equal diffusivity, the diffusivity of 0 adatoms on Ti0 2 (11 0) was found
to be inequivalent. While the 0 adatoms filling the vacancy are locked in the bridging
oxygen row, 0 adatoms in the Ti trough are relatively free to move. A majority of 0
ada toms on the Ti trough (",81 %) were found separated from the 0 adatoms in the
previous vacancy sites by two lattice constants.
167
•
681
3 In Situ STM Studies of Model Cata lysts
Zhang et al. [4811
on Ti0 2 (1 10) usi n
suggest that bridgi
slow diffusion of ~
agreement with D
chemistry ofTi01 (
for approximately 1
However, there
of trimelhylacetic
Ti0 2 (1 1 0) at roo
AA toformT
of the hydroxyl gr
hydrogen atom wa
the adjact'nt TMA
coverage, TMAA ~
Wendtet al. [501 r
in detail and sugge
account for 0) diss
approximately 10%
a few Langmuirs of
the influence of bri
"perfect" Ti0 2 (J 1
temperature. H),dr
oxygen vacancies,
Figure 3.10c and
groups and create
previously suggest
oxygen adatoms 0
However, tJle incr
hydroxyl group h
......•
..
•
•
CH 3
•
• • • OH
• .
CH3 •
CH
O-H 6 3
/e
'~
[001) .
•
o
•
Figure 3.9 STM images of s am e are a be fore
an d after adsorpti o n of m etha nol o n reduced
Ti0 2 (1 1 0) at 300 K (V, = 1. 0 ± 0. 3 V and
I, <0. 1 nA): (a) bare su rface; (b) afte r 80 s
exposure to m e tha nol; (c) after 110 s ex pos ure
to metha nol; (d) taken on (c) after spo nta neous
tip change; (e) after h igh bia s (3.0 V) sweep of
(c); (f) schematic m odel of th e adso rpti on
Ti io n
dissociation chSTM results co
state ofTi0 2(1 1 0)
leading to a perfe
state (Pigme 3, lOe)
plo the evolution
and sugg sts the Ti
The authors suggl:
the reduction of
responsible for the
of O 2,
Indeed. the imp
situ STM studies ~
[110]
V
Bridging-bond 0 ion
0 from methanol
p rocess. Insets s how m ag ,fled areas ma rked
by sq ua res . Yel low circl es show the pos itio n of
brid gi ng oxygen vaca ncies. Bl ue ci rcles sh ow th e
m thoxy group s o n oxygen vacan ies. Red
squares s how H atoms diffUS ing o n bridging
oxy en rows. (R pri nted with . pe rmission from
Ref. [45 ]. Copyrigh t 2006 , The Ame rican
Chem ical Society.)
- - , .........II!I!! 3.3 Visualizing the Pathway of Catalytic Reactions
Zhang et al. [48] have recently measured the stability of bridging oxygen vacancies
on Ti0 2 (11 0) using in situ STM. Sequences of STM images between 340 and 420 K
suggest that bridging oxygen vacancies migrate along the bridging oxygen row via the
slow diffusion of bridging oxygen atoms with a diffusion barrier of 1.15 eV, in
agreement with DFT calculations. All the above studies suggest that the surface
chemistry ofTi0 2 (1 1 0) is dictated by bridging oxygen vacancies, which can account
for approximatelyl0% of the bridging oxygen sites.
However, there are disagreements. Lyubinetsky et al. [49] studied the adsorption
of trimethylacetic acid ((CH 3 bCCOOH, TMAA), a photoreactive molecule, on
TiO z(1 1 0) at room temperature. In situ STM found that the deprotonation of
TMAA to form TMA does not necessarily occur at bridging oxygen vacancies. None
of the hydroxyl groups was found during the adsorption of TMAA. instead, the
hydrogen atom was bound to a pair of bridging oxygen atoms and stabilized by
the adjacent TMA groups sitting on the five-coordinated Ti trough. At saturation
coverage, MAA formed a (2 x 1) overlayer on the Ti 0 2 (1 1 0) surface.
Wendt et al. [501recently studied the interaction between O 2 and Ti0 2 (11 0) surface
in detail and suggested that bridging oxygen vacancies are only the minor sites that
account for O 2 dissociation. Even though bridging oxygen vacancies account only for
approximately 10% of surface bridging oxygen sites, exposing the clean Ti0 2 (11 0) to
a fe w Langmuirs of O 2 could not fully remove all bridging oxygen vacancies. To isolate
the influe nce of bridging oxygen vacancies in O 2 dissociation, the authors created a
"p rfeet" Ti0 2 (1 1 0) surface by exposing the TiO z(1 1 0) surface to water at room
tem perature. Hydroxyl groups, formed via water dissociation, covered all bridging
oxygen vacancies, yielding a vacancy-free Ti0 2 (1 1 0) surface (Figure 3.1 0a).
Figure 3.lOc and d illustrates that O 2 exposure can fully remove surface hydroxyl
groups and create a Ti0 2 (I 1 0) surface with perfect bridging oxygen rows, as
previously suggested in T PD studies [51 ]. With the titration of hydroxyl groups,
oxygen adatoms on the five-coordinated li row (Ootl also increase (Figure 3.lOb) .
However, the increase in oxygen adatoms does no t seem to stop even after all the
hydroxyl groups have be -' n replaced with oxygen (Figure 3.IOe and d). Paired Oot
atoms start to appear on the five-coordinated Ti row during extended O 2 exp osure. On
the basis of these observations, the authors showed that a second and primary O 2
dissociation channel is operative on the five-coo rdinated Ti row.
STM results combined with photoelectron spectroscopy (PE ) on the valence
state ofTi0 2 (11 0) further show that the removal of all hydroxyl grou ps by oxygen,
leadi ng to a perfect Ti0 2 (1 1 0) surface, only slightly attenuates the Ti 3d defect
state (Figur 3.10e). The full attenuation of Ti 3d state requires 420 Lof0 2 . Figure 3.9f
plots tlte evolutions of the Ti 3d defect state and the 0 I-I 30" state over O2 exposure
and suggests tll e Ti 3d defect stOltI:' is not mainly caused b bridging oxygen acancies .
he aut hors suggest that other types of defects, Ti3 ' interstitials thal form during
the reduction of Ti0 2 (1 1 0) and are hidden beneath the surface, a.re primarily
responsible for the formation of the Ti 3d defect stat and the dissociative ad orption
of O2 ,
Inde d, the importance of T? ~ interstitials has also been realized in previous in
situ STM studies of the reoxidation of Ti0 2 (1 1 0) [52- 54]. It is noted that Ti 3 +
169
70
I
3 In Situ STM Studies oj Model Catalysts
Ti 3 + interstitiaJs ar
oxygen vacancies are
surprising to expect
dissociation of adsor I
worth noting that sue
diffuse to the Ti0 2(
Considering PES Wi
attenuation of the Ti 3
have been oxidized a
inlerstitials involves .
eveliheless, the abo'
active si tes, hidden 0
will stimulate more il
their surface chemis
14 12 10 8 6 4 2
Binding Energy (eV)
I
gj 0.8
::c
-gN
0.6
';ij 0.4
E
00.2
z
•
J
*
" .
- il_ _ _
-l
I
Visualizing Reaction h
•
- a Ti3d
__
\ OH3"
...
o 2
3.3 .4
h-~i02(110) : oxygen exposur~ at room temperature
4 6
I
J
-~
8 10
50
--
100
02 Exposure (L)
Figure 3.10 (a-d) STM image s (105 A >< 105 A)
of the Ti02 (1 1 0) surface cove red with
hydroxyls [h-Ti02 (1 1 O)J and then expo se d to
in c rea sing amounts o f O 2 at room tempe ratu re.
(e) Selected PES va lence- band spectra recorded
on a n h-Ti02(1 10) s urface that was exposed to
O 2 at RT. Arrows indicate t he representative
STM im age s. (f) Normalized integra ted •
- - 8
200
300
400
intensities of the O H 30 (red) and Ti 3d (blue)
features for O 2 exposures up to 420 L from PES
spectra; c ircl es indicate intensity values that
were o bta ined from the spectra s how n in (e).
(Repri n ted with permission from Ref. [50]. Copyright 2008, The Am erican Associati o n for the Advancement o f Science.) interstitials diffuse to the surface in the presence ofOz and form TiO x species, which
serve as the building blocks for the regr O\vth ofT iO z(l l 0) plane. Using in situ STM,
Bowker and cowo rkers [52] measured the reoxidation kinetics of reduced TiO z (ll 0)
surface at temperatures b etween 573 and 1000 K and oxygen pressures of5 x 10- 8 to
2 X 10- 6 mba r. By mo nitoring the T iO x species that diffused and coalesced on the
Ti 2(1 10) surface, the sur£1Ce growth rate co uld be measured th rough the change of
island morphology. This growth rate was found to be linear with re sp ct to the oxygen
partial pressure. The regrowth of T iOz(l 1 0) has a low activation energy of
approximately 25 kl/mo], suggesting a high m obility of
~ interstitials in the
presence of oxygen. Previous studies on the r eduction 0[1i0 2 (1 1 0) [37J have shown
the ru tile TiO l( l 1 0) b ulk serves as a huge r se rvoir for T? I inte rstit ials during
red uction, so that the surface is maintained near-stoichio m e try and is thermody
nam ically stable.
Te
In situ STM ha alsa
measure their kineti
metals to produce w
reaction is still a core I
the oldest of catalytic 1
unclear, especially fc
temperature ("- 170 ~
combination of disso
hydroxyl groups tha
formation of hydroxy.
the h ydroxyl group,
surface spectroscopic
situ STM, that the r
oxidation reaction or
Wintterlin and co
Pt(l 1 1) using in sitl
which the surface v.
subsequently expose
prepared byexposi ng
22S K to di:;sociate 0
and monitored by ST
by a (2 x 2)·O,dlayer,
exposure to H2, a
(Figure 3.11b). 'The!
ordered layer with 1
phases are character l
bonding. Additional
3.3 Visualizing the Pathway of Catalytic Reactions
Ti 3 + interstitials are essentially oxygen vacancies within the bulk whereas bridging
oxygen vacancies are basically undercoordinated Ti ions at the surface. It is not
surprising to expect that Ti 3 + interstitiaIs in the subsurface playa role in the
dissociation of adsorbed molecules. In the above STM study by Wendt et aI., it is
worth noting that subsurface Ti 3 + interstitials were neither visualized nor seen to
diffuse to the Ti0 2 (1 1 0) surface during O2 adsorption at room temperature.
Considering PES usually probes the top few layers at the surface, the complete
attenuation ofthe Ti 3d defect state suggests that Ti 3 + interstitials within those layers
have been oxidized and therefore quenched. Details ofhow the excess charge ofTi 3 +
interstitials involves in the bond breaking of O2 molecules remain to be elucidated.
