Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Jahn–Teller effect wikipedia , lookup
Metalloprotein wikipedia , lookup
Ring-closing metathesis wikipedia , lookup
Metal carbonyl wikipedia , lookup
Spin crossover wikipedia , lookup
Hydroformylation wikipedia , lookup
Coordination complex wikipedia , lookup
Stability constants of complexes wikipedia , lookup
Evolution of metal ions in biological systems wikipedia , lookup
STUDIES ON TRANSITION METAL NITROSYL CHEMISTRY
by
JOHN T. MALITO
B.Sc. (Hons.) U n i v e r s i t y of B r i t i s h Columbia,
1972.
A THESIS SUBMITTED IN PARTIAL FULFILMENT OF
THE REQUIREMENTS FOR THE DEGREE OF
DOCTOR OF PHILOSOPHY
in the Department
of
Chemistry
We accept t h i s t h e s i s as conforming to
required standard
THE UNIVERSITY OF BRITISH COLUMBIA
August, 1976
(3
John T. M a l i t o , 1976
the
In
presenting
this
an a d v a n c e d d e g r e e
the
I
Library
further
for
agree
in p a r t i a l
fulfilment
of
at
University
of
Columbia,
the
make
it
freely
that permission for
this
representatives.
thesis
for
It
financial
gain
of
The U n i v e r s i t y
of
British
2075 Wesbrook Place
Vancouver, Canada
V6T 1W5
6
for
extensive
by
the
Columbia
shall
not
the
requirements
reference
copying of
Head o f
is understood that
written permission.
Department
British
available
s c h o l a r l y p u r p o s e s may be g r a n t e d
by h i s
of
shall
thesis
I agree
and
be a l l o w e d
that
study.
this
thesis
my D e p a r t m e n t
copying or
for
or
publication
without
my
- ii
-
ABSTRACT
The reactions of . n i t r o s y l c h l o r i d e with a v a r i e t y of anionic
and neutral metal carbonyl and n -cyclopentadienylmetal
5
compounds of C r , Mo, W, Mn, Re, Fe and Co are d e s c r i b e d .
carbonyl
In most cases
n i t r o s y l - c o n t a i n i n g complexes are formed in reasonable y i e l d s .
The
advantages and l i m i t a t i o n s of n i t r o s y l c h l o r i d e as a general s y n t h e t i c
reagent f o r the preparation of t r a n s i t i o n metal n i t r o s y l complexes
are discussed.
The high y i e l d syntheses of CpM(C0) (N0) (M = C r , Mo or W)
2
from NaCCpMCCO)^] and Diazald are d e t a i l e d .
Subsequent r e a c t i o n of
the d i c a r b o n y l n i t r o s y l species with n i t r o s y l c h l o r i d e affords the
corresponding CpM(N0) Cl complexes in e x c e l l e n t y i e l d s .
2
The CpM(N0) R
2
(M = Cr or Mo; R = Me, E t , i-Bu or Ph: M = W; R = Me or-Ph) complexes
are obtained in the reactions of CpM(N0) Cl with a l k y l - or arylaluminum
2
reagents.
Some f u r t h e r d e r i v a t i v e s of the CpM(N0) Cl complexes and
2
the high y i e l d preparation of [ C p C r ( N 0 ) ]
2
2
are also d e s c r i b e d .
Previously unreported compounds are c h a r a c t e r i z e d by i n f r a r e d and
nuclear magnetic resonance spectroscopy and by mass spectrometry.
ACKNOWLEDGEMENT
I wish to thank the technical s t a f f and f a c u l t y of t h i s
department f o r t h e i r assistance and support.
In p a r t i c u l a r , I am
indebted to Dr. B.R. James, Dr. R. Stewart and Dr. A. Storr who read
t h i s t h e s i s and made suggestions f o r improvement.
A l s o , the aid and
thoughtful d i s c u s s i o n offered by Dr. A . E . Crease and Mr. B.W.S.
Kolthammer are g r e a t l y appreciated.
I also thank Miss L. Hon who
typed t h i s t h e s i s .
I extend my gratitude to Dr. P. Legzdins, whose unshakable
optimism, s p i r i t e d humor and companionship provided both encouragement
and guidance.
- iv -
TABLE OF CONTENTS
Page
ABSTRACT
i i
ACKNOWLEDGEMENTS
iii
TABLE OF CONTENTS
iv
LIST OF TABLES
vi i i
LIST OF FIGURES
ix
ABBREVIATIONS AND COMMON NAMES
x
CHAPTER I,
1
CHAPTER I I ,
GENERAL INTRODUCTION
THE REACTION OF NITROSYL CHLORIDE WITH SOME
TRANSITION METAL CARBONYL ANIONS
4
2.1
INTRODUCTION
4
2.2
EXPERIMENTAL
5
2.2a
Preparation of n i t r o s y l c h l o r i d e
6
2.2b
Preparation of bis(triphenylphosphine)iminium
c h l o r i d e , bromide and i o d i d e , (PPh ) NX (X = C l ,
Br or I)
Preparation of bis(triphenylphosphine)iminium
pentacarbonylhalotungstates, (PPh-) NW(CO)r-X
(X = C l , Br or I)
f.t
11
Reactions of n i t r o s y l c h l o r i d e with some anionic
and neutral complexex of manganese, rhenium and
iron
13
Reactions of n i t r o s y l c h l o r i d e with t r i c a r b o n y l (n -cyclopentadienyl) anions of chromium,
molybdenum and tungsten, [CpM(C0)o]~ (M = C r ,
Mo or W)
15
Reaction of sodium n i t r i t e and a c e t i c a c i d with
sodium t r i c a r b o n y l ( n - c y c l o p e n t a d i e n y l ) t u n g s t a t e ,
Na[CpW(C0) ]
15
3
2.2c
2
9
2.2d
2.2e
8
5
2.2f
5
3
- v Page
2.2g
Reactions of n i t r o s y l c h l o r i d e with b i s ( t r i p h e n y l phosphine)iminium pentacarbonylhalotungstates,
(PPh3) NW(C0) X (X = C l , Br or I ) , and with t e t r a ethylammonium pentacarbonylchloro(or methyl)
tungstate, Et NW(C0) X (X = Cl or Me)
2
5
4
2.2h
Reactions of
5
16
tetracarbonylhalonitrosyltungstens,
W(C0) (N0)X (X = Cl or B r ) , w i t h tetrahydrofuran
4
...
16
2.3
RESULTS AND DISCUSSION
18
2.3a
Bis(triphenylphosphine)iminium c h l o r i d e , bromide
or i o d i d e , (PPh ) NX (X = C l , Br or I)
Bis(triphenylphosphine)irninium pentacarbonylhalotungstates, (PPh ) NW(C0) X (X = C l , Br or I)
18
20
2.3c
Reactions of n i t r o s y l c h l o r i d e with metal carbonyl
anions
22
2.3d
Reactions of t e t r a c a r b o n y l h a l o n i t r o s y l t u n g s t e n s ,
W(C0) (N0)X (X = Cl or B r ) , with t e t r a h y d r o f u r a n . .
30
3
2.3b
2
3
2
5
4
CHAPTER I I I ,
REACTIONS OF NITROSYL CHLORIDE WITH NEUTRAL METAL
CARBONYL COMPLEXES
-
32
3.1
INTRODUCTION
32
3.2
EXPERIMENTAL
35
Reactions of n i t r o s y l c h l o r i d e with neutral
complexes of iron and cobalt
35
3.3
RESULTS AND DISCUSSION
37
3.3a
Reactions of n i t r o s y l c h l o r i d e with pentacarbonyli r o n , Fe(C0)
Reaction of n i t r o s y l c h l o r i d e with b i s [ d i c a r b o n y l (n -cyclopentadienyl)iron], [CpFe(C0) ]
Reactions of n i t r o s y l c h l o r i d e with t r i c a r b o n y l n i t r o s y l c o b a l t , Co(C0)3(N0), and d i c a r b o n y l ( n c y c l o p e n t a d i e n y l ) c o b a l t , CpCo(C0)
3.3b
5
5
3.3c
2
2
37
39.
