Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Molecular evolution wikipedia , lookup
Silencer (genetics) wikipedia , lookup
Community fingerprinting wikipedia , lookup
Gene expression wikipedia , lookup
Endogenous retrovirus wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
Gene expression profiling wikipedia , lookup
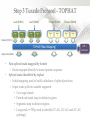
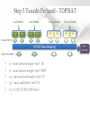
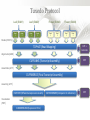
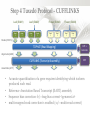
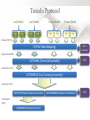
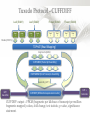
Expression Analysis of RNA-seq Data Manuel Corpas Plant and Animal Genomes Project Leader [email protected] Generation of Sequence Mapping Reads Identification of splice junctions Assembly of Transcripts Statistical Analysis 1 Summarization (by exon, by transcript, by gene) 2 Normalization (within sample and between sample) 3 Differential expression testing (poisson test, negative binomial test) The Tuxedo Tools • • Developed by Institute of Genetic Medicine at Johns Hopkins University / University of California, Berkeley / Harvard University 157 pubmed citations Tophat Fast short read aligner (Bowtie) Spliced read identification (Tophat) Cufflinks package Cufflinks – Transcript assembly Cuffmerge – Merges multiple transcript assemblies Cuffcompare – Compare transcript assemblies to reference annotation Cuffdiff – Identifies differentially expressed genes and transcripts CummeRbund Visualisation of differential expression results RNA-seq Experimental design • Sequencing technology (Solid, Illumina) – Hiseq 2000, 150 million read pairs per lane, 100bp • Single end (SE) Paired end (PE), strand specific – SE Quantification against known genes – PE Novel transcripts, transcript level quantification • Read length (50-100bp) – Greater read length aids mapping accuracy, splice variant assignment and identification of novel junctions • Number of replicates – often noted to have substantially less technical variability – Biological replicates should be included (at least 3 and preferably more) • Sequencing depth – Dependent on experimental aims RNA-seq Experimental design Toung et al. RNA-sequence analysis of human B-cells. Genome Research (2011) . Labaj et al. Characterization and improvement of RNA-Seq precision in quantitative transcript expression profiling. Bioinformatics (2011) • Extrapolation of the sigmoid shape suggests 20 % of transcripts not expressed • First saturation effects set in at ~40 million read alignments • ~240 Million reads achieve 84 % transcript recall RNA-seq Experimental design General guide • Quantify expression of high-moderatly expressed known genes • ~20 million mapped reads, PE, 2 x 50 bp • Assess expression of alternative splice variants, novel transcripts, and strong quantification including low copy transcripts • in excess of 50 million reads, PE, 2 x 100 bp Example • Examine gene expression in 6 different conditions with 3 biological replicates (18 samples) • Multiplexing 6 samples per lane on 3 lanes of the HiSeq (50 bp PE) • Generates ~25 M reads per sample • Assuming ~80% of reads map/pass additional QC (20 M mapped read per sample) • Cost – 3 lanes (£978 x 3 ) 18 libraries (£ 105 x 18), total £4824 Step 1 – Preprocessing reads • Sequence data provided as Fastq files • QC analaysis – sequence quality, adapter contamination (FASTQC) • Quality trimming, adapter removal (FASTX, Prinseq, Sickle) Step 2 – Data sources • Reads (Fastq, phred 33) • Genomic reference (fasta TAIR10), or pre built Bowtie index • GTF/GFF file gene calls (TAIR10) • http://tophat.cbcb.umd.edu/igenomes.html Tuxedo Protocol Leaf (SAM1) Read 1 Read 2 Leaf (SAM2) Read 1 Read 2 Flower (SAM3) Read 1 Read 2 Flower (SAM4) Read 1 Read 2 Reads (FASTQ) TOPHAT (Read Mapping) GTF + Genome CUFFLINKS (Transcript Assembly) GTF Alignments (BAM) Assemblies (GTF) CUFFMERGE (Final Transcript Assembly) Assembly (GTF) CUFFDIFF (Differential expression results) Visualisation (PDF) CUMMERBUND (Expression Plots) CUFFCOMPARE (Comparion to reference) GTF Step 3 Tuxedo Protocol - TOPHAT Leaf (SAM1) Read 1 Read 2 Leaf (SAM2) Read 1 Read 2 Flower (SAM3) Read 1 Read 2 Flower (SAM4) Read 1 Read 2 Reads (FASTQ) TOPHAT (Read Mapping) Alignments (BAM) • • Non-spliced reads mapped by bowtie – Reads mapped directly to transcriptome sequence Spliced reads identified by tophat – Initial mapping used to build a database of spliced junctions – Input reads split into smaller segments • Coverage islands • Paired end reads map to distinct regions • Segments map in distinct regions • Long reads >=75bp used to identify GT-AG, GC-AG and AT-AC splicings) GTF + Genome Step 3 Tuxedo Protocol - TOPHAT Leaf (SAM1) Read 1 Read 2 Leaf (SAM2) Read 1 Read 2 Flower (SAM3) Read 1 Read 2 Flower (SAM4) Read 1 Read 2 Reads (FASTQ) TOPHAT (Read Mapping) Alignments (BAM) • • • • • -i/--min-intron-length <int> 40 -I/--max-intron-length <int> 5000 -a/--min-anchor-length <int> 10 -g/--max-multihits <int> 20 -G/--GTF <GTF/GFF3 file> GTF + Genome Tuxedo Protocol Leaf (SAM1) Read 1 Read 2 Leaf (SAM2) Read 1 Read 2 Flower (SAM3) Read 1 Read 2 Flower (SAM4) Read 1 Read 2 Reads (FASTQ) TOPHAT (Read Mapping) GTF + Genome CUFFLINKS (Transcript Assembly) GTF Alignments (BAM) Assemblies (GTF) CUFFMERGE (Final Transcript Assembly) Assembly (GTF) CUFFDIFF (Differential expression results) Visualisation (PDF) CUMMERBUND (Expression Plots) CUFFCOMPARE (Comparion to reference) GTF Step 4 Tuxedo Protocol - CUFFLINKS Leaf (SAM1) Read 1 Read 2 Leaf (SAM2) Read 1 Read 2 Flower (SAM3) Read 1 Read 2 Flower (SAM4) Read 1 Read 2 Reads (FASTQ) TOPHAT (Read Mapping) GTF + Genome CUFFLINKS (Transcript Assembly) GTF Alignments (BAM) Assemblies (GTF) • Accurate quantification of a gene requires identifying which isoform produced each read. • Reference Annotation Based Transcript (RABT) assembly • Sequence bias correction -b/--frag-bias-correct <genome.fa> • multi-mapped read correction is enabled (-u/--multi-read-correct) Tuxedo Protocol Leaf (SAM1) Read 1 Read 2 Leaf (SAM2) Read 1 Read 2 Flower (SAM3) Read 1 Read 2 Flower (SAM4) Read 1 Read 2 Reads (FASTQ) TOPHAT (Read Mapping) GTF + Genome CUFFLINKS (Transcript Assembly) GTF Alignments (BAM) Assemblies (GTF) CUFFMERGE (Final Transcript Assembly) Assembly (GTF) CUFFDIFF (Differential expression results) Visualisation (PDF) CUMMERBUND (Expression Plots) CUFFCOMPARE (Comparion to reference) GTF Tuxedo Protocol - CUFFDIFF Leaf (SAM1) Read 1 Read 2 Leaf (SAM2) Read 1 Read 2 Flower (SAM3) Read 1 Read 2 Flower (SAM4) Read 1 Read 2 Reads (FASTQ) TOPHAT (Read Mapping) Alignments (BAM) CUFFLINKS (Transcript Assembly) CUFFMERGE (Final Transcript Assembly) Assembly (GTF) GTF mask file CUFFDIFF (Differential expression results) CUFFDIFF output – FPKM (fragments per kilobase of transcript per million fragments mapped) values, fold change, test statistic, p-value, significance statement. GTF + Genome CUFFDIFF - Summarisation 1 (A) 1 (B) (C) 2 1 1. 2. 3. 4. 3 4 2 4 2 4 A + B + C Grouped at Gene level B + C Grouped at CDS level A + C Grouped at Primary transcript level A, B, C No group at the transcript level • Cuffdiff output (11 files) – FPKM tracking files (Transcript, Gene, CDS, Primary transcript) – Differential expression tests (Transcript, Gene, CDS, Primary transcript) Look at – Differential splicing tests – splicing.diff difference in distribution – Differential coding output – cds.diff (rather than total – Differential promoter use – promoters.diff level) A test case – Ricinus Communis (Castor bean) • 5 tissues – Aim : identify differences in lipid-metabolic pathways A test case – Ricinus Communis (Castor bean) • Cufflinks – Cuffcompare Results – RNA-Seq reads assembled into 75090 transcripts corresponding to 29759 ‘genes’ – Compares to the 31221 genes in version 0.1 of the JCVI assembly – 35587 share at least one splice junction (possible novel splice variant). – 2847 were located intergenic to the JCVI annotation and hence may represent novel genes – 218147 splice junctions were identified, 112337 supported by at least 10 reads, >300,000 distinct to the JCVI annotation Visualisation • Bam files can be converted to wiggle plots • CummeRbund for visualisation of Cuffdiff output Bam, Wiggle and GTF files viewed in IGV CummeRbund volcano and scatter plots Thanks • David Swarbreck (Genome Analysis Team Leader, TGAC) • Mario Caccamo (Head Bioinformatics Division, TGAC)