Nevertheless, the above studies demonstrate the power of in. situ STM in tracing the
active sites, hidden or unhidden. The finding of mobile defects in a rigid Ti0 2(1 1 0)
will stimulate more investigations on other reducible oxides and encourage revisiting
their surface chemistry.
3.3 .4
Visualizing Reaction Intermediates and the Mechanism of H ydrogen Oxidation
In situ STM has also been used to study the pathway of surface reactions and to
measure their kinetics. The hydrogen oxidation reaction, catalyzed by Pt group
metals to produce water, was the first catalytic reaction discovered in 1823. This
reaction is still a core catalytic reaction at the heart of fuel cell technologies. Although
the oldest ofcatalyti.c reactions, the mechanism ofca talytic hydrogen oxidation is still
unclear, especially for its surprising reactivity at or below the water desorption
temperature (~170K). It has been proposed that the reaction proceeds via the
combination of dissociatively adsorbed 0 atoms (Oad) and H atoms (Had), forming
hydroxyl groups that subsequently bind with another Had to form water. The
formation ofhydroxyl groups has been postulated as the rate-limiting step. However,
the hydroxyl group, as a reaction intermediate, could not be confirmed in early
surface spectroscopic studies. It has not been until the past decade, with the help of in.
situ STM, that the reaction mechanism has become apparent for the hydrogen
oxidation reaction on metal surfaces.
Wintterlin and coworkers [5 5, 56J studied the hydrogen oxidation reaction on
Pt(l 1 1) using in. situ STM. The study was conducted as a titration experiment, in
which the surface was precovered with Oad and then the O-terminated surface
subsequently exposed to H2 molecules. The O·terminated Pt(l 1 1) surface was
prepared by exposing the clean Pt(111) surface to 10 L of O2 , followed by annealing at
225 K to dissociate O2 , The surface was then exposed to 8 x 10- 9 mbar H2 at 131 K
and monitored by STM , as shown in Figure 3.11 . The Pt(111) surface was precovered
by a (2 x 2)·O"d layer, where Oad atom s were imaged as da rk dots (FiguTe 3.lla). Upon
exposure to H 2 , a few bright islands form on top of the (2 x 2)-Oad layer
(Figure 3.11 b). These islands continue to grow and eventually develop into an
ordered layer with hexagonal and honeycomb phases (Figure 3.l1c). These two
pha ses are charact rized as a surface hydroxyl (OHad) overlayer, fo=ed by hydrogen
bonding. Additional diffusi ng islands seen in Figure 3.11 c are attributed to adsorbed
171
721
3 In Situ STM Studies oj Model Catalysts
below the water de:;
H 2 0 starts to desori
cycle by stopping tl
The autoca lytic
apply to catal)'lic hy(
the fir st to use S1'
m eso scopic level, v.
in surface reactions
situ STM to visual i2
atomic resolution.
3.3.5
Measuring the Reac
o
OH
Figure 3.11 Series of success ive STM images,
recorded during dos in g of the O-covered
Pt(l 1 1) su rface with Hz. (a-c) Frames
(17 nm x 17 nm) from an experiment at 131 K
[P(H 2 ) = 8 X 10- 9 mbarJ. The hexagonal pattern
in (a) is the (2 x 2) -0 structure; 0 atoms appear
as da rk dots and bright features are the initial
OH is lands. In (c), the area is mostly covered by
OH, wh ich forms ordered structures. The white,
fuzzy features are H2 0 -covered areas.
(d-f) Frames (220 nm x 220 nm) from an
experiment at 112 K [P(H z) = 2 x 10- 8 mbarJ.
In (d), the surface is mostly O-covered
(not resolved). The bright dots are sma ll OH
island s, most of which are concentrated in the
expanding ring. H2 0 in the interior of the ring is
not re so lved here. Thin , most ly vertical line s are
atomic steps. (Reprinted with perm ission fro m
Ref. [55J Copyrigh t 2001, The American
Associat io n for the Advancement of Scie nce.)
H 20 islands (H 2 0"d)' Figure 3.11a-c shows the formation of hydroxyl group as the
reaction intermediate and reveal the atomic details ofhydrogen oxidation catalyzed by
Pt(l 1 1).
Figure 3.1 Id- f presents snapshots of STM images of the Oad-te rmin ated Pt(ll 1)
surface exposed to 2 x 10- 8 mbar H 2 at 112 K. The imaged area includes several
surface terrace s covered with numerous small bright dots and a bright r ing that
expands with tim . These small bright dots h ave been assigned to small OH. d islands
that appear after H ]. exposure . The white ring, termed as the rea ction fr ont, is
concentrated with s mall O H ad islands and contin u s to grow as the reaction
progress s, suggesting a fa st rea ction at the boundaries b etween Oad atoms and
the diffusing Z . d· The fast reaction between ad and [-{ZOad produces two O H ad,
which in tum forms HZOod through a rapid reaction with Had atom. Since the
combination of Oad and H ad to form OHad is the r ate-lim iting s tep, th e presence of
H ZO ad removes this kinetic limit and promotes an autocatalyti c cycl e until depletion
of Oad atoms. STM images thus illustrate an autocatalytic reaction mechanism that
accounts for the low activation barrier and high reactivity of hydrogen oxidation at or
Another importa nt
oxidation catalyzed
on m eas uring the
study ofll)Tdrogen (
as a titration expen
then removed by e~
kinetics of surface'
from STM images
Win tterlin et aJ.
Pt(111) using in si
by exposing the S1.
di ssociate 02' The
exposed to 5 x 10
struc tures as a fun
form an ordered (2
mainly covered by
islands, scattered 0
surface oxygen ate
The adsorbed CO .
As time progresse:
of the (2 x 2)-0 i'
oxidation ca n be E
(2 x 2)-0 islands.
Figure 3.13 pic
perimeter of oxyg(
is linear with resp'
oxidation main!: (
on the Pt(l 1 1
temperatures be
of 0.49 eV and a
parameters oblaill
3.3 Visualizing the Pathway of Catalytic Reactions
below the water desorption tem perature. At temperatures higher than 170 K, where
HzO starts to d ' sorb, the shortened lifetime of HZO ad breaks down the autocatalytic
cycle by stoppin g the fast reaction between H 2 0 0u and Oad to form OH ad .
The autocatalytic reaction mechanism apparent at low temperatures is expected to
apply to catalytic hydrogen oxidation at high pressures. In addition, the above tudy is
the first to use STM to observe the formation of dynamic surface patterns at the
mesoscopic level, which had previously been observed by other imaging techniques
in surface rea ti ons with no nlinear kinetics [57]. This study illustrates the ability of in
situ STM to visualize rea tion intermediates and to reveal the reaction pathway with
atomic resolution.
3.3 .5
Measuring the Reaction Kinetics of CO Oxidation
Another important catalytic reaction that has been most exten sively studied is CO
oxidation catalyzed by noble metals. In situ STM studies of CO oxidation have focused
on measuring the kinetic parameters of this surface reaction. Similar to the above
study ofhydrogen oxidation, in situ STM studies of CO oxidation are often cond ucted
as a titration experiment. Metal surfaces are precovered with oxygen atoms that are
then removed by exposure to a constant CO pr ssure. In the titration experiment, the
kinetics of surface reaction can be simplified and the reaction rate directly measured
fro m STM images.
Wintterlin et al. [58] investigated the catalytic oxidation of carbon monoxide on
Pt(J. 11) using in situ STM. Oxygen atoms were preadsorbed on the Pt(lll) surface
by exposing the surface to 3 L Oz at 96 K, followed by a short anneal at 293 K to
dissociate O2 , The oxygen·cov red Pt(1 1 1) surface was then cooled to 247 K and
exposed to 5 x 10-8 Torr CO. STM was used to follow the change of surfac adsorbate
stlUctures as a function of the CO exposure time (Figure 3.12). At 247 K, Oad atoms
form an ordered (2 x 2) overlayer, imaged as dark dots . At t = 0, the Pt(11 1) surface is
mainly covered by the (2 x 2)·0 layer together with empty Pt sites, imaaed as bright
islands, scattered on the surlace. 'The addition ofCO molecules lowers the mobility of
surface oxygen atom s and slowly compresses the (2 x 2)-0 layer into large islands.
The adsorbed 0 molecules form ordered c(4 x 2) domains on the Pt(11 1) surface.
As time progresse ,the areas of c(4 x 2) CO domains continue to grow at the expense
of the (2 x 2)-0 island s. From the series of in situ STM images, the rate of CO
oxidation can be estimated based on the reduction rate of the surface areas of the
(2 x 2)-0 islands.
Figure 3.13 plots the dependence of the reaction rate on the 'u rface a.rea or
perimet r of oxygen domains, as a function of time. ApprOximately, the rea tion rate
is linear with respect to th e perimeter ofthe surface oxygen domains, sugges tin g 0
oxidation mainly occurs along the boundary between the oxygen and the CO domains
on the Pt(1 1 1) surface. The titration experiments were repeated at various
temperatures between 237 and 274 K. An Arrhenius plot gives an activation energy
of 0.49 eV and a prefactor of 3 x lOZl crn - 2 - \ in good agreem ent with the kinetic
parameters obtained from m acroscopic m easurements.
173
•
741 3
In Situ STM Studies of Model Catalysts
30.,-----i
C 20
~
i5
15
+
..J
10
~
~ ~(ll
a:
au
~..
.~
~
5
°°
" ' - -""------'-
--:!
400
Figure 3.13 Reactio n rate.
the change in the size of
between successive pa nel
Figure 3.12, norma lized t
length of the boundary b
CO doma ins (the fu ll line
Figure 3.12 Series of STM images, recorded
during reaction of adsorbed oxygen atoms
with coadsorbed CO molecules at 247 K. all
from the same area of a Pt(l 1 1) crystal.
Before the experiment. a subm onolaye r of
oxygen atoms was prepared and co was
con tinuously supplied from th e gas phase
(Peo = 5 x 10- 8 mbar). The times refer to the
start of the CO exposure. The structure at the
upper left corner is an atomic step of the Pt
surface. Image sizes. 180A x l70A; V,=0.5 V;
I, = 0 .8 nA . (Reprinted with permission from
Ref. [58J. Copyright 1997, The American
Association for the Advancement of Science.)