5
9
40
- vi -
Page
CHAPTER IV,
CYCLOPENTADIENYLNITROSYL COMPLEXES OF CHROMIUM,
MOLYBDENUM AND TUNGSTEN
42
4.1
INTRODUCTION
42
4.2
4.2a
EXPERIMENTAL
Preparation of d i c a r b o n y l ( n - c y c l o p e n t a d i e n y l ) n i t r o s y l complexes of chromium, molybdenum and
tungsten, CpM(CO) (NO) (M.= C r , Mo or W)
46
Preparation of c h l o r o ( n - c y c l o p e n t a d i e n y l ) d i n i t r o s y l
complexes of chromium, molybdenum and tungsten,
CpM(N0) Cl (M = C r , Mo or w)
48
Preparation of ( n - c y c l o p e n t a d i e n y l ) i o d o d i n i t r o s y l
complexes of chromium, molybdenum and tungsten,
CpM(N0) I (M = C r , Mo or W)
50
Preparation of b i s [ ( n - c y c l o p e n t a d i e n y l ) e t h o x o nitrosylchromium], [CpCr(NO)(0Et)]
50
Preparation of b i s [ c h l o r o ( n - c y c l o p e n t a d i e n y l ) nitrosylchromium], [CpCr(N0)Cl]
51
Preparation of a l k y l - and a r y l - ( n - c y c l o p e n t a d i e n y l ) d i n i t r o s y l complexes of chromium, molybdenum and
tungsten, CpM(N0) R (M = C r , Mo or W; R = a l k y l or
aryl)
52
Preparation of b i s [ ( n - c y c l o p e n t a d i e n y l ) d i n i t r o s y l chromium], [ C p C r ( N 0 ) ]
55
4.3
RESULTS AND DISCUSSION
58
4.3a
The d i c a r b o n y l ( n - c y c l o p e n t a d i e n y l ) n i t r o s y l complexes
of chromium, molybdenum and tungsten, CpM(C0)~(N0)
(M = C r , Mo and W)
7
58
The c h l o r o ( n - c y c l o p e n t a d i e n y l ) d i n i t r o s y l complexes of
chromium, molybdenum and tunqsten, CpM(N0) Cl (M = C r ,
Mo or W)
7
59
Bis[(n -cyclopentadienyl )ethoxonitrosylchromium,
[CpCr(NO)(0Et)]2, and b i s [ c h l o r o ( n - c y c l o p e n t a d i e n y l ) nitrosylchromium, [CpCr(N0)Cl]
66
2
4.2b
5
2
4.2c
5
2
4.2d
5
2
4.2e
5
2
4.2f
5
9
4.2g
5
2
4.3b
2
5
5
9
4.3c
46
5
5
5
9
- vii
-
Page
4.3d
The a l k y l - and a r y l - ( n - c y c l o p e n t a d i e n y l ) d i n i t r o s y l
complexes of chromium, molybdenum and tungsten,
CpM(N0) R (M = C r , Mo or W; R = a l k y l or a r y l )
70
Bis[(n -cyclopentadienyl)dinitrosylchromium,
[CpCr(N0) ]
78
CONCLUSION
83
5
2
4.3e
5
2
CHAPTER V,
2
- vi i i -
LIST OF TABLES
Table
I
Page
Elemental Analyses and Physical Properties of
(PPh ) NW(C0) X (X = C l , Br or I)
12
Reactions of N i t r o s y l Chloride with some Complexes
of Rhenium and Iron
14
3
II
III
2
5
Reactions of N i t r o s y l Chloride with (PPh ) NW(C0)rX
3
2
(X = C l , Br or I) and Et NW(C0) X (X = Cl or
4
IV
V
5
CH r...
3
Reactions of N i t r o s y l Chloride with some Neutral
Complexes of Iron and Cobalt
36
Elemental Analyses and Physical Properties of
CpM(N0) X (M = C r , Mo or W; X = Cl or I)
49
Reaction Conditions and P u r i f i c a t i o n Methods f o r
CpM(N0) R
53
2
VI
2
VII
Elemental Analyses and Physical Properties of
CpM(N0) R
56
2
VIII
IX
X
IR and H NMR Data f o r CpM(N0) R Complexes
57
]
2
High Resolution Mass Spectral Data f o r CpCr(N0) Cl
XII
XIII
63
2
Low Resolution Mass Soectral Data f o r CpM(N0) Cl
9
(M = Mo or W)
XI
17
64
7
Low Resolution Mass Spectral Data f o r [CpCr(N0)X]
2
••
69
Mass Spectra Fragmentation Data f o r CpM(N0) R
74
Low Resolution Mass Spectral Data f o r [CpCr(NO),], . .
81
2
- ix -
LIST OF FIGURES
F l
>re
Page
1.
2.
Apparatus f o r P u r i f y i n g N i t r o s y l Chloride
]
H NMR of (n -C H )Mo(N0) CH CH
5
5
5
2
2
3
7
72
-
X -
ABBREVIATIONS AND COMMON NAMES
The following abbreviations and common names, some of which
are presently used i n the s c i e n t i f i c l i t e r a t u r e , are employed throughout
this thesis.
o
Angstrom
A
A1R
t r i a l k y l ( o r aryl)aluminum
3
amount
Amt.
butyl
Bu
n-Bu 0
2
calcd.
cm
compd.
di-n-butyl
ether
cal culated
wave numbers i n r e c i p r o c a l centimeters
compound
pentahapto-cyclopentadienyl, n -CgHg
5
Cp
dec.
Diazald
diglyme
DME
Et
EtpO
EtOAc
EtOH
h
decomposes
N-methyl-N-nitroso-p-toluenesulfonami de
bis(2-methoxyethyl)ether
1,2-dimethoxyethane
ethyl
diethyl
ether
ethyl acetate
ethanol
hour(s)
Hz
Herz, cycles per second
IR
infrared
J
magnetic resonance coupling constant i n c y c l e s per second
3,4-lutidine
3,4-dimethyl pyridine
m
medium
Me
methyl
m/e
mass to charge r a t i o
min.
minute(s)
mm
m i l l i m e t e r s of mercury
- xi abbreviations and common names (cont'd)
NMR
:
nuclear magnetic resonance
4-picoline
:
4-methylpyridine
Ph
:
phenyl
PPh.j
:
triphenylphosphine
:
pyrazoyl
R
:
a l k y l or aryl
R.p
:
perfluoroal kyl
s
:
strong
Temp.
:
temperature
THF
:
tetrahydrofuran
w
:
pz
-
weak
n
3
:
trihapto-
n
5
:
pentahapto-
v
:
s t r e t c h i n g frequency
T
:
NMR chemical s h i f t i n parts per m i l l i o n
- 1 -
CHAPTER I
GENERAL INTRODUCTION
Numerous t r a n s i t i o n metal n i t r o s y l compounds are known and
the development of t h e i r chemistry, which has been the subject of
several reviews ( 1 , 2, 3 ) , has c l o s e l y p a r a l l e l e d that of the metal
carbonyl complexes.
Consequently, i t has been recognized f o r some time
that n i t r i c oxide can bond to metals in a manner quite d i f f e r e n t from
carbon monoxide.
The s t r u c t u r e s o f a number of n i t r o s y l compounds (4)
s t r i k i n g l y demonstrate the v a r i a b l e nature of the m e t a l - n i t r o s y l bond,
which has a l s o received considerable a t t e n t i o n from a t h e o r e t i c a l
of view (5).
point
In p a r t i c u l a r , two bonding modes have been observed which
are best represented by the Lewis structures I and II.
Structure I i s
characterized by a l i n e a r M-N-0 bond and an M-N bond distance which
M=N = 0
i
M-_N
n
strongly suggests the existence of m u l t i p l e bond c h a r a c t e r .
In t h i s
instance the n i t r i c oxide ligand formally behaves as a t h r e e - e l e c t r o n
donor.
This type of bonding i s by f a r the most common among the complexes
studied to date.
In structure II the n i t r o s y l group formally acts as
- 2 -
a one-electron donor and the M-N-0 bond angle i s 120°.
M-N bond i s longer than i n the l i n e a r bonding mode, i t
Although the
is
still
somewhat shorter than the expected s i n g l e M-N a-bond d i s t a n c e .
While
both of these l i m i t i n g structures have been observed, most of the
complexes have M-N-0 bond angles which range from 120° to 180°.
The use of metal n i t r o s y l complexes as. s p e c i f i c homogeneous
c a t a l y s t s has generated much recent i n t e r e s t .
For example, F e ( C 0 ) ( N 0 )
2
2
c a t a l y t i c a l l y dimerizes butadiene to 4 - v i n y l c y c l o h e x - l - e n e , and isoprene
to a mixture of 1 , 4 - d i m e t h y l - 4 - v i n y l 1-ene (6).
Interestingly,
and
1,5-dimethyl-5-vinylcyclohex-
both of these dimerizations occur even i n the
presence of other o l e f i n s , Lewis a c i d s , or Lewis bases.
the Rh(NO)(PPh^)^ complex has been found to c a t a l y t i c a l l y
Furthermore,
hydrogenate
both terminal and c y c l i c o l e f i n s under ambient conditions (7).
If
this
hydrogenation i s performed i n the absence of peroxides or 0 , accompanying
2
o l e f i n isomerization does not occur and the c a t a l y s t may be recovered
unchanged.
Metal n i t r o s y l complexes f i n d t h e i r greatest a p p l i c a t i o n i n
the f i e l d of o l e f i n d i s p r o p o r t i o n a t e
the M ( N 0 ) X L
2
2
2
(8).
(M = Mo or W; X = C l , Br or I;
Most commonly used are
L = phosphines, phosphites,
arsines and amines) complexes i n conjunction with an organoaluminum
c o c a t a l y s t such as M e A l C l
3
2
3
or A l C l .
3
Bearing i n mind the i n t e r e s t i n g bonding, s t r u c t u r a l ,
catalytic
and r e a c t i v i t y properties of metal n i t r o s y l complexes, the development
of new s y n t h e t i c routes to these compounds must surely be welcomed.
A
- 3 -
perusal of a review a r t i c l e (9) which deals e x c l u s i v e l y with the
preparation of metal n i t r o s y l compounds reveals the obvious lack of a
general preparative route.
This f a c t i n i t s e l f i s not s u r p r i s i n g
since a large v a r i e t y of metals i s encompassed.
Unfortunately, many
of the e x i s t i n g synthetic routes produce the desired product i n low
y i e l d and/or with much expenditure of e f f o r t .
The work i n t h i s t h e s i s
i s presented i n the s p i r i t of developing new synthetic techniques which,
i n a d d i t i o n to y i e l d i n g e x i s t i n g compounds more c o n v e n i e n t l y , may also
be applied to the synthesis of new metal n i t r o s y l complexes.
studies presented i n Chapter II and III
The
are exploratory i n nature.
For
i n s t a n c e , the scope of the reaction of n i t r o s y l c h l o r i d e with a v a r i e t y
of anionic and neutral organometal1ic complexes i s o u t l i n e d .
Chapter IV
describes i n d e t a i l the successful a p p l i c t i o n of t h i s and other
s y n t h e t i c methods during the preparation of a v a r i e t y of h a l o - , a l k y l and a r y l - ( n - c y c l o p e n t a d i e n y l ) n i t r o s y l
5
and tungsten.
complexes of chromium, molybdenum
- 4 -
CHAPTER II
THE REACTION OF NITROSYL CHLORIDE WITH SOME
TRANSITION METAL CARBONYL ANIONS
2.1
Introduction
The f i r s t and only reported r e a c t i o n of n i t r o s y l
chloride
with a t r a n s i t i o n metal carbonyl anion involves the synthesis of
(RB(pz) )M(C0) (N0) from [ ( R B ( p z ) ) M ( C 0 ) ] ~ (RB(pz) =
2
3
3
3
borate; R = H or p z ; M = C r , Mo or W) ( 1 0 ) .
3
tris(pyrazoyl)-
In s p i t e of the large
number of r e a d i l y obtainable metal carbonyl anions ( 1 1 ) , t h i s type of
r e a c t i o n has been completely ignored.
to which n i t r o s y l
A study to determine the extent
c h l o r i d e can be employed as a general reagent f o r
the synthesis of neutral n i t r o s y l
complexes, from the corresponding
carbonyl anions, was therefore undertaken.
The g e n e r a l i t y of
this
s y n t h e t i c approach as well as i t s l i m i t a t i o n s , are herein c l e a r l y
delineated.
nitrosyl
In many i n s t a n c e s , t h i s new route affords the desired
complexes more conveniently and in higher y i e l d s than was
previously p o s s i b l e .
Furthermore, the need f o r organometallic anions
which are soluble i n non-donor solvents has l e d , during the course of
t h i s work, to the e f f i c i e n t
(PPh ) NW(C0) X
3
2
5
and high y i e l d synthesis of (PPh ) NX and
(X = C l , Br or
3
I).
2
- 5 -
S p e c i f i c a l l y , t h i s chapter describes the reactions of
n i t r o s y l c h l o r i d e with the f o l l o w i n g s p e c i e s :
(PPh ) NMn(C0) ,
3
2
5
Na[Re(C0) ], Ph SnRe(C0) , N a [ F e ( C 0 ) ] , Na[CpFe(C0) ], CpFe(CO) SnPh ,
5
3
5
2
4
2
2
3
Na[CpM(C0) ] (M = C r , Mo or W) and A[W(C0) X] (A = ( P P h ) N ; X = C l , Br or
3
A = E t ^ N ; X = Cl or Me).
5
3
2
The chemical and physical properties of the
products in the above reactions are considered where appropriate.
2.2
Experimental
A l l chemicals used were of reagent grade or comparable
p u r i t y and were e i t h e r purchased from commercial s u p p l i e r s or prepared
according to reported procedures.
Their p u r i t y was ascertained from
elemental analyses and/or melting point determinations.
melting points were taken in c a p i l l a r i e s i n a i r or under
nitrogen using a Gallenkamp Melting Point Apparatus.
The uncorrected
prepurified
A l l solvents were
dried according to known methods (12) and thoroughly purged with
p r e p u r i f i e d nitrogen p r i o r to use.
A l l manipulations, unless otherwise
s t a t e d , were performed on the bench (using conventional techniques f o r
handling a i r s e n s i t i v e compounds (13)) or in a Vacuum Atmospheres
Corporation Dri-Lab model HE-43-2 dry box f i l l e d with p r e p u r i f i e d
nitrogen.
Infrared spectra were recorded on Perkin Elmer 457 or 71 OA
spectrophotometers and c a l i b r a t e d with the 1601 cm" absorption band
1
of a polystyrene f i l m .
Proton magnetic resonance spectra were recorded
on a Varian Associates T-60 spectrometer using tetramethylsi 1ane as an
I:
- 6 -
i n t e r n a l standard.
The l o w - r e s o l u t i o n mass spectra were taken at
70 eV on an A t l a s CH4B spectrometer and the h i g h - r e s o l u t i o n mass
spectrum was obtained on an Associated E l e c t r i c a l Industries MS902 spectrometer with the assistance of Dr. G. Eigendorf and Mr. G. Gunn.
Elemental
analyses were performed by Mr. P. Borda of t h i s Department.
Two reagents used frequently throughout t h i s work were
n i t r o s y l c h l o r i d e (C1N0) and the bis(triphenylphosphine)iminium halides
((PPhg^NX; X = C l , Br or I ) .
2.2a
Preparation of n i t r o s y l
Their syntheses are described below.
chloride.
N i t r o s y l c h l o r i d e was prepared i n the f o l l o w i n g manner, the
procedure employed being a m o d i f i c a t i o n of an e a r l i e r report
(14).
A
100 ml three-necked f l a s k was equipped with a nitrogen i n l e t , a
dropping funnel and a drying tower (2 x 20 cm) packed from top to
bottom with equal volumes of anhydrous CaClp, KCI
and NaN02-
The top
of the tower was f i t t e d with a 5 ml graduated cold trap equipped with
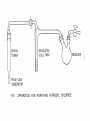
a stopcock on the trap i n l e t , as shown in Figure 1 [ClNO i s a highly
c o r r o s i v e substance which necessitates the use of apparatus constructed
e x c l u s i v e l y from g l a s s ] .
The trap o u t l e t was connected to a 100 ml
two-necked f l a s k equipped with a nitrogen i n l e t secured with a s i l i c a gel drying tube.
A f t e r f l u s h i n g the e n t i r e apparatus with n i t r o g e n ,
the reaction f l a s k was charged with concentrated aqueous HC1 (32 ml).
An aqueous s o l u t i o n (10 ml) of NaNOp (5.54 g) was added dropwise to
the r a p i d l y s t i r r e d a c i d s o l u t i o n at room temperature.
The gaseous C1N0
which formed i n s t a n t l y was c a r r i e d by a slow stream of N
7
(ca. 20 ml min
)
FIG 1
A P P A R A T U S FOR
PURIFYING
NITROSYL
CHLORIDE
- 8 -
i n t o the cold trap held at -78°C.