On the Pd(l 1 1) s urface, Wintterlin and coworkers [59J have shown that CO
oxidation goes through a different reaction pathway at low temperatures. Similar
titration experiments were performed by exposing the Pd(l 1 1) surface precovered
by (2 x 2)-0 overlayer to 2 x 10- B Torr CO at 143 K (Figure 3.14). Using STM to
follow the same area of the Pd(l 1 1) surface, these authors found that CO does not
react with surface oxygen at this temperature. Instead, CO molecules slowly occupy
the surface sites of Pd(l 1 1) and compresses the (2 x 2) oxygen domains into the
(2 x 1)-0 phase. This phase was imaged with a stripe pattern and exhibited an
oxygen density twice that of the (2 x 2)-0 structure. The reaction kjnetics of CO
titration was then measured on these (2 x 1)-0 islands between 144 arid 185 K. The
(2 x 1)-0 phase shows a superior reactivjty over the (2 x 2)-0 phase that does not
react with CO up to
accelerated when the
0.3 ML (Figure 3.l5a)
surface, the titration r
linear relation with t
islands, as shown in I
occupation of CO on
reaction. There was n
phase based on the
study unambiguously
islands, especially wh
The adsorption of 0
as is the case of (110)
surfaces would also i
accompanyi ng the oX)
considerably reduced
this reason, the CO tit
of fcc metals in the el
surface by Leibsle et a
The experimen ts I I '
surface "'1th CO. Chen
pha ses. wb idl. ill tur
By monitoring the s
that oxygen was reI
1.1. 1 OJdirection. Later.
fcc(l 1 0) sy tems.511
3.3 Visualizing the Pathway of Catalytic Reactions
30
+
,
~ 25
c
~
g
20
.
:0 15
... .
~
..J
ill
0;
II
10
\
5
+ ' 't
+
- ~:!
0
0
-
.
f--
•
.•• + •
• e_
~
•
•
•• -!t :; •
I
400
• • •••i •
_.__ ++.. ;_1-
.
800
#
.
..
I
'.
~
'c
0.35
C
:J
:-7
::J
040
0 .30
0 .25
0. 20
sg
:0
g
'0
(l)
0
~
OJ
0.15
ro
II
1200
1600
Time (5)
Figure 3.B Reaction rates, determined from
the change in the size of the (2 x 2) area
between successive panels of the data of
Figure 3.12, normalized to (squares) the
length of the boundary between oxygen and
CO domains (the full line is a linear fit) and
(crosses) divided by eo (1 - eo), which is equal
to eoe co if e = 1 implies maximum coverage
of the respective phase (the broken line is only
to guide the eye). (Reprinted with permission
from Ref. [58J. Copyright 1997, The American
Association for the Advancement of Science.)
react with CO up to 180 K. More interestingly, the removal of oxygen islands
accelerated when the surface coverage of (2 x 1)·0 islands decreased to below
0.3 ML (Figure 3.15a). Below 0.3 ML, unlike the previous study on the Pt(1 1 1)
surface, the titration reaction rate with the (2 x 1)·0 islands on Pd(1 1 1) shows a
linear relation with the surface area of oxygen, instead of the perimeter of oxygen
islands, as shown in Figure 3.15b and c. The authors speculated that a transient
occupation of CO on the oxygen island causes all 0 atoms to be accessible for the
reaction. There was no direct evidence for the existence of this kind of mixed O/CO
phase based on the STM images or other spectroscopic studies. Nonetheless, this
study unambiguously illustrated the superior reactivity of compressed oxygen
islands, especially when they become very smalL
The adsorption of oxygen atoms often induces the reconstruction ofmetal surfaces
as is the case of (110) surfaces of fcc metals. It is expected that CO titration on such
surfaces would also involve the local transformation of metal substrates. Indeed,
accompanying the oxygen· induced reconstruction, the mobility of surface oxygen is
considerably reduced so that they can be resolved by STM at room temperature. For
this reason, the CO titration experiments using STM were initiated on (110) srnfaces
of fcc metals in the early 1990s. 0 oxidation was first visualized on a Rh(l 1 0)
surface by Leibsle et al. [60J where a pronounced reaction anisotropy was observed.
The experiments were carried out by titrating the oxygen precovered Rh(1 1 0)
surface with CO. Chemisorption of oxygen on Rh(l 1 0) forms several reconstructed
phases, which, in turn, were imaged as striped patterns along the [1 I OJdirection.
By monitoring the surface changes during CO exposure, STM images revealed
that oxygen was removed on the elongated stripes of the added rows in the
[1 TOJ direction. Later, similar one-dimensional reactivi ty was also found on other
fcc(1 1 0) systems, such as Cu [61-6 3], Ni [64], and Ag [65, 661.
175
761
3 In Situ STM Studies of Model Catalysts
(a)
0.:1
:l •
,
0.2 CD
0.1
..
0.0
(a)
250 s (b)
-200
430 s
(b)
500
s (d)
Figure 3.14 Se ries of STM images reco rded
during CO dosing on the (2 x 2) -0-covered
Pd (1 1 l)surface. T = 143 K, Pco = 2 X 10--8 Torr,
all images are from the same area. Indicated
is th e time elapsed since the start of the CO
-6
,
dosing. Vt = 0.3 V, I, = 2.2 nA , 240 A x 240 A.
The two close-ups in (d) show details from the
marked areas in frames (a) and (c). (Repr inted
with permission from Ref (59J. Copyright 2005,
The American Physical Soc ie ty.)
While most in situ studi"s on the one-dimensional reactivity of fcc(I 1 0) metals
remain qualitative, recent studies on the reactivity of oll.'Ygen-induced added rows of
Ag(I 1 0) have provided quantitative measurement of one-dimensional reactivity.
akagoe et al. [65, 66J conducted CO ti tration experiments on the added row
recon structed Ag(I 1 O)(n x 1)-0 surfaces, where one-dimensional-Ag-O - chains
arrange periodically along the [0 0 11direction. Figure 3.16 shows two series of in situ
STM images, where Ag(l 10)(2 x 1)-0 surfaces were exposed to 1 x 10- 8 Torr CO at
room t mperature. As a fun ction of time, Figure 3.16a-g depicts the continuous
segmentation of AgO chains on the clean or carbon-containing Ag(l 1 0)(2 x 1)-0
surfaces. Figure 3.16h plots the remaining cov('nge of surface oxygen as a function of
CO exposure_ Clearly, the reaction rate accompanying the segmentation of AgO
chains is significantly accelerated. The authors have gone furth er to study the
structure fluctuation at various temperatures. Below 230 K, the AgO chains were
found to be straight while the removal of AgO chains only occurs at the end of the
-10+--..--
-4
(e)
:::
M
-I
-2
CD
;:
-3
-4 4----,.--""",
o
Figure 3.15 (a) Time
(b) plotofl n[ - d8(2,
t ?:: 0; (c) plot ofln
(Reprinted wi th permi
The American Phys ical
3.3 Visualizing the Pathway of Catalytic Reactions
O. 0 .I...-~---,-----.---t---'-----'-----"T----'
200
400
-200
o
Time (s)
(b)
-6
.;;)
01
~
~
-8
-10
-3
-4
-I
-2
In6
(c)
::
~
( 2.d)
-\
-2
<P
E
-3
_4 +---~--~--~--~--~--~~~
·L----,
o
JOO
200
300
400
Time (s )
Figure 3.15 (a) Time evolution of the (2 x 1)-0 coverage;
(b) plot ofln[ - d8(2), 1)/dtl versus In 8(2 " 1) for the data from (a) for t ? 0; (c) plot of In 8(2x1) versus t for the data from (a) for t ? 0 (Reprinted with permission from Ref. [59J . Copyright 2005, The American Physical Society.) 177
781
3 In Situ STM Studies of Model Catalysts
r I
IL
[001]
(e)so
",0
~O. 4
i!1
~ 0.3
8
(6x 1)
0 0 .2
,.. .~
<ii
c:
Q
-.
]01
°0
20
40
CO
Figure 3.16 Two series of STM images of
37nm x 27 nm continuously taken at RT under
a nominal CO pressure of 1 x 1O- 8 Torr for
clean (a-d) and C-containing (e-g) Ag(l 1 0)
(2 x 1)-0 surfaces (i, = 0.2nA, V"p= l.4V).
Schematic models of the regio ns are als o shown
for (a-d). (h) Titration curves obtained for both
clean (red solid circles) and C-containing
60
80 100
exposure (L) 12g
(empty circles) Ag(l 1 0) (2 x 1)-0 surfaces.
Thick red and black curves are the least square
fit s o btained by assuming second-o rder
kinetics . The relative number of segments for
the clean surface is also pl otted (blue triangles
and curve) . (Reprinted with permission from
Ref. [65J. Copyright 2003, The American Physical
Society.)
Figure 3.17 Evolution (j
o su rface during expos
chains and exhibits zero-order kinetics to CO exposure. The Arrhenius plot gives an
activation barrier of 41 kJ mol- 1 and a prefactor of l.7 x 10 3 cm - 2 S-1 The results
below 230 K agree with the previous study by Wintterlin et al. on Pt(1 1 1), where the
reaction takes place at the periphery of oxygen domains. On the contrary, at room
temperature , the reaction rate is drastically accelerated as the AgO chains become
segmented and the shape of the AgO chains begins to fluctuate. It is clear there is a
direct correlation between the surface structure and the reaction kinetics , although
such correlation cannot be quanti.tatively described by the first- or second-order
kinetic models.
The nonlinear kinetics of CO titration on oxygen precovered surface has been
found to take place not only on the oxygen-reconstructed one-dimensional wires
but also on the two-dimensional surface oxides. [(lust and Madix [67J have recently
studied the reduction of the Ag(1 1 1)-p(4 x 4)-0 surface by CO titration. The Ag
(1 1 1)-p(4 x 4)-0 surface was prepared by exposing Ag(1 1 1) to NO z at 500 K. The
reduction ofthi s surface was mon itored at room temperature by S M in the presence
of 10- 8 mbar CO (Figure 3.17). With time, the surface areas covered by p(4 x 4)-0
continue to shlink while the bright (1 x 1) islands continue to grow on top of the p
(4 x 4)-0 overlayer. Figure 3.18a-c illustrates the atomic structures of p(4 x 4)-0
phase, the oxygen-free (1 x 1) Ag islands, and the remnant dots of Ag surface oxide .
The authors found that the reaction rate does not correlate with the perimeter of the
temperature. The imagl
surface area exposed to
CO durin g imaging (aJ
befo re CO exposure an ,
boundary layers but
nonlinear increase il
led the authors to s
either at the bound
oxygen atoms re l ea~
speculation of this I
Jn sitl' CO ti tratic
systems, that is , ir
compared the CO ti
(Ill) surface coven
surface covered with
the three surface \
temperature. III litll
10- 8_10 - 7 mbar CC
surface with large V
3. 3 Visualizing the Pathway of Catalytic Reactions
179
•
Figure3 .17 Evolution oftheAg(l 11) -p(4
>(
4)
o sunface during exposure to CO at room
temperature. The images show the same
sunface area exposed to increasing amounts of
CO during imaging. (a) Shows the surface
before CO exposure and (b). (c) , and (d) at 15,
30, and 45 min after exposure start, respectively.
Image (d) shows the final state of the sunface, no
changes were observed after 45 min exposure to
CO. (Reprinted with permission from Ref. [67J.
Copyright 2007, The American Institute of
Physics.)
boundary layers but increases more rapidly with CO exposure (Figure 3.18d). The
nonlinear increase in the reaction rate approximately scales with the reacted area and
led the authors to speculate that CO reacts with undercoordinated oxygen atoms,
either at the boundary between the Ag surface and the p(4 x 4) -0 phase or with
oxygen atoms released onto the Ag surface. Due to the invisibility of such species,
speculation of this kind is difficult to verify_
In situ CO titration experiments have also been conducted on multicomposition
systems, that is, inverse model catalyst. Schoiswohl et al. [68J in their studies
compared the CO titration reaction on three surfaces: clean Rh(1 1 1) surface, Rh
(Ill) surface covered with large 2D V309 islands (mean size >50 nm), and Rh(1 1 1)
surface covered \-vith sma112D V30 9islands (mean size <15 nm) . Prior to CO titration,
the three surfaces were exposed to 10-- 7 mbar O2 to form a (2 x 1)-0 phase at room
temperature. In situ STM was used to follow the titration reaction in the presence of
10- 8_10--7 mbar CO . CO titration on the clean Rh(1 1 1) surface or the Rh(1 1 1)
surface vv'ith large V309 islands exhibits similar reaction kinetics. Figure 3.19 shows
80
I3
In Situ STM Studies of Model Catalysts
1.0
(d)
~08
m
OJ
~
06
OJ
to
al
~
0.4
o
Figu re 3.18 (a) Ag(l 1 1) islands and pits
surrounded by the Ag(l 1 1)-p(4 x 4)-0
structure. The white arrow points to the
remnants of the surface oxide that are occasion
ally observed in the pits. The atomically resolved
STM images show (b) the Ag(l 1 1)-p(4 x 4)-0
surface and (c) a small area of the (1 x 1)
structure obtained on the island shown in (a)
that appeared during CO oxidation. The black
15
30
time (min)
45
square on the island shown in (a) marks
the approximate scan area of image (c).
(d) Development of the reacted surface area
during the titration reaction. The curve shows
an exponential function fitted to the data.
(Reprinted with permission from Ref [67J.
Copyright 2007, The American Institute of
Physics.)
the adsorption of CO on the Rh(111)-(2 x 1)-0 surface occupies the on-top sites and
reacts with half of the oxygen in the (2 x 1)-0 phase, leading to the formation of a
coadsorbed (2 x 2) 0 + CO phase. The islands of (2 x 2) 0 + CO phase grow at the
expense of the (2 x 1)-0 layer upon CO exposure. The titration reaction stops at
approximately 30 L of CO and removes half of the surface oxygen atoms. Further
removal of the adsorbed oxygen is kinetically inhibited on these two surfaces at room
temperature. In contrast, CO titration reaction on the Rh(lll) surface covered with
small V309 islands is significantly accelerated and could proceed further to remove all
chemisorbed oxygen atoms on the Rh(lll) surface (Figure 3.20). The fuzzy edges of
the V309 islands in Figure 3.20 suggest participation of the periphery of small V309
islands in CO oxidation via promotion of the CO oxidation reaction at the metal-oxide
phase boundary.
In summary, in situ STM studies of CO titration on the oxygen precovered metal
surfaces have demonstrated atomic details of CO oxidation on metal surfaces and
have shown excellent agreement with macroscopic kinetic measurements. Moreover,
in situ studies have revealed an interesting but not well-understood, nonlinear
behavior of reaction kinetics. The accelerated reaction rate observed takes place
only when surface oxygen islands, either compressed oxygen islands or surface oxide
islands, are reduced to the nanometer size. The nonlinear reactivity of these
nanoislands is in stark contrast with the large adsorbate layer and requires further
investigations.
Figu re 3.1 9 Series of 5
0 1 "A) recorded du
su rface covered wi th la
(a) 1 L; (b) 8 l; (c) 15 L;
large V30, island is seer
(Repri nted with pennls
El sev,er.)
I,~
3.4
Metal Surfaces at Hi
The above studies ;
composition and str
has played a critica i I
UH V to atm osp heri
The pioneering}ll
surface restructure
pressUJ s and at 42 ~
surface using a flow
surface ofPI(11 0) ex
of CO lifts this recoil
STM shows the expo
to a bul k-like (1 x l
Figure 3.21 shows s
al 425 K. t he local [(
phase upon CO exp,
3.4 Metal Surfaces at High Pressures
Figure 3.19 Series ofSTM images (25 nm x 25nm, V, = 1.5 V,
I, = 0.1 nA) recorded during dosing CO on the (2 x l)O-Rh (1 1 1)
surface covered with large (mean size or ~5 0 nm) V3 0 9 islands:
(a) 1 L; (b) 8 L; (c) 15 L; (d) 22 L; (e) 30 L; (f) 600 L. A fraction or a
large V30 9 isla nd is seen in the upper right-hand side orthe image.
(Rep rinted with permission rrom Ref. [68J. Copyright 2005,
Elsevier.)
3.4
Metal Surfaces at High Pressures
The above studi s show that the chemisorptions on metals could often alter the
composition and structure of metal surfaces. 10 bridge the pressure gap, in situ STM
has played a critical role in observing the dynamic behavior of catalytic surfaces from
UI:-IV to atmospheric pressures .
The pioneering high-pressure STM study by dntyre et al. [69Jshows the Pt(l 1 0)
surface res tmctures in single-component gases of H2 , O2 , and CO at atmospheric
pressures and at 425 K. Hendriksen et al. [22, 23J took one ste p further to view tbjs
su rface using a flow-reactor STM under high-pressure 0 or a COj02 m ixture. The
surface of Pt(ll 0) exhibits a (1 x 2) missing·row reconstruction in HV The exposure
ofCO lifts this reconstruction even at low pressures. In the presence of1 bar 0 , in situ
STM shows the exposure ofhigh-pressure CO not only lifts the (1 x 2) reconstruction
to a bulk-like (1 x 1) phase but also causes the coarsening of Pt(l 1 0) surface.
Figure 3. 21 shows sequences of snapshots on the Pt(l 1 0) surface in 1.25 bar CO
at 425 K. The local rearrangement caused by the transition from the (1 x 2) to (1 x 1)
phase upon CO exposure leads to the fragmentation of Pt(l 1 0) terraces and a high
181
3.4 Metal Surfaces at High Pressures
Figure 3.1 9 Series of STM images (25 nm x 25 nm, V, = 1.5 V,
',= 0.1 nA) recorded du ring dosing CO on the (2 x l)O-Rh(l 1 1)
surface covered wi th l arge (mean size of ~ 50 nm) V309 islands:
(a) 1 L; (b) 8 L; (c) 15 L; (d) 22 L; (e) 30 L; (f) 600 L. A fraction of a
large V109 isla nd is seen in the upper righ t-ha nd side of the image.
(Reprinted with perm issio n from Ref. [68J. Copyright 2005,
Elsevier.)
3.4
Metal Surfaces at High Pressures
The above sht dies show that the chemisorptions on m etals could often alter the
composition and struc:ture of m etal surfaces. To bridge the pressure gap. in situ STM
has played a critical role in observi ng the dynamic behavior of catalytic surfaces £i-om
!-IV to atmospheric preSSLU('s.
The pioneering high-pressure STM study by Mdntyre et al. [69J shows the Pt(l 10)
surface restructures in single-com ponent gases of H 2 , O2 , and CO at atmospheric
pressures and at 42 K. Hendriksen et al. l22, 23J took one step further to view this
SUIface using a flow-reactor STM under high-pressure C or a COI0 2 mixture. The
surface ofPt(11 0) exhibits a (1 x 2) missing-row reconstruction in U H V. The e"'Posure
of CO lifts Lhis reconstruction even at low pressures. In the presence ofl ba r CO, in situ
STM shows the exposure ofhigh· pressure CO not only lifts the (1 x 2) reconstruction
to a bulk·lik (1 x 1) phase but also causes the coarsening of Pt(l 1 0) : urface.
Figure 3.21 shows sequences of snapshots on the Pt(l 10) surface in 1.25 bar CO
at 425 K. The local rearran gement caused by the transition from the (1 x 2) to (1 x 1)
phase upon CO exposure leads to the fragmentation of Pt(l 1 0) terraces an d a high
181
821 3
In Situ STM Studies of Model Catalysts
Cd )
Figure 3.21 Series of
(140n m x 140 nm)
immediately after in
in the reactor·STM at
Figure 3.20 Series of STM images
(25 nm x 25 nm, V, = 15 V, I, = 0.1 nA)
recorded during dosing CO on the (2 x l)O-Rh
(1 1 1) surface covered with small (mean size of
~ 15 nm) Vj 0 9 is lands: (a) 1 L; (b) 21 L; (c) 31 L;
(d) 108 L; (e) 204 L; and (f) 270 l. Several small
irreg ular shaped Vj 0 9 is lands phase are visible .
The areas labeled (2 x 2)-A, (2 x 2) -8, and COs",
correspond to the (2 x 2)-0 + CO, (2 x 2)-CO,
and CO saturation layer phase s, respectively.
(Reprinted with permission from Ref. [68]
Copyright 2005 , Els evier.)
density ofsurface steps (Figure 3.21a). To reduce the total surface energy, coarsening of
the surface steps takes place (Figure 3.21b-f) whereas a much slower (1 x 1) phase
transition occurs upon CO exposure . With time the curved Pt islands are smoothed,
forming rounded large islands covered by CO on the Pt(l 1 0) surface.
The CO-covered Pt(l 1 0) surface \vas then exposed to a mixture of CO/0 2 gases,
with the ratio of CO/0 2 adjusted by flow meters. The pressure changes ofCO , O 2 , and
the reaction product, CO 2 , were monitored by leaking the gases from the flow reactor
to a quadrupole mass spectrometer (QMS) attached to the flow-reactor STM. The
surface structure and reactivity of Pt(l 1 0) could be measured simultaneously with
the combination of STM and QMS.