ClNO were generated.
In t h i s manner, 2.5 ml (50 mmol) of
This was d i s t i l l e d under vacuum i n t o the 100 ml
f l a s k and 50 ml of C H C T , THF or Et^O were added to the cold ClNO
2
2
to y i e l d deep red s o l u t i o n s which were found to be thermally
at 20°C.
stable
The chemical and physical properties of C1N0 have been
e x t e n s i v e l y described (15).
All
subsequent reactions i n v o l v i n g C1N0 were performed by the
dropwise a d d i t i o n of one of these s o l u t i o n s to the appropriate
reaction
mixture while monitoring the course of the r e a c t i o n by i n f r a r e d
spectroscopy.
2.2b
Preparation of bis(triphenylphosphine)iminium
and i o d i d e , ( P P h J J l X (X = C l , Br or
The compounds ( P P h ^ N X
I).
(X = C l , Br or I) were synthesized
according to a modification of a published procedure
2.2bl
Preparation of
c h l o r i d e , bromide
(16).
(PPhJpNCl.
A three-necked 300 ml f l a s k was equipped with a nitrogen
i n l e t , a gas i n l e t , a magnetic s t i r r e r
and a Dry-ice r e f l u x condenser
secured with a s i l i c a gel drying tube.
The f l a s k was charged with
PPh
3
(76.8 g , 293 mmol; Matheson Coleman and B e l l ) and
chloroethane (100 m l ; Fisher reagent).
1,1,2,2-tetra-
The r e s u l t a n t s o l u t i o n was
cooled to -78° i n a Dry-ice-isopropanol bath and c h l o r i n e (14.2 g ,
200 mmol; Matheson of Canada) was bubbled through the reaction
mixture
_
over a period of 10 min.
9
-
The gas i n l e t was replaced with a stopper
and the Dry-ice condenser with a water-cooled r e f l u x condenser.
NHpOH-HCl (6.9 g , 100 mmol; M a l l i n c k r o d t a n a l y t i c a l reagent grade)
was added and the mixture was allowed to warm to room temperature.
F i n a l l y , the r e a c t i o n mixture was heated under r e f l u x u n t i l the
evolution of HCl(g) ceased (about 6 h) and then was allowed to cool to
room temperature.
[Subsequent manipulations were performed i n
air.]
The orange s o l u t i o n was poured i n t o EtOAc (400 ml) whereupon
a large quantity of tan c r y s t a l s formed over a period of 18 h.
The
c r y s t a l s were c o l l e c t e d by suction f i l t r a t i o n , r e c r y s t a l 1 i z e d from
b o i l i n g water
and dried i n a vacuum d e s i c c a t o r .
d i s s o l v e d i n CH C1
2
The s o l i d was
(260 ml) and E t 0 (500 ml) was added, thereby
2
2
producing the white c r y s t a l l i n e product which was c o l l e c t e d by suction
filtration.
Final drying i n vacuo (5 x 10
49.1 g (85.5 mmol, 85% y i e l d ) of
(PPh ) NCl.
3
2
Anal. Calcd. for [ P ( C H ) ] N C 1 :
6
Found:
5
3
2
C, 75.56; H, 5.42; N, 2.22.
Proton NMR, r ( i n CDC1 ), - C ^ :
3
2.2b2
mm) at 125°C f o r 70 h afforded
C, 75.32; H, 5.27; N, 2.44.
m.p. (under N ) :
2
275.5-276.5°C.
2.33(m).
Preparation of (PPh-J^NBr.
The (PPh ) NBr was prepared in a manner comparable to that
3
2
described above f o r (PPh ) NC1.
2
The required amount of bromine (Fisher
reagent grade) was d i s s o l v e d in CHC1 CHC1
2
2
(20 ml) and added
dropwise, over a period of 1 h, to the vigorously s t i r r e d r e a c t i o n
mixture cooled in an ice-water bath.
The i n i t i a l crude product was
- 10-
obtained by pouring the cooled reaction mixture i n t o E t 0 (600 ml)
2
and was r e c r y s t a l l i z e d from b o i l i n g water (750 ml).
The product was
f i n a l l y r e c r y s t a l ! i z e d by the a d d i t i o n of EtOAc (500 ml) to a CH^Clg
s o l u t i o n (150 ml) of the s a l t .
Drying in vacuo (5 x 10
mm) f o r 70 h at
125°C produced 51.0 g (83 mmol, 83% y i e l d ) of the a n a l y t i c a l l y pure
(PPh ) NBr.
3
2
A n a l . Calcd. f o r [ P ( C H ) ] N B r :
5
5
Found:
2
C, 69.69; H, 4.92; N, 2.58.
Proton NMR, x ( i n CDC1 ), - C H :
3
2.2b3
3
g
C, 69.91; H, 4 . 8 9 ; N, 2.26.
m.p. (under N ) :
251.0-252.5°C.
2
2.52(m).
5
Preparation of (PPhJoNI.
[The f o l l o w i n g manipulations were performed in a i r . ]
To a
s o l u t i o n of (PPh ) NCl (18.8 g , 32.8 mmol) in EtOH (200 m l , "100%" not
3
2
f u r t h e r p u r i f i e d ) was added f i n e l y pulverized Nal (15.0 g , 100 mmol;
M a l l i n c k r o d t a n a l y t i c a l reagent).
The mixture was s t i r r e d r a p i d l y
1 h at room temperature and then taken to dryness i_n_ vacuo.
product was extracted into a t o t a l of 200 ml of CH C1
2
2
for
The
and the e x t r a c t
was reduced to ca_. 100 ml in vacuo.
The addition of E t 0 (300 ml) produced the white
2
crystalline
product which was c o l l e c t e d by suction f i l t r a t i o n and dried at 125°C
(5 x 10
mm) f o r 70 h.
This procedure produced 20.6 g (31.0 mmol,
95% y i e l d ) of a n a l y t i c a l l y pure
(
p p h
3
)
N I
2
A n a l . Calcd. f o r [ P ( C g H ) 3 N I :
5
Found:
C, 65.09; H, 4.67; N, 2.17.
3
2
g
5
C, 64.97; H, 4 . 5 3 ; N, 2.10.
m.p. (under N )
Proton NMR, i-(in CDC1 ), - C H : 2.50(m).
3
•
2
251.0-252.3°C.
_ n_
The (PPh ) NX (X = C l , Br or I) compounds
3
2
which are soluble in C H C 1 , CHC^CHC^, EtOH
2
2
i n s o l u b l e in EtOAc, E t 0 , THF, benzene
2
are white s o l i d s
and b o i l i n g water, but
and hexanes.
Although stable
i n dry a i r i n d e f i n i t e l y , these compounds are hygroscopic and are
therefore best stored in a d e s i c c a t o r .
2.2c
Preparation of bis(triphenylphosphine)iminium pentacarbonylhalotungstates, (PPh ) NW(C0) X (X = C l , Br or
3
2
I).
5
These complexes were conveniently prepared by the reaction
of W(C0) with the appropriate (PPh ) NX s a l t .
6
3
2
The experimental
procedure, using the iodide complex as a representative example, was
as f o l l o w s .
A DME (90 ml) s o l u t i o n containing W(C0) (7.39 g, 21.0 mmol;
6
Pressure Chemical Co.) and (PPh ) NI (13.3 g , 20.0 mmol) was heated under
3
r e f l u x for 6 h.
2
The r e s u l t a n t c l e a r yellow s o l u t i o n was allowed to
cool to room temperature and the volume reduced in vacuo u n t i l the f i r s t
traces of c r y s t a l s appeared.
Slow a d d i t i o n of hexanes (120 ml) produced
yellow c r y s t a l s which were c o l l e c t e d by suction f i l t r a t i o n and washed
with hexanes (2 x 40 ml).
f o r 18 h.
The product was d r i e d at 60°C (5 x 10 mm)
A q u a n t i t a t i v e y i e l d of a n a l y t i c a l l y pure (PPh ) NW(C0) I
3
was thus obtained (Table
2
5
I).
The (PPh ) NW(C0) X (X = Cl or Br) complexes were s i m i l a r l y
3
2
5
obtained in greater than 95% y i e l d s and t h e i r elemental analyses and
physical properties are compiled in Table
I.
Table I.