Figure 3.22 plots the real-time pressure of CO , O 2 , and CO 2 , as well as the
corresponding sna pshots of in situ STM images on Pt(l 1 0). The production of CO 2
starts right away with the presence of both CO and O 2 in the reactor. As the reaction
proceeds, two stages of reaction rates exist, as evidenced by the step increase in CO 2
pressure in Figure 3.22. At the stage of low reaction rate (see B, F, and H in
Figure 3.22), the corresponding STM images show no apparent change in the surface
structure, indicating the Pt(l 1 0) surface remains metallic. At the stage of high
reaction rate (see D and G in Figure 3.22, about three times higher than the
low reaction rate) , the corresponding STM images suggest a rough ened surface,
indicating the
reaction rate (Figure
in surface roughnesi
Subsequent high-pr
group [70] verified
assumed to be resp
suggest that the th
periodicity. It is 11'01
only during the CO
Combined with tl'
gas phase could dim
relatively high reacti
"Mars-Van Krevelen
on Pt group metals i!
proceeds via (1) the
(2) surface diffusion
The authors furth,
STM and SXRD Al
oxidation. The gradl
surface oxides and a
high pressure is in I
shows a strong orie :
3.4 Metal Su1aces at High Pressures
Figure 3.21 Series of STM snapshots
(140 nm x 140 nm) taken on Pt(l 10) , starting
immediately after introduction of 1.25 bar CO
in the reactor-STM at 425 K. The "tiger skin"
pattern in the fir st panel shows that the (1 x 2)
to (1 x 1) transition has divided the s urface in
two levels, each 50%, and a high density of
steps. Subsequent images show the progressive
reduction of the step density by coarsening of
the s tep pattern. The elapsed time in minute s
is indicated in each panel. The two ball
models indicate the atomic-scale geometries
characteristic for the starting and end
situations . (Reprinted with permission from
Ref. [22]. Copyright 2005, Springer.)
indicating the formation of surface Pt oxide. At the point of the step increase in
reaction rate (Figure 3.22C), the STM image suggests a modest and uniform increase
in surface roughness, which the authors assigned as a commensurate Pt oxide film.
Subsequent high-pressure surface X-ray diffraction (SXRD) studies by the same
group [70] verified the formation of this commensurate oxide film, which was
assumed to be responsible for the increased reaction rate. Further SXRD results
suggest that the thickness of this oxide film is one monolayer with a (1 x 2)
periodicity. It is worth noting that roughening of the oxide surface was obs erved
only during the CO oxidation reaction but not in 1 bar pure O2,
Combined with their kinetic measurements, the authors proposed CO from the
gas I hase could directly react with oxygen atoms in the surface oxides, accounting for
relativ ly high reactivity of this phase for CO oxidation. This mechanism, termed as
"Mars-Van Krevelen mechanism," challeng s the general concept that CO oxidation
on Pt group metals is dominated by the Langmuir-Hinshelwood mechanism, which
proceeds via (1) the adsorption of CO and the dissociative adsorption of O2 and
(2) surface diffusion of COad and Oad atoms to ultimately form CO 2,
The auU10rs further tested the Pt(1 1 1) and Pd(1 10) surfaces [71, 72Jusing in situ
STM and SXRD. All these single crystals show a similar kinetic behavior in CO
oxidation. The gradual rough ening of the surface corresponds to the formation of
surface oxides and a higher CO oxidation rate. The structure insensitivity observed at
high pressure is in contrast with the results obtained in UHV, where the reactivity
shows a strong orientational dependence.
183
841
3 In Situ STM Studies of Model Catalysts
,. '
ro
.~
..
~
..... -.. ..
. ... . ......
~.
,'
~
_. - - - -
-
-- -
"
~
0.1
;£
~
:::J
en 0.01
en
~
Q
ro
ro
1: 1E-3
Q
1E-4
0
1000
2000
3000
4000
5000
6000
7000
time [S]
(A )
can lead to uncerta l
global reaction rate.
STM measnreme
ing the surface stm
further prevents pre
application and devi
provide more precis
theless. the above sh
studies in model cat
effect of lowering
rep rod uce the same
the restricted difTus
surface structures UJ
UHVexperiments.
3.5
In Situ Studies ofSu
Figure 3.22 QMS signa ls and STM images
sim ultaneously measured during CO oxidation
onthe Pt(ll 0) surfaceatatemperatureof425 K
in a 3.0 ml min - 1 flow of mixtures of CO and/or
O 2 at 0.5 bar. (Upper panel) QMS signals of
O 2 , CO, and CO z, measured directly from the
reactor cell. Label s (A) - (H) correspond to the
STM images in the lower panel. R,ow and Rhigh
denote the low and high CO z production rate
branches. P'h indica tes the threshold value
of the CO press ure at which the rate switched
from R ,ow to R high and the surface changed
simultaneously from smooth to rough . (Lower
panel) STM images (210 nm x 210 nm) from an
STM movie (65 s/image, I, = 0.2 nA. V, = 80 mV).
The images were differentiated to enhance the
contrast. Images (A). (B), (E). (F), and (H) show
flat terra ces separated by steps of the Pt lattice.
This co rres ponds to the metallic, CO-covered
surface. Image (C) shows the change in the
surface accompanying the step in activity at
t = 2109 s (the image was built up from bottom
to top). Images (D) and (G) show the rough
surface consisting of protrusions. with heights of
0.2-0.4 nm , and pits (see inset) . (Reprinted with
permission from Ref [23]. Copyright 2002.
The American Phys ical Society.)
Due to the increased complexity at high pressure (e.g.. the impinging reactant
gases are no longer under molecular flow, causing the gas environment near the
surface to be markedly different from the ambient gases). much more work is
required before establishing a firm correlation between the surface structure and the
surface reactivity. The difficulty in u sing STM to measure the reaction rate directly
In an effort to brid
catalysts have emel
clusters supported a
STM. The tremendc
remarkable catalytic
ago [73-801. It was g'
The TiO l support G
clusters in the 2-4n
reactivity for low-ter
Although the cause
supported model A
understandil1g of tt
support. In this secti
the synthesis of SUP!
metal support inter;
supported Au calaly.
3.5 .1
Monitoring the Grow
Supported model ca
atoms onto a planar (
metal clusters depeJ
deposition rate. and,
model catalysts, it i,
-
-
--
--
---- -
-
-
3.5 In Situ Studies of Supported Model Catalysts
can lead to uncertainties in correlating local surface structure with the measured
global reaction rate.
STM measurements during high-pressure reaction present challenges for resolv
ing the surface structures at high resolution. Rapid diffusion of surface adsorbates
further prevents precise characterization of surface-active species. In this case, the
application and development of the fast-scanning technique is essential and could
provide more precise surface information during the high-pressure reaction. None
theless, the above study demonstrates the necessity ofexpanding high-pressure STM
studies in model catalysis. The effect of high pressure intuitively is equivalent to the
effect of lowering the temperature. Low-temperature UHV experiments might
reproduce the same adsorbate structures as do high pressures. However, due to
the restricted diffusion of metal atoms at low temperature, the dynamic change of
surface structures under catalytically realistic conditions could not be observed in the
UHVexperiments .
3.5
In Situ Studies of Supported Model Catalysts
In an effort to bridge the material gap, in situ STM studies of supported model
catalysts have emerged in recent years. Among supported model catalysts, Au
clusters supported on Ti0 2 (1 1 0) are the most investigated model system by in situ
STM. The tremendous interest in studying this model system originates from the
remarkable catalytic reactivity of supported Au clusters discovered a few decades
ago [73-80J. It was generally believed that Au, the noblest metal, is inert as a catalyst.
The Ti0 2 support alone also exhibits limited catalytic reactivity. In contrast, Au
clusters in the 2-4 nm size range supported on Ti0 2 exhibit a remarkable catalytic
reactivity for low-temperature CO oxidation and selective hydrogenation reactions.
Although the cause of the extraordinary activity of Au clusters is still under debate,
supported model Au catalysts are playing a key role in the development of our
understanding of the relative importance of cluster morphology and the cluster
support. In this section, we highlight a few recent in situ STM studies in monitoring
the synthesis of supported model catalysts , revealing the mechanism of the "Strong
metal support interaction" (SMSI) effect and measuring the sintering kinetics of
supported Au catalysts.
3.5.1
Monitoring the Growth Kinetics of Supported Metal Catalysts
Supported model catalysts are frequently prepared by thermally evaporating metal
atoms onto a planar oxide surface in UHV The morphology and growth ofsupported
metal clusters depend on a number of factors such as substrate morphology, the
deposition rate , and the surface temperature . For a controlled synthesis of supported
model catalysts, it is necessary to monitor the growth kinetics of supported metal
-
-
-
-
- - --
- -
-
185
•
86 1 3
In Situ STM Studies of Model Catalysts
Figure 3.23 STM imagesofthesameareaofaTi0 2 (11 0) surface
after the deposition of (a) 0.17 M L Au; (b) 0.34 M L Au; (c) 0.51 M L
Au; (d) 0.69 M L Au; (e) 0.86 M L Au; and (f) 1.3 M L A u. All images
have the same d imensions of 100 nm x 100 nm. The circle
highlighted in white in each image indicates the identical area.
(Reprinted with permission from Ref. [811 . Copyright 2003,
The Japan Society of Applied Physics .)
clu sters. Figure 3.23 shows a series of STM images as a function of increased Au
coverage obtained on the same area of a Ti0 2 (1 1 0) surface at room temperature [81].
At the very early stages of growth, Au clusters preferentially decorate the step edges
(Figure 3.23a). With an increase in Au coverage, the prevailing role of the step edges
as major nucleation sites decreases , whereas new clusters begin to nucleate on
terraces (Figure 3.23b-f). Au clusters grown at step edges grow much faster than
those at terraces. A quantitative analysis of Au cluster distribution suggests a bimodal
size distri bution for the growth of Au clusters at step edges and terraces. In situ STM
studies allow the growth mode of Au clusters to be followed on a cluster-to-cluster
basis. The results demonstrate that step edges playa dominant rol in the growth of
Au clusters on Ti0 2 (1 1 0) at room temperature.
Using in situ STM also makes it possible to monitor the growth of supported alloy
model catalysts. The simplest way to synthesize supported alloy model catalysts is to
Figure 3.24 STM images
deposition on an Ag preco
after deposi tion of (b) 0.1
precovered (0.033 ML) TiC
(e) 0.17MLAu, (f)O.S 1 M I
(e) show the appearance·
permission from Ref [821
evaporate both metal a
this method does not g
clusters or separate to
this problem to be add
catalysts.