Compound
Elemental Analyses and Physical Properties of (PPh ) NW(C0) X (X = C l , Br or
3
Color
Analyses, %
Mp. ,°C
(under N )
C
H
I
Calcd:
54.84
3.37
1.56
Found:
54.44
3.51
1.38
Calcd:
52.25
3.21
1.49
Found:
52.04
3.31
1.33
2
(PPh ) NW(C0) Cl
3
2
5
(PPh ) NW(C0) Br
3
2
5
Yellow
Yellow
145(dec)
150(dec)
2
5
Proton NMR,
T(in CDClJ
" 6 5
C
H
2.52(m)
I).
-1
v ( C 0 ) , cm
(in CH C1 )
2
2
2065(w), 1915(s),
1837(m)
2.50(m)
2065(w), 1915(s),
1842(m)
ro
(PPh ) NW(C0) I
3
2
5
Yellow
178(dec)
Calcd:
49.77
3.06
1.42
Found:
49.74
3.20
1.22
2.51(m)
2063(w), 1918(s),
1850(m)
i
- 13 -
A l l three complexes are yellow s o l i d s which may be exposed to
a i r for several days without noticeable decomposition but are best
stored under n i t r o g e n .
They are very s o l u b l e in C r ^ C ^ , diglyme
DME, less soluble in Et^O, THF and EtOAc
and
and i n s o l u b l e in hexanes.
The s o l u b i l i t y increases markedly with i n c r e a s i n g atomic weight of the
halogen.
2.2d
Reactions of n i t r o s y l c h l o r i d e with some anionic and neutral
complexes of manganese, rhenium and i r o n .
Reaction with
bi s (triphenylphosphine)imini um pentacarbonylmanganate, (PPh,,),,NMn (CO)
To a l i g h t yellow s o l u t i o n of (PPh ) NMn(C0) (17) (0.81 g ,
3
1.1 mmol) in CH C1
2
2
2
5
(30 ml) was added dropwise with s t i r r i n g at room
temperature a s o l u t i o n of ClNO i n C H C 1 .
2
2
Immediately, the reaction
s o l u t i o n became deep red and gas was evolved.
A f t e r the a d d i t i o n of ClNO
was complete, the s o l u t i o n was s t i r r e d f o r an a d d i t i o n a l 10 min.
D i s t i l l a t i o n in vacuo y i e l d e d a CH^Cl^ s o l u t i o n containing only
Mn(C0) (N0) which was i d e n t i f i e d by i t s i n f r a r e d spectrum (18).
4
The y i e l d
was estimated to be 70% from the r e l a t i v e i n t e n s i t i e s of the carbonyl
absorptions i n the reactant and product i n f r a r e d s p e c t r a .
The reactions of C1N0 with some organometallic complexes of
rhenium and iron were performed s i m i l a r l y and the experimental procedures
are summarized in Table
II.
Table II.
T r a n s i t i o n metal compd.
(mmol)
Na[Re(C0) ] (4.0)
19
I;
Reaction of N i t r o s y l Chloride with some Complexes of Rhenium and Iron.
Amt. of
C1N0
(mmol)
4.0
Solvent
(ml)
THF(50)
Temp, and
reaction time
0 ° , 15 min.
Products(yields)
Re (C0)
2
I s o l a t i o n and
Identification
Sublimation at 40-60°
1Q
infrared spectrum
Ph SnRe(C0)5°(1.5)
3
2.0
CH C1 (10)
2
2
20°, 15 min.
Re(C0) SnCl Ph _
5
x
3
x
(x = 0,1,2,3)
Na [Fe(C0) ] ' (10)
20
Et 0(150)
,24
Na[CpFe(C0) ]"(14)
14
THF(50)
2 1
2
2 2
4
2
9
- 7 8 ° , 30 min.
0°,. 20 min.
Sublimation at 60°
infrared spectrum*
[CpFe(C0) ] M0%)
Identification
2
2
(5xl0 mm):
2
3
3.0
CrLCl (15)
?
20°, 15 min.
CpFe(C0) 9
x 3-x
(x = 0,1,2,3)
S n C 1
P h
THF e x t r a c t s ;
.
25
spectrum
a.
By comparison with the i n f r a r e d spectrum of the authentic compound.
b.
Inferred from the c h a r a c t e r s t i c s h i f t of the carbonyl absorptions to higher wave numbers.
infrared
in s o l u t i o n by
infrared spectroscopy
24,
CpFe(C0) SnPh (2.0).
-,j
3
Vacuum d i s t i l l a t i o n ;
23
spectrum
9
J
9
Fe(C0) (N0) (30%)
9
(5xlO" mm);
9
infrared
- 15 -
2.2e
Reactions of n i t r o s y l c h l o r i d e with t r i c a r b o n y l ( n - c y c l o p e n t a 5
d i e n y l ) anions of chromium, molybdenum and tungsten, [CpM(C0) ]~
3
(M = C r , Mo or W).
A THF s o l u t i o n of C1N0 was added dropwise, with r a p i d
stirring
at room temperature to Na[CpCr(C0) ] (27) (4.89 g , 22.0 mmol) in THF
3
(120 m l ) .
Gas was evolved and a dark s o l i d p r e c i p i t a t e d during the
course of the r e a c t i o n .
The a d d i t i o n of C1N0 was continued u n t i l the
s t a r t i n g anion was completely consumed.
The i n f r a r e d spectrum of the
f i n a l reaction mixture revealed the presence of CpCr(C0) (N0) (1) and
2
[CpCr(C0) ]
3
2
(28) as the major carbonyl-containing compounds.
The
mixture was taken to dryness i n vacuo and sublimation of the residue
at 60° (5 x 10
mm) onto a water-cooled probe produced 1.01 g (5.0 mmol,
23% y i e l d ) of CpCr(C0) (N0) which was. i d e n t i f i e d by i t s
2
spectrum.
infrared
The reactions of the molybdenum and tungsten containing
anions proceeded s i m i l a r l y .
If an excess of C1N0 was employed i n the
above syntheses, s i g n i f i c a n t amounts of CpM(N0) Cl and attendant
2
decomposition products were formed.
The physical and chemical properties
of the CpM(C0) (N0) complexes have been previously described (28).
2
2.2f
Reaction of sodium n i t r i t e and a c e t i c a c i d with sodium t r i c a r b o n y l ( n - c y c ! o p e n t a d i e n y l ) t u n g s t a t e , Na[CpW(C0) ].
5
3
To a s o l u t i o n of Na[CpW(C0) ] (27) (1.80 g , 5.1 mmol) and
3
NaN0
2
(0.35 g , 5.1 mmol) i n nitrogen-saturated water (30 ml) was added
dropwise with s t i r r i n g an aqueous s o l u t i o n (10 ml) of a c e t i c a c i d (5 mmol).
Immediately, gas was evolved and a dark yellow s o l i d formed.
The reaction
- 16 -
mixture was f u r t h e r s t i r r e d for 0.5 h and then f i l t e r e d .
s o l i d was d r i e d in vacuo and sublimed at 60°C (5 x 10
The r e s u l t a n t
mm) onto a
water-cooled probe producing 1.01 g of a mixture of CpW(C0) (N0) and
2
CpW(C0) H which were i d e n t i f i e d by t h e i r i n f r a r e d spectra (1, 27, 29).
3
2.2g
Reactions of n i t r o s y l c h l o r i d e with
bis(triphenylphosphine)iminium
pentacarbonylhalotungstates, (PPh ) NW(C0) X (X = C l , Br or
3
2
5
I),
and with tetraethylammonium pentacarbonylchloro (or methyl)
tungstate, Et NW(C0) X (X = Cl or Me).
4
5
A C h ^ C ^ s o l u t i o n of C1N0 was added dropwise with s t i r r i n g
(PPh ) NW(C0) Br (5.5 g , 5.8 mmol) in CH C1
3
2
5
2
2
(50 ml) at -78°C.
to
Gas was
evolved and the a d d i t i o n of C1N0 was continued u n t i l the carbonyl
i n f r a r e d absorptions due to the s t a r t i n g material disappeared.
mixture was taken to dryness in vacuo.
45°C (5 x 10
The
Sublimation of the residue at
mm) onto a water-cooled probe produced 0.82 g (2.0 mmol,
35% y i e l d ) of W(C0) (N0)Br which was i d e n t i f i e d by i t s
4
infrared
spectrum (30).
The reactions of ClNO with the remaining tungsten complexes
were performed s i m i l a r l y and the experimental d e t a i l s are presented i n
Table
2.2h
III.
Reactions of t e t r a c a r b o n y l h a l o n i t r o s y l t u n g s t e n s , W(C0) (N0)X
4
(X = Cl or Br), with t e t r a h y d r o f u r a n .
A THF s o l u t i o n (20 ml) containing W(C0) (N0)C1 (0.50 g,
4
1.4 mmol) was s t i r r e d at room temperature u n t i l the i n f r a r e d absorptions
Table
III.
Reactions of N i t r o s y l
Chloride with (PPh ) NW(C0) X (X = C l , Br or
3
2
5
I)
and Et NW(C0) X (X = Cl or Me).
4
Metal Complex
(mmol)
Solvent
(ml)
Reaction
temp.
(PPh ) NW(C0) Cl(5.1)
CH C1 (50)
-78°
W(C0) (N0)C1(25%)
A
(PPh ) NW(C0) BY(5.8)
CH C1 (50)
-78°
W(C0) (N0)Br(35%)
A
(PPh ) NW(C0) I(9.O)
CH C1 (50)
-78°
W(CO) (NO)I(2535)
A
Et NW(C0) Cl
THF(75) or
-78°
W(C0) and
CH C1 (100)
25°
3
3
3
4
2
5
2
5
2
5
(7.7)
31
5
2
2
2
2
Et NW(C0) CH (3.6)
2
4
5
3
2
2
2
2
CH C1 (50)
2
2
-78° or 25°
Products(yields)
4
4
4
g
W(C0) (N0)C1 (<v8%)
W(C0) , W(C0) (N0)C1,
6
4
Sublimation at 45°C (5x10" mm), i n f r a r e d spectrum.
B
I d e n t i f i c a t i o n in s o l u t i o n by i n f r a r e d spectroscopy.
A
4
4
Et NW(C0) Cl
A
I s o l a t i o n and
Identification
5
B
i
- 18 -
due to the i n i t i a l reactant disappeared (ca. 24 h).
taken to dryness in vacuo (5 x 10
The s o l u t i o n was
mm) to give a q u a n t i t a t i v e y i e l d of
an orange complex i d e n t i f i e d as •[W(C0) (N0)(THF)C1] .
2
2
A n a l . Calcd. for W(C0) (N0)C1(C Hg0):
2
N, 3 . 7 1 ; C l , 9.39.
v(C0) cm"
1
Found:
(in THF):
4
C, 19.10; H, 2 . 1 2 ;
C, 19.20; H, 2 . 1 1 ; N, 3.56; C l , 9.64.
2020(s); 1907(s).
Proton NMR, ( i n CgDg), THF:
v(N0) cm"
(in THF):
1
1636(s).
5.60(m); 7.85(m).
T
S i m i l a r l y , W(C0) (N0)Br was q u a n t i t a t i v e l y converted to
4
[W(C0) (NO)(THF)Br]
2
as evidenced by the s i m i l a r i t y of i t s
2
spectrum with that of
v(C0) cm"
[W(C0) (N0)(THF)C1] .
2
2
(in THF):
1
infrared
2010(s); 1905(s).
v(N0) cm"
1
(in THF):
1635(s).
The [W(C0) (N0)(THF)X]
2
2
(X = C l o r B r ) complexes are y e l l o w -
orange s o l i d s which are stable in a i r f o r at l e a s t 10 h but are best
stored under n i t r o g e n .
i n s o l u b l e in hexanes.
They are soluble i n THF, CH C1
2
2
and benzene and
However, s o l u t i o n s containing these compounds
decompose r a p i d l y upon exposure to a i r .
2.3
2.3a
Results and Discussion
Bis(triphenylphosphine)iminium c h l o r i d e , bromide and i o d i d e ,
( P P h J N X (X = C l , Br or
2
I).
The (PPh ) NX (X = Cl or Br) compounds can best be prepared
3
2
in high y i e l d s according to eq. 1.
- 19 -
3PPh + 2X + NH 0H-HC1
3
2
2
° - ( P P h ^ N X + Ph PO + HC1 + 3HX
1.
3
This synthesis has been previously u t i l i z e d only in the preparation of
(PPh ) NCl (16), but we have subsequently found that the above reaction
3
2
can also be d i r e c t l y applied to the high y i e l d synthesis of
(PPh^NBr.
This method, however, f a i l s f o r the preparation of ( P P h ) N I .
3
2
The
l a t t e r compound i s conveniently obtained q u a n t i t a t i v e l y by the
d i s p r o p o r t i o n a t i o n reaction between (PPh ) NCl and Nal in ethanol
3
2
according to the equation
(PPh ) NCl + Na + I"
3^- (PPh ) NI + NaCl(s)
+
3
2
3
2.
2
i n which the e q u i l i b r i u m l i e s well to the side of ( P P h ^ N I by v i r t u e
of the low s o l u b i l i t y of NaCl in ethanol.
The syntheses of
(PPh ) NX
3
2
(X = Br or I) have been previously reported although a cumbersome and
i n e f f i c i e n t method was used (16).
The (PPh ) NX compounds are stable i n d e f i n i t e l y in a i r and
3
water.
2
However, contrary to a previous report (16), we f i n d them to be
quite hygroscopic as shown by elemental analyses and proton NMR
spectroscopic i n v e s t i g a t i o n s .
i s detrimental
Consequently, i f the presence of water
in subsequent reactions i n v o l v i n g these s a l t s , they must
be stored in a d e s i c c a t o r .
The compounds are thermally s t a b l e even at
t h e i r r e l a t i v e l y high melting points which are in excess of 250°C.
chemical and physical properties are not u n l i k e those of
Their
tetraethyl-
ammonium s a l t s except t h a t , s u r p r i s i n g l y , the (PPh ) NX compounds are
3
very soluble in c h l o r i n a t e d s o l v e n t s .
2
The geometric and e l e c t r o n i c
- 20 -
structures of the [ ( P P h ) N ]
3
T
2
cation have been the subjects of
considerable study and both a bent and l i n e a r arrangement of P-N-P
atoms have been found (33, 34).
The use of [ ( P P h ) N ]
3
2
+
as a counterion in the i s o l a t i o n of
t r a n s i t i o n metal carbonyl anions i s of immense value.
Not only are
c r y s t a l l i n e samples of the carbonyl anions r e a d i l y o b t a i n a b l e , but
these s a l t s are often a i r stable in the s o l i d state f o r many hours.
A l s o , unlike the a l k a l i metal s a l t s of carbonyl anions, the [ ( P P h ) N ]
3
d e r i v a t i v e s are very soluble and stable i n c h l o r i n a t e d s o l v e n t s .
+
2
In
f a c t , t h i s enhanced s t a b i l i t y has been highly b e n e f i c i a l i n the x-ray
s t r u c t u r a l determinations of many i n t e r e s t i n g mono- and p o l y - n u c l e a r
metal carbonyl complexes (33).
2.3b
Bis(triphenylphosphine)iminium pentacarbonylhalotungstates,
(PPh ) NW(C0) X (X = C l , Br or
3
2
5
I).
The (PPh ) NX (X = C l , Br or I) s a l t s react with W(C0)
3
2
r e f l u x i n g DME with the evolution of gas to give v i r t u a l l y
g
in
quantitative
y i e l d s of the (PPh ) NW(C0) X complexes, as i n eq. 3.
3
2
(PPh ) NX + W(C0)
3
2
g
6
(PPh ) NW(C0) X + CO
3
2
5
3.
These syntheses are comparable to those reported f o r the preparation of
the Et NW(C0) X complexes i n diglyme at 120°C (31).
4
5
However, the reactions
reported here are performed with great f a c i l i t y at 85°C and are complete
in s i x hours.
The lower reaction temperature and the shorter reaction
_ 21 _
time serve to eliminate any accompanying decomposition products which
may a r i s e from the p y r o l y s i s of the desired m a t e r i a l s .
Unlike the Et NW(C0) X compounds, the [ ( P P h ) N ]
4
5
3
+
2
derivatives
are not p h o t o l y t i c a l l y decomposed by ordinary l i g h t i n g , and they are
also more stable toward atmospheric o x i d a t i o n .
In f a c t , as s o l i d s
they are stable in a i r f o r several days and t h e i r s o l u t i o n s may be
exposed to a i r f o r a short period of time without noticeable
decomposition.