In our study, Ag- Au
and the grOWtll kinetic
Au clusters in the prest
carried out as a funct
A particular precover~
saturate the step edge ~
in the absence of their
3.5 In Situ Studies of Supported Model Catalysts
187
•
Figure 3.24 STM images (100 nm x 100 nm) during Au
depos ition on an Ag precovered (0.08 ML) Ti0 2 (1 1 0) surface (a)
after deposition of (b) 0.17 ML Au. (c) 0.85 ML Au; or on an Ag
precovered (0. 033 ML) Ti0 2 (1 10) surface (d) after deposition of
(e) 0 17 M L Au. (f) 0.51 M L Au . T he white circles in image (d) and
(e) show the appearance of new Au cluste rs. (Reprinted wi th
permission from Ref [82J. Copyr ight 2004 , Elsevier BV)
evaporate both metal atoms in UHV directly onto an oxide support surface. However.
this method does not give information on whether the t\'vo metals mix and form alloy
clusters or separate to form tvvo types of monometallic clusters. In situ STM allows
this problem to be addressed by investigating the gro\oV1:h kinetics of supported alloy
catalysts.
In our study, Ag-Au alloy clusters ofvaryin g ratios were synthesized on Ti0 2 (11 0)
and the gro\vth kinetics for the clusters determined [82J . To understand the grO\oV1:h of
Au clusters in the presence ofAg clusters, sequential in situ STM mea surements were
carried out as a fun ction of Au coverage on a Ag precovered Ti0 2 (J I 0) surface.
A particular precoverage of 0.08 ML Ag (Figure 3.24a) was chosen to essentially
saturate the step edge sites with Ag clusters, allowing the investigation of Au growth
in the absence of their preferred nuclea tion sites. The series of in situ ST M images
881
3 In Situ STM Studies of Model Catalysts
(Figure 3.24a-c) show a preferential growth of Au on the existing Ag clusters. As the
Au coverage is increased, new Au clusters begin to appear on the surface. In
another set of experiments, the Ti0 2(1 1 0) surface was pre covered with less Ag
so that a fraction of empty step edge sites remain open for the nucleation of new
Au clusters (Figure 3.24d). The series of in situ STM images (Figure 3.24d-f) show
even a small amount of Au deposition (0.17 M L) leads to the nucleation of new Au
clusters.
These systematic experiments of Au growth on Ti0 2 (1 1 0) precovered with Ag
clusters show the versatility of in situ STM experiments in controlling the synthesis of
supported alloy model catalysts. Simply by blocking step edge sites to a varying extent,
either monometallic clusters or bimetallic clusters could be selectively synthesized
and monitored. By changing the sequence of metal deposition, the surface structure
of alloy clusters can also be altered. Adopting the same in situ approach, the
synthesis of supported alloy model catalysts has been extended to Pdj Au clusters
on Ti0 2 (1 1 0) [83] and PtjRh clusters on Ti0 2 (1 1 0) [84]. It is expected that this
method will be generally adopted for the preparation and characterization of
supported model catalysts.
3.5.2 Studies of the SMS I Effect [n the late 1970s, Tauster et al. [85-87] discovered the unusual properties of group
VII[ metal. for example, Pt and]Y, when supported on Ti0 2 and reduced at relatively
high temperatures. The Pt and [r clusters supported on Ti0 2 show a suppressed CO
and H2 chemi sorption and an increased methanation catalytic activity. "S trong
metal support interaction" has since been introduced to describe the unusual
properties of group VIll metal supported on reducible oxides such as Ti0 2 . A few
reasons for the SMS[ effect have been postulated, including alloy formation,
altered electronic effects at the interface, or encapsulation of supported metal
clusters.
Bowker et al. [88 ,89] have investigated the SMSI effect using insitu STM. As shown
in Figure 3.25, Pd clusters with a mean si ze of approxima tely 4 nm were supported
on Ti0 2 (11 0) and heated to 673 K. The surface was then exposed to 5 x 10- 8 mbar O 2
and monitored by STM as a function of time. In the presence of oxygen, the
reoxidation of the Ti0 2 (1 1 0) surface proceeds. Since the dissociation probability
of O 2 on Pd is about three magnitudes higher than on Ti0 2 (1 1 0), O 2 dissociation
mainly occurs on supported Pd clusters. The dissociated oxygen atoms subsequently
diffuse onto the adjacent surface, that is, spillover, and coalesce with Ti interstitials
from the bulk, causing the enrichment ofTiOx species surrounding the Pd clusters.
The regrowth of Ti0 2 layers thus accelerates at the periphery of Pd clusters.
Eventually, the growth of Ti0 2 layers surpass the height of the Pd clusters and
fully encapsulates them v.rithin the Ti0 2 overlayers, that is, in situ STM study
verifies that encapsulation is a major contributor to the SMSI effect. This is also
the first study to have directly observed in situ the spillover, an elementary step in
catalytic process.
Figure 3.25 Asequence
at 673 K of oxygen spillovt
an amb ient pressu re of 5
(doubled before image (f):
to oxygen from Image (a)·
of L): 114, 178.237, 282,
spillove r begins With the fc
3.5.3
Sintering Kinetics of SL
Although supported A
such as removing CO
for the commercializal
rapidly [78, 90, 91]. S
consisting of Au cluste
has been shown to con
the sintering of suppor
Ti0 2 (1 1 0) in UHV ar
The first issue in si
process. The sinterin e
modes: (a) the migrati
monomers (single 1m
cluster migration, ent,
coalesce with neigh bor
monomers dissociate
clusters [93, 94).
3. 5 In Situ Studies of Supported Model Catalysts
189
•
Figure 3.25 A sequence of STM images taken
at 673 Kof oxygen spillover from Pd clusters in
an ambient pressure of 5 x 10- 8 mbar of O 2
(doubled before image (f)). The total exposures
to oxygen from image (a}-(f) were (in units
of L): 114, 178, 237, 282. 344, and 53l. The
spillover begins with the formation of one layer
around the Pd clusters, which then becomes
multilayer growth, eventually burying the Pd
cluster with Ti0 2 . In image (f), a total of seven
layers ofTi0 2 have grown around the Pd on top
of the original Ti0 2 surface. (Reprinted with
permission from Ref. [89J. Copyright 2000,
Elsevier)
3.5.3
Sintering Kinetics of Supported Au Clusters
Although supported Au catalysts have great potential in technological applications ,
such as removing CO selectively from hydrogen fuel for fuel cells, a major problem
for the commercialization of supported Au catalysts is that these catalysts deactivate
rapidly [78, 90. 91J. Similar deactivation has been observed for model catalysts
consisting of Au clusters deposited on a reduced Ti0 2 (11 0) where rapid deactivation
has been shown to correlate with sintering of the Au clusters [91. 92J. To understand
the sintering of supported Au catalysts. we have studied the sintering kinetics of Aul
Ti0 2 (1 1 0) in UHV and in the presence of reactant gases.
The first issue in sintering kinetics is to determine the primary mass transport
process. The sintering of supported nanoclusters can occur in either one or two
modes: (a) the migration and coalescence of whole clusters or (b) the migration of
monomers (single metal atoms or metal complexes). The first mode, known as
cluster migration, entails the migration of whole clusters on the surfa ce that then
coalesce with neighboring clusters. In the second mode, known as Ostwald ripening,
monomers dissociate from small clusters, diffuse to, and coalesce with large
clusters [93. 94J.
90
I3
In Situ STM Studies of Model Catalysts
In situ STM allows one to monitor individual clusters under realistic conditions
and to directly determine the mass transport mode controlling the kinetics. Mitchell
et al. studied Au clusters supported on Ti0 2 (1 1 0) at approximately 750 K as a
function of time [95]. Their STM results show Au clusters are mostly static while
some small clusters with size below 3 nm diffuse along the surface. The diffusion of
small clusters does not behave in a continuous manner. Instead, the clusters stick for
a relatively long time before taking a sudden jump. This unconventional diffusion of
clusters, termed as Levy flight, leads to the speculation that Au clusters sinter in the
mode of cluster migration. Such results are in agreement with the in situ ST M study
by Kolmakov et al. [12], who examined the stability of AujT i0 2(1 1 0) after thermal
treatment at 950 K. Dramatic morphological changes were observed. Accompanying
the decomposition of surface steps, the density of Au clusters decreased by 20%,
whereas the volume of the Au clusters increased. Most Au clusters changed their
positions on the Ti0 2 (11 0) surface. Clearly, cluster migration has played a role in the
thermally induced sintering of supported Au clusters under UHV. Interestingly, the
above studies were both conducted at the temperature under which the Ti0 2 (1 1 0)
substrate becomes unstable and starts to decompose. To fully explore the sintering
mode induced by thermal treatment, it is necessary to conduct such in situ experi
ments at intermediate temperatures where the Ti0 2 (1 1 0) lattice remains stable.
The sintering of AujTi02(1 1 0) was also studied by our group in the presence of
reactant gases [96J. Figure 3.26 shows the morphological changes of 0.5 ML Au
clusters supported on Ti0 2 (1 1 0) in the presence of COj02 at 300 K. Under UH V
conditions, the Au clusters remain immobile and show no detectable change ofeither
cluster size or shape for more than 4 h. Sintering of Au clusters began immediately
upon the introduction ofa 0.1 Torr COj02 gas mixture (1: 1 ratio). Figure 3.26 shows
no apparent change of cluster positions before and after gas exposure; however, the
small Au clusters gradually decay whereas the large Au clusters grow in size. The
gradual change in the cluster size and static cluster positions are consistent with an
Ostwald ripening process. Most Au clusters with a height around 4.6 A or less
(assuming the height of Au clusters of two-layer thickness is 4.6 A [97]) disappear
within 2 h of CO oxidation reaction. Gold clusters supported on Ti0 2 (1 10) were also
exposed to O 2 and CO separately to investigate the influence of the chemisorbed
reactants. Minimal changes were observed for 0.5 ML Au clusters supported on
Ti0 2 (11 0) after an exposure of 0.1 Torr O 2 or 3 Torr CO for more than l.5 h at 300 K.
Therefore, a synergetic effect of COj02 was found to induce and accelerate the
sintering of Au clusters on Ti0 2 (1 1 0).
The synergetic effect of a CO and O 2 mixture can be explained by a reaction
induced mechanism, which might involve hot electrons generated by CO oxida
tion [98- 100], inducing the detachment ofAu monomers from supported Au clusters
and ini tiating the sinterin g process. On the basis of the sintering model of Ostwald
ripening, the activation energy for Au clusters with diameters around 3 nm is
estimated to be approximately 10kJmol- 1 , a value that closely tracks the activation
energy of CO oxidation on supported Au clusters. The large difference in comparing
this number with the activation energy ofsintering for supported Ali clusters in UH V,
approximately 280 kJ mol 1 [101, 102], demonstrates that CO oxidation itself does
(e)
Figure 3.26 75 nm x :
of0.5 MLAudusterssu
surface in the presenc
mixture at 300 K. (a- f
at the same surface ar
indeed influence (
oxidation over oth(
3.6
Outlook
This chapter re iel
From revealing rea
UHVa nd on exten r
questions in catal;
3. G OlAtlook
(e)
(f)
Figure 3.26 7S nm x 7S nm in situ STM images
of 0.5 MLAu ciusterssupported ontheTi0 2 (1 1 0)
surface in the presence of 0.1 Torr CO and O 2
mixture at 300 K. (a-f) are consequently taken
at the same surface area. The time intervals are
(a) 0 min; (b) 28 min ; (c) 42 min ; (d) 63 min; (e)
120 min ; and (f) 280 min . Tunneling parameters
are V, = 2 V and = 0.1 nA. (Reprinted with
permission from Ref. [96J. Copyright 2009, The
American Chemical Society.)