In c o n t r a s t , the [Et^N]" " containing compounds are so
1
unstable i n s o l u t i o n that the a c q u i s i t i o n of c o n s i s t e n t l y accurate
i n f r a r e d data i s d i f f i c u l t .
The s o l u t i o n i n f r a r e d spectra of the (PPh ) NW(C0) X
3
2
5
complexes d i s p l a y the expected three-band pattern s i m i l a r to the i s o structural
neutral compounds Mn(C0)gX (X = C l , Br or I)
W(C0)^L (L = amine or phosphine) (36)-.
(35) and
Of course, the C-0 s t r e t c h i n g
frequencies of the i o n i c species occur at much lower frequencies due to
the enhanced metal-carbonyl backbonding.
Because of t h e i r greater s o l u b i l i t y in common organic
s o l v e n t s , the [ ( P P h ) N ]
3
+
2
s a l t s are more useful than the [Et^N]" " analogs
1
as r e a c t i v e synthetic precursors (17, 37-40).
In p a r t i c u l a r , they can
be r e a d i l y converted to the W(C0) (N0)X (X = C l , Br or I) complexes as
4
described subsequently.
In summary i t should be noted that a wide
v a r i e t y of (PPh ) NM(C0) X complexes, where X i s a halogen or pseudo3
2
5
halogen, have been previously prepared by diverse means (37-41).
However, the preparative methods described in t h i s t h e s i s are f a r
superior simply because the desired products are obtained much more
- 22 -
conveniently and i n q u a n t i t a t i v e
2.3c
Reactions of n i t r o s y l
It
yields.
c h l o r i d e with metal carbonyl anions.
is a well known f a c t that t r a n s i t i o n metal carbonyl anions
are s u f f i c i e n t l y n u c l e o p h i l i c to d i s p l a c e a halide from both organic
and inorganic halides according to the general reaction scheme
Cp M(C0) R + X
[Cp M(C0) ]" + RX
m
m
n
4.
n
where m = 0 or 1; n = 1-5 depending on M (11).
The success of the above
reaction i s dependent, among other v a r i a b l e s , on the n u c l e o p h i 1 i c i t y of
the a n i o n , and the rate of such a reaction has been used as a measure
of the n u c l e o p h i l i c strength of some metal carbonyl anions
(42).
In a conversion analogous to the above reaction we f i n d that
some of these anions w i l l also d i s p l a c e the c h l o r i d e from n i t r o s y l
c h l o r i d e thereby providing a convenient synthetic route to neutral
nitrosyl
compounds.
The method can a-lso be applied to the d i - a n i o n i c
complex,[Fe(C0) ] ".
Except f o r the preparation of
2
4
(RB(pz) = t r i s ( p y r a z o y l ) b o r a t e ;
3
[(RB(pz) )M(C0) ]"
3
ignored.
(RB(pz) )M(C0) (N0)
3
2
R = H or p z ; M = C r , Mo or W) from
(10), t h i s c l a s s of reactions has l a r g e l y been
3
Typical examples of such reactions are summarized in eq. 5-8.
(PPh ) NMn(C0) + C1N0
3
2
5
N a [ F e ( C 0 ) ] + 2C1N0
2
3
(M = C r , Mo or W)
4
Fe(C0) (N0)
4
Na[CpM(C0) ] +C1N0 •
Mn(C0) (N0)
2
THF
* ~ CpM(C0) (N0)
2
2
5.
6.
7.
- 23 -
THF or CH C1
9
A[W(C0) X] + C1N0
L
5
(A = ( P P h ) N ;
3
2
L
9
*~ W(C0) (N0)X
8.
4
X = C l , Br or I:
A = Et^N;
X = Cl or Me)
A l l these conversions are accompanied by gas evolution and they
proceed r e a d i l y i n reasonable y i e l d s .
The attendant formation of an
inorganic c h l o r i d e undoubtedly provides a strong thermodynamic
driving
force f o r these r e a c t i o n s .
It appears that n i t r o s y l
c h l o r i d e behaves as a nitrosonium
s a l t in i t s reactions with anions even though the molecule i t s e l f
bent and the N-Cl bond i s l a r g e l y covalent.
is
For i n s t a n c e , microwave
spectroscopy of i s o t o p i c a l l y l a b e l l e d ClNO molecules shows the 0-N-C1
angle to be 113.3° (43), and an N-0 bond order of 1.9 i s obtained from
force constant c a l c u l a t i o n s u t i l i z i n g i n f r a r e d data (15).
Furthermore,
the N-0 s t r e t c h i n g frequency of C1N0 i n a dichloromethane s o l u t i o n occurs
at 1845 cm" while i n nitrosonium s a l t s , which contain the t r i p l y bonded
1
nitrosonium c a t i o n , the values f a l l w i t h i n the 2150-2400 c m
These observations therefore suggest that the n i t r o s y l
?
i s mainly covalent containing an sp
double bond.
-1
range (44).
c h l o r i d e molecule
hybridized nitrogen atom and an N-0
Nevertheless, the anomalously large N-Cl distance of
O
1.975 ± 0.005 A (43) has been explained in terms of the
of i o n i c resonance forms such as
0=
[:O=N:] CI
+
contribution
- 24 -
In the gaseous s t a t e , at 20°C, n i t r o s y l
NO and C l
2
to the extent of about 0.5% (44).
c h l o r i d e disproportionates
Although the extent of
t h i s e q u i l i b r i u m in s o l u t i o n s of dichloromethane or tetrahydrofuran
not known, evidence f o r i t i s presented i n Chapter IV.
is
Chemically,
n i t r o s y l c h l o r i d e often behaves as a strong o x i d i z i n g agent, being
reduced to C l " and NO or N
2.3cl
(15, 45, 46).
2
Reactions of n i t r o s y l c h l o r i d e with organometallic anions of
manganese, rhenium
and i r o n .
The reaction of n i t r o s y l c h l o r i d e with (PPh^^NMn(C0)
g
in
dichloromethane proceeds smoothly and c l e a n l y at room temperature to give
Mn(C0) (N0) i n good y i e l d s .
However, the analogous reaction
4
involving
Na[Re(C0) ] affords R e ^ C O ) - ^ as the only carbonyl containing compound.
g
It appears that the [Re(C0) ]~ anion i s o x i d i z e d to the dimer by
g
n i t r o s y l c h l o r i d e according to eq. 9.
Na[Re(C0) ] +.C1N0
5
yte (C0)
2
1 0
+ NaCl + NO
9.
In t h i s connection the use of n i t r o s y l c h l o r i d e as an oxidant has already
been mentioned and indeed, nitrosonium s a l t s have been used p r e v i o u s l y
i n the oxidation of organometallic anions.
An example of such an
oxidation i s the formation of the paramagnetic C r ( C 0 ) I complex as
g
indicated i n eq. 10 (45).
[ C r ( C 0 ) I ] " + NOPFg
5
C r ( C 0 ) I + P F " + NO
5
g
10.
to
- 25 -
The d i a n i o n i c N a [ F e ( C 0 ) ] complex reacts with n i t r o s y l
2
4
c h l o r i d e to form F e ( C 0 ) ( N 0 ) as the only n i t r o s y l containing species.
2
2
The p o s s i b i l i t y that t h i s reaction proceeds through the well known
[Fe(C0) (N0)]~ anion was not i n v e s t i g a t e d i n our work.
3
However, the
i n f r a r e d spectrum of the f i n a l reaction mixture confirms the presence of
small amounts of F e ( C 0 ) - | , l i k e l y produced by the n i t r o s y l
3
chloride
2
o x i d a t i o n of [ F e ( C 0 ) ]
4
.
This i s not s u r p r i s i n g i n l i g h t of the f a c t
that a s i m i l a r oxidation of [HFe(C0) ]
4
an important synthetic route to F e ( C 0 )
3
by manganese dioxide c o n s t i t u t e s
1 2
(29, 47, 48).
The r e l a t i v e n u c l e o p h i l i c i t i e s of a v a r i e t y of metal carbonyl
and cyclopentadienylmetal
carbonyl anions, i n the presence of Bu^NClO^,
have been compiled and the f a c t o r s which determine n u c l e o p h i l i c
character have been discussed (42).
The reactions of n i t r o s y l
chloride
with the anions described in t h i s chapter suggest a r e l a t i o n s h i p between
the r e a c t i o n products and the n u c l e o p h i l i c i t y of the anions.