't
indeed influence cluster sintering. This effect may very well be general fo r CO
oxidation over other supported metal catalysts.
3.6
Outlook
This chapter reviews the recent progress in in. situ STM studies of model catalysts.
From revealing reaction pathways to delineating active sites, in. situ STM studies in
UHVand on extended surfaces have demonstrated their power to solve fundamental
questions in catalysis and enhance our understanding of the elementary steps of
191
92 / 3 In Situ STM Studies of Model Catalysts
surface catalytic processes. Recent efforts in expanding in situ STM studies to high
pressures and to supported model catalysts illustrate the need to study model
catalysts under realistic conditions.
The studies discussed in this chapter have shown surface structures are extremely
dynamic under reaction conditions, consistent with the hypothesis promoted by
Somorjai [103-107]. The h ypothesis, termed as the "flexible surface," supposes that
metal surfaces with low coo rdinated metal atoms or curved cluster surfaces are highly
fluxional during reaction. Those atoms with high diffusivity are responsible for
high activity of metal surfaces. To this end, it is critical to compare the timescale and
energetics of metal atom diffusion with their catalytic reactivity. With the develop
ment of fast scan techniques, in situ STM now provides a great opportunity to test this
hypothesis. The studies to date have shown that small can be quite different and
less can sometimes mean mo re. In the coming years, there is no doubt that in situ
STM studies on well-defined nanostructures will exponentially increase and
contribute invaluably to an atomic-level understanding of reactions catalyzed by
solid surfaces.
Acknowledgments
We gratefully acknowledge the support for this work by the Department of Energy
(DOE) , Office of Basic Energy Sciences, Division of Chemical Sciences , and the
Robert A. Welch Foundation.
14 Melmed, A.) . (199
9, 601.
15 Oliva, A. I. , Rom
Anguiano, E., an
Sci. hlstnml.. 67,
16 Weinstein, v..Sl u
and Ben jacob, E.
66, 307 5.
17 Ren. B., Picardi, a
(20 04) Rev. Sci. [;1
18 Fried, G.A. , Wang
(1 993 ) Rev. Sci. In
19 lwami , M.. Ueh'
(1998) Rm Sci. In
20 Schroder. U., Mci
M.. and Somorjai,
333. 33 7.
21 Reiw el.t. R.. Gun
Win tterlin, )., Ku
Schlogl , R. (2007)
Phys.. 9. 35 90.
22 Hendr iksen. B. L.
and Frenkel1, ).\V.
36, 43 .
23 Hendriksen, B.L. \
(2002) Phl'S. Rev. L
24 Wintterlin. j. (20
References
25 Hahn , j.R. and Ho
Lett.. 87. 1661 02.
Langmuir, I. (191 5) Phys. Rev., 6, 79.
2 Bartels, L , Wang. F., Moll er. D.. Knoesel,
E.•and Hein z, TF. (2004) Science. 305, 648 .
3 Che. M. and Bennett, C O. (1989) Adv.
Cata!.. 36. 55 .
4 Wintterhn, )., Trost, j .. Renisch. S..
Schu ster. R., Zambelli, T . and Ert!. G.
(1997) Suif. Sc i , 394, 159.
Kuipe rs . L. . Hoogeman. M.S. , and
Frenken. j.W.M . (199 3) Phys. Rev. Lett., 71.
3517.
6 Ludwig, C, Gompf, B.. Glatz. w..
Peterse n. ) .. Eisenmenger. w.. Mobus,
M.. Zimmermann, U., and Karl, N. (1992)
Z . Phys. B. 86, 397 . 7 Rost, M.).. Crama, L.. Schake\. P., Van To!.
E., Van Ve\zen -Williams, G. B.E.M .,
Overgauw, CF.. Ho rst. H. , Dekker, H.,
Okhuijs en, B.. Seynen , M., Vi jfti gschild,
A. , Han, P., Katan . A. ) ., Schoots, K. ,
Schumm, R., Van Loo, w.. Oosterkamp,
TH. , and Frenken, ). W.M . (200 5) Rev. Sc i.
instrum., 76 , 053710.
8 Rossler, M. , Geng, P., and Wintterlin , ).
(2005) Rev. Sci. Instrum., 76. 023705.
9 La egsga ard , E.. Osterlund. L , Thostrup.
P., Rasmu ssen. P.B. , Stensgaard , 1.. and
Besenbacher, F. (2001) Rev. Sc i. IItSt rum.,
72, 353 7.
10 Rasmussen, P.B., Hendriksen, B.L.M. ,
Zeijlemake r, H., Ficke . H.G .. and
Frenken. j.W.M. (1998) Rev. Sci. Instrum ..
69 . 3879.
11 Mcintyre, B.) ., Salmeron. M.. and
Somorjai. GA (199 3) Rev. Sci. instrum. ,
64 . 687.
12 Kolmakov, A. and Goodman, D.W. (2002)
Chern. Rec., 2, 446.
13 Kolmakov. A. and Goodman, D.W. (2003)
Rev. Sci. Instrum., 74, 2444 .
26 Hla, S.w. and Riedl
Rev. Ph ys. (hem ., 5,
27 Rieder, K.H.. Meye l
Moresco, F., Braun
K., Repp, ). , Foelscl
(20 04) Phi/os. Tram.
1207
28 Wong, K. L.. Rao, B.
Ulin-Avila, E., and .
J Chern. Pill'S., 123.
29 Longwitz, S.R.. Sch
EX, Yang, R.T. La ,
gaa rd, I., Brune, H.
(2004) J Phys. Chu
30 Vestergaard, EX , 1
Laegsgaa rd. E., S t~r
B., and Besenbache
Lett., 88, 259601.
31 Osterlund, L. , Rasn
Thostrup, P., Laegs
References
14 Melmed, A. J. (1991)). Vac. Sci. Technol. B,
9, 60l.
lS Oliva, A.I. , Romero, A. , Pena, j.L. ,
Anguiano, E., and Aguilar, M. (1996) Rev.
Sci. Instrum. , 67, 1917.
16 Weinstein, v., Slutzky, M., Arenshtam, A. ,
and Benjacob, E. (1995) Rev. Sci. Instrum .,
66, 3075.
17 Ren, 8. , Picardi, G., and Pettinger, B.
(2004) Rev. Sci. Instrum., 75, 837.
18 Fried, G.A ., Wang, X.D., and Hipps, KW
(1993) Rev. Sci . Ins trum., 64, 1495 .
19 iwami , M., Uehara, Y., and Ushioda, S.
(1998) Rev. Sci . instrum., 69 , 4010.
20 Schroder, U., Mcintyr e, B.j. , Salmeron,
M. , and Somorj ai, G.A. (1995) Surf Sci.,
333,337.
21 Reichelt, R. , Gun the r, S., Rossler, M.,
Wintterlin, j., Kubias, B. , jakobi, B., and
Sch log! , R (2007) Phys. Chem . Chem.
Phys., 9, 3590.
22 Hendriksen, B.L.M. , Bobaru, S.c. ,
and Frenke n , j.WM. (2005) Top . Ca tat.,
36, 43.
23 Hendriksen , B.L.M. and Frenken, j.WM.
(2002) Phys. Rev. Lett., 89, 04610l.
24 Wintterlin, J. (2000) Adv. Catal., 45 , 131.
2S Hahn, j.R and H o, W (2001) Phys. Rev.
Lett., 87, 166102.
26 Hla, S.W. and Rieder, KH. (2003) Annu.
Rev. Phys. Chem., 54, 307.
27 Rieder, KH. , Meyer, G., Hla, S.W,
Moresco, F. , Braun, KF., Morgenstern,
K., Repp , j., Foelsch, S., and Bartels, L.
(2004) Philos. Tra ns. Roy. Soc. , 362,
1207.
28 Wong, KL. , Rao, BV, Pawin, G.,
Ulin·Avila, E.. and Bartels, L. (2005)
). Chem. Phys., 123, 201102.
29 Longwitz, S.R., Schnadt, j., Ves terga ard,
E.K. , Yang, R.T. , Laegs gaard , E. , Stens·
gaard , I., Brune, H ., and Besenbacher, F.
(2004)). Phys. Chem . B, 108, 14497.
30 Vestcrgaard, E.K. , Thostrup, P. , An, 1.,
Laegsgaard , E. , Stensgaard, L, Hammer,
B., and Besenbacher, F. (2002) Phys. Rev.
Lett., 88, 25960 l.
31 Osterlund, L. , Rasmussen, P.B. ,
Thostru p, P., Laegsgaard, E. , Stensgaard,
I., and Besenbacher, F. (2001) Phys. Rev.
Lett. , 86, 460.
32 Thostrup , P. , Ves tergaard, E.K , An , 1.,
La egsgaard , E., and Besenbacher, F.
(2003)). Chem. Phys., 118, 3724.
33 Yang, R.T., Wang, j .G ., Knudsen, j. ,
Schnadt, j., La egsgaard , E., Stensgaa rd , I. ,
and Bese nbacher, F. (2005 )). Phys. Chem.
B, 109 , 14262.
34 Mi tsui, 1., Rose , M.K , Fomin, E.,
Ogletree , D.F. , and Salmeron, M. (2003)
Nature, 422, 70S.
35 Salm eron, M. (2005) Top. Catat. , 36, 55.
36 Mits ui, 1., Rose, M.K, Fomin, E. ,
Ogletree, D.F. , and Salme ron, M. (2003)
Surf Sci ., 540, 5.
37 Diebold, U. (2003) Surf Sci. Rep.,
48, 53.
38 Diebold , U. , Lehman , j., Mahmoud, 1.,
Kuhn, M., LeonardeIli, G., Hebenstreit,
W, Sc hmid , M., and Varga, P. (1998)
Surf Sci., 411, 137.
39 Schaub, R. , Thostrup , R, Lopez , N.,
Laegsgaard, E., Stens gaard , L, Norskov,
).K , and Besenbacher, F. (2001) Phys. Rev.
Lett., 87, 266104.
40 We ndt, S., Schaub , R , Mattbiesen, j.,
Ves tergaard, E.K. , Wa hlstrom, E.,
Rasmu ssen , M.D. , Thostrup, P. , Molina ,
L.M., Laegsgaard, E., Stensgaard, I. ,
Ha mmer, B., and Besenbacher, F. (2005)
Surf Sci. , 598, 226.
41 Bikondoa , 0 ., Pang, c.L., lthnin, R.,
Muryn, c.A. , O nishi , H ., and Thornton ,
G. (2006) Nat. Mater., 5, 189.
42 Wendt, S., Matthiesen, )., Schaub, R.,
Veste rgaard , E.K., Laegsgaard , E.,
Besenbacher, F., and H ammer, B. (2006)
Phys. Rev. Lett., 96, 066107.
43 Zhang, Z., Bondarchuk, 0 ., Kay. B.D.,
White, j.M .. and Dohnalek, Z. (2006)
J. Phys. Che m. B, 110, 21840.
44 Ii, S.c., Zhang, Z. , Shepp ard, D., Kay,
B.D., White, j.M., Du, Y., Lyubioe tsky, I. ,
Henkelman, G., and Dohnalek, Z. (2008)
). Am. Chem. Soc. , 130, 9080.
45 Zhang, Z.R , Bondarchuk, 0., White ,
j .M. , Kay, B.D. , and Dohn alek, Z. (2006)
). Am. Chem. Soc., 128, 4198.
193
941
3 In Situ STM Studies of Model Catalysts
46 Zhang, Z.R" Bondarchuk, '., Kay, B.D.,
White, J .M., and Dohnalek, Z (2007)
J. Phys. Chem. C, 111, 302l.
47 Du, YG. , Dohnalek, Z., and Lyubinetsky,
I. (2008) J. Phys. Che m. C, 112, 2649.
48 Zhang, Z , Ge, Q., Li, S.C, Kay, B.D.,
White, ).M., and Dohnalek, Z. (2007)
Phys. Rev. Lett., 99, 126105.
49 Lyubine tsky, I. , Yu, Z.Q. , and Henderson,
M.A. (2007) J. Phys. Chem. C, 11 1, 4342.
50 Wend t, S., Sprunge r, PT, Lira, E.,
Madsen, G.KH ., Li , Z.S ., Hansen , ).0 .,
Matthiese n, )., Bfekinge-Rasmussen. A.,
Laegsgaard, E. , Hammer, B., and
Bese nbacher, F. (2008) Science, 320, 1755.
51 Henderson, M.A., Epling, WS., Peden,
CH.F., and Perkins, CL (2003) J. Phys.
Chem. B, 107, 534.
52 Smi th , R.D., Bennett, RA., and Bowker,
M. (2002) Phys. Rev. B, 66, 7.
53 Park, KT, Pan, M., Meunier, V., and
Plumm er, E.W (2007) Phys. Rev. B, 75,
245415.
54 Onishi, H. and [wasawa, Y (1996) Phys.
Rev. Lett., 76, 79l.
55 Sachs, C, Hilde brand, M., Volkening, S.,
Wintterlin . I. , and Ertl, G. (2001) Science,
293, 1635.
56 Volkening, S., Bedurftig, K , Jacobi , K ,
Wintterlin, j. , and Ertl, G. (1999) Phys.
Rev. Lett., 83. 2672.
57 1mbihl, R. and Ertl, G. (1995) Chem. Rev. ,
95, 697.
58 Wintterlin, J., Volkcning, S., Janssens,
TV.W , Zambelli, T, and Ertl, G. (1997)
Science, 278, 1931.
59 Kim, S.H., Mendez, )., Winttcrlin , J., and
Frt! , G. (2005) Phys. Rev. B, 72, 155414.
60 Leibsle, F.M., Murray, P.W, Francis, S.M ..
Thornton , G .. and Bowker, M. (1993)
Nature, 363, 706.
61 Le ibsle, F.M., Francis, S.M. , Hag, S. , a nd
Bowker, M. (1994) Surf Sci., 318, 46.
62 Le ibsle. F.M. , Francis, S.M. , Da vis, R.,
Xiang. N., Hag, S., and Bowker, M. (1994)
Phys. Rev. Lett .. 72, 2569.
63 Crew, WW and Madix, R.J. (1996) Surf
Sci., 349, 275.
64 Ruan, L . Stensgaard, I. , Laegsgaard, E..
and Bese nbache r. F. (1994) Surf Sci..
314, L873 .
65 Nakagoe, 0 .. Watanabe, K, Takagi, N..
and Mats umoto, Y (2003) Phys. Rev. Lett.,
90 .226105.
66 akagoe. 0., Watanabe , K., Takagi. N.,
and Mats um oto. Y (2005) J. Phys. Chem.
B, 109. 14536.
67 Klus t, A. and Madix, R.). (2007) J. Chem.
Phys., 126, 084707.
68 Schoiswohl, I., Eck, S., Ramsey, M.G. ,
Andersen, I.N ., Surnev. S., and Netzer,
F.P. (2005) Surf Sci.. 580, 122.
69 Mcintyre. B.)., Salmeron, M., and
Somorjai, G.A. (1993) J. Vac. Sci. Tech., 11 ,
1964.
70 Acke rma nn. M.D .. Pedersen, TM.,
Hendriksen, B.L.M., Robach. 0 .. Bobaru,
S.C. Popa, I., Quiros, C, Kim, H.,
Hammer, B.. Ferrer. S., and Frenken.
JWM. (2005) Phys. Rev. Lett., 95, 255505.
71 Hendrikse n, B.L.M., Bobaru. S.C, and
Fre nken, IWM. (2004) Surf Sci.. 552.
229 .
72 He ndriksen, B.LM., Bobaru, S.C . and
Fre nke n, I.WM. (2005) Catal. Today, 105,
234.
73 Freund , H .J., Baumer, M., and
Kuhlenbeck, H. (2000) Adv. Catal., 45 ,
333.
74 Henry. CR (1998) Sulj Sci. Rep. , 31, 235.
75 Campbell, CT (1997) Surf Sci. Rep. , 27, l.
76 Rainer, D.R. and Goodman, D.W (1998)
J. Mol. Catal. A, 131, 259.
77 Campbell, CT, Grant, A.W., Starr. D.E. ,
Parker, S.C, and Bondzie, VA. (2001)
Top . Catal., 14, 43.
78 Haruta, M. and Date, M. (2001) Appl.
Catal. A. 222, 427.
79 Corti, CW , Hollida y. R.) ., and
Thompson, DT (2007) Top. Catal. , 44,
33l.
80 Bond . G.C and Thompson, DT (1999)
Catal. Rev., 41, 319.
81 Santra, A.K, Kolmakov, A., Yang, F. , and
Goodman, DW (2003)jpn. J. Appl. Phys.
Part 1. 42, 4795.
82 Sanrra, A. K. , Yang. J
DW (2004) Sur}, Sc
83 Han, P. and Goodm
j. Phys. ehem. 11
84 Park. J.B .. Ratliff. J. !
Chen, DA (2006) 5
85 Tausler.S .J.( 1987} At
86 Ta uster. S.)., Fung. :
and Horsley, ).A. (1'
c.
1121.
87 ·[auster. S.J .. Fung. S
(1978) JAm. Cktm
88 Bowker. M (2007) I
Phys., 9, 3514.
89 Bowker, M .. Bowker
Stone, P.. and Ra mi
J Mol . Catal. A. 16:
90 Haruta, M. (1997) (
91 VaIden. M., Lai. X.•
(1998) Scimc<. 281.
92 lAi. X., St Clair. TP
Goodman, D.W (19'
25.
93 Chakravc rty, B.K. (1
Sol. 28, 2401
94 Wynbl att. P. and Gj
Prog. Solid Stare ell
References
82 $anlra, A. K.. Yan g, F., and Goodman,
D.W. (2004) Surf Sci.. 548, 324.
83 Han, P. and Goodman, D.W. (2008)
j. Phys. Chern. C, 112, 6390.
84 Park, ).B., Ratliff. ).5., Ma. 5. , and
Chen, D.A. (2006) Surf. Sci. , 600, 2913.
85 Tau$ter, S.). (1987) Acc. Chern. Res., 20, 389 .
86 auster. 5. )., Fung. S.c., Baker, RT K.,
and Horsley, ) A. (1981) Science, 211 ,
1121.
87 Tau ster, 5.)., f ung, S.c. , and Garten, R.L.
(19 78) j. Arn. Chern. Soc, 100, 170.
88 Bowker, M. (2007) Phys. Chern Chern.
Phys. , 9, 3514.
89 Bowker, M. , Bowker, L.). , Be nnett, R. A..
Stone, P., and Ramirez-Cuesta, A. (2000)
J. Mol. Catal. A, 163, 221.
90 Haruta, M. (1997) Catal. Today, 36, 15 3.
91 Vaiden , M., Lai. X., and Goodman, D.W.
(1998) Science, 281, 1647.
92 Lai , X., St Clair, TP .. Vaid en, M .. and
Goodman , D.W. (1998) Prog. Suif. Sci ., 59,
25 .
93 Chakraverty, B.K. (1967) J. Phys. Chern.
Sot, 28, 2401.
94 Wynblatt, P. and Gjostein, NA (1975)
Prog. Solid State Chern., 9, 21..
95 Mitchell, C.E.)., Howard , A., Carney, M.,
and Egdell , R.G. (2001) Suif. Sci., 490,
196 .
96 Yang, F. , Chen. M.S., and Goodman, D.W.
(2008) J. Phys. Chern. C. 113, 254.
97 Cosandey, F. and Madey. TE. (2001) SUI!
Rev. Lett ., 8, 73.
98 ji, X. Z. , Zuppero, A.. Gidwani, j.M.,
and Somorja i, G.A. (2005) Nan o Lett .,
5, 753.
99 Ii, X.Z., Zuppero, A.. Gidwani , ).M. , and
Somorjai , GA (200 5) J. Arn. Chern. Soc. ,
127, 5792.
100 Ii , X.Z. and omorjai, G.A. (2005 ) J. Ph)'s
Chern. E, 109, 22530.
101 Parker, S.c. and Campbell, CT (2007)
Phys Rev. E, 75, 035 430.
102 Campbell. C.T, Parker, S.c.. and Starr,
D.E. (2002) Science, 298,811.
103 Somorjai, GA (1991) Langmuir, 7. 3176.
104 Somorjai, G.A. (1992) CataL Lett .. 12, 17.
lOS Somo rj ai . GA (1994) Anlm. Rev. Phys.
Chern., 45 , 721.
106 Somorjai, G A (1996) J. Mol. CataL A,
107, 39.
107 Somorjai, G .A. and Rupprechte r, G.
(1998) J. Chern. Educ., 75, 161.
195