Hence, the
m i l d l y n u c l e o p h i l i c [Mn(C0)g]~ anion r e a d i l y y i e l d s Mn(C0) (N0) while the
4
more n u c l e o p h i l i c [Re(CO)^]
Re (C0).|Q.
anion simply gives the o x i d a t i o n product,
Not s u r p r i s i n g l y t h e r e f o r e , the most n u c l e o p h i l i c anion thus
2
f a r i n v e s t i g a t e d , [CpFe(C0) ]~> i s o x i d i z e d nearly q u a n t i t a t i v e l y
2
to
[CpFe(C0) l .
2
2
In an e f f o r t to reduce the n u c l e o p h i l i c behavior of [Re(C0)g]
and-[CpFe(C0) ]
2
the Ph^Sn d e r i v a t i v e s of these anions were prepared.
G e n e r a l l y , complexes of the type Ph^SnM, where M i s a t r a n s i t i o n metal
carbonyl moiety, contain a predominantly covalent tin-metal bond.
Consequently, i t
i s not unreasonable that the t r a n s i t i o n metal carbonyl
- 26 -
group w i l l
possess a g r e a t l y reduced n u c l e o p h i l i c i t y .
However, the
subsequent reactions of n i t r o s y l c h l o r i d e with Ph SnRe(C0)
3
CpFe(C0) SnPh
2
5
and
in dichloromethane y i e l d only the mixed R e ( C 0 ) S n C l P h _
3
5
and C p F e ( C 0 ) S n C l P h _
2
x
3
x
complexes, where x = 1, 2 or 3.
the tin-carbon bond rather than the tin-metal
x
3
x
The cleavage of
bond p a r a l l e l s the reactions
of halogens and hydrogen halides with Ph SnMn(C0) , Ph SnRe(C0) , and
3
CpFe(C0) SnPh
2
5
3
5
(24, 49-51).
3
The success of the reaction of n i t r o s y l c h l o r i d e with
(PPh ) NMn(C0)
3
2
5
prompted the attempted i s o l a t i o n of the [ R e ( C 0 ) ] " and
5
[CpFe(C0) ]~ anions as t h e i r [ ( P P h ) N ]
2
3
+
2
salts.
Unfortunately,
all
e f f o r t s were thwarted by the p e r s i s t e n t formation of Re (C0)-jQ,
2
[CpFe(C0) ]
2
and attendant decomposition products.
2
Even though
[Mn(C0)g]
and HMn(C0) react with Diazald to y i e l d Mn(C0) (N0) (18), the same
5
4
reaction could not be effected with e i t h e r [Re(C0) ]~ and [CpFe(C0) ]~
5
or t h e i r respective hydrido d e r i v a t i v e s .
reason f o r t h e i r inherent i n s t a b i l i t y
While there i s no
4
Reactions of n i t r o s y l
a priori
under ambient c o n d i t i o n s , the
Re(C0) (N0) and CpFe(C0)(N0) complexes s t i l l
2.3c2
2
c h l o r i d e with
remain unknown.
tricarbonyl(n -cyclopentadienyl)
5
anions of chromium, molybdenum and tungsten, [CpM(C0) ]~
3
(M = C r , Mo or W).
Treatment of Na[CpM(C0) ] with n i t r o s y l
3
chloride in t e t r a -
hydrofuran y i e l d s a mixture of the CpM(C0) (N0), CpM(N0) Cl and [ C p M ( C 0 ) l
2
compounds.
2
Even though the anions of chromium and molybdenum are
3
slightly
2
- 27 -
less n u c l e o p h i l i c than [Mn(C0)g]~ (42), a s i g n i f i c a n t amount of oxidation
to [CpM(C0) ] occurs.
3
The formation of CpM(N0) Cl a r i s e s from the
2
2
o x i d a t i v e s u b s t i t u t i o n of n i t r o s y l c h l o r i d e on the i n i t i a l CpM(C0) (N0)
2
products according to eq. 1 1 .
CpM(C0) (N0) + C1N0
«~ CpM(N0) Cl + 2C0
2
11.
2
(M = C r , Mo or W)
This i s a p a r t i c u l a r example of the broad c l a s s of reactions
between n i t r o s y l c h l o r i d e and neutral metal carbonyl complexes to be
discussed i n Chapters III
and IV.
The preparation of F e ( C 0 ) ( N 0 ) by the treatment of
2
2
Na[Fe(C0) N0] or Na[Fe(C0) H] with aqueous sodium n i t r i t e and a c e t i c a c i d
3
4
has been previously reported (52, 53).
The reaction of Na[CpW(C0) ]
3
with these reagents proceeds analogously whereupon a mixture of
CpW(C0) (N0) and CpW(C0) H i s formed.
2
3
The l a t t e r complex i s undoubtedly
produced by the d i r e c t protonation of [CpW(C0) ]~ by a c e t i c a c i d , a well
3
known synthesis (27).
However, d i f f i c u l t y
in separating these two
products renders t h i s synthetic route i m p r a c t i c a l .
The very high y i e l d
syntheses of a l l three CpM(C0) (N0) complexes developed during the course
2
of t h i s work are d e t a i l e d in Chapter IV.
- 28 -
2.3c3
Reactions of n i t r o s y l
c h l o r i d e with
iminium pentacarbonylhalotunqstates,
bis(triphenylphosphine)(PPhp)QNW(C0)^X
(X = C l , Br
or I) and tetraethylammonium pentacarbonylchloro (or methyl)
tungstate, Et NW(C0) X ( X = C l o r M e ) .
4
5
A l l three (PPh ) NW(C0)gX complexes react with n i t r o s y l
3
2
c h l o r i d e i n dichloromethane at -78°C to give moderate y i e l d s of
W(C0) (N0)X according to eq. 12.
4
( P P h ) N W ( C 0 ) X + C1N0
3
2
5
If excess n i t r o s y l c h l o r i d e i s used,
*-W(C0) (N0)X + ( P P h ^ N C l + CO
12.
4
or i f the reaction i s attempted at room temperature, the y i e l d s of the
desired products are reduced markedly.
The W(C0) (N0)X complexes have been previously synthesized by
4
the action of p-toluenesulphonic acid and p e n t y l n i t r i t e ,
gensulfate acid on Et NW(C0)^X (30).
4
or nitrosonium hydro-
Our repeated attempts to obtain the
desired products i n y i e l d s which even approximate those reported met
with f a i l u r e .
Another reported route to the c h l o r i d e and iodide
containing compounds involves the treatment of HW (C0)g(NO) with n i t r o s y l
2
c h l o r i d e and iodine r e s p e c t i v e l y (54).
However, the required
HW (C0)g(N0) complex can be obtained from a two-step synthesis i n y i e l d s
2
of only 40% (55, 56).
Unlike a l l of the previous syntheses discussed above, the
present reactions of n i t r o s y l
c h l o r i d e with (PPh ) NW(C0)gX a f f o r d the
3
2
W(C0) (N0)X complexes without the attendant formation of W(C0)g.
4
Thus,
the d i f f i c u l t task of separating the p h y s i c a l l y s i m i l a r W(C0) (N0)X
4
and W(C0) compounds i s avoided.
fi
A l s o , since the (PPh,) NW(C0) X
9
R
- 29 -
complexes are r e a d i l y obtained in q u a n t i t a t i v e y i e l d , as discussed
e a r l i e r i n t h i s chapter, t h i s preparative route i s indeed a t t r a c t i v e .
I n t e r e s t i n g l y , the choice of countercation i n the reactions
between the [W(C0)gX]
s a l t s and n i t r o s y l c h l o r i d e has a profound
e f f e c t on product d i s t r i b u t i o n .
Unlike the [ ( P P h ^ N ] * s a l t s , the
Et NW(C0) X compounds r e a d i l y y i e l d W(C0)g as the major product regard4
5
less of the solvent employed.
Et NW(C0)gMe, when treated with n i t r o s y l c h l o r i d e in d i c h l o r o 4
methane, produces a mixture of W(C0) (N0)C1, W(C0) and Et NW(C0) Cl.
4
6
4
5
The l a t t e r compound i s l i k e l y formed in a manner s i m i l a r to the
reaction of hydrogen c h l o r i d e with Me NW(C0) R, where R = Me or CHpPh.
4
g
For example, treatment of Me^VKCO^CHpPh with a s l i g h t excess of
hydrogen c h l o r i d e y i e l d s toluene (94%) and Me NW(C0) Cl (32).
4
5
Once the
primary product, Et NW(C0)gCl, i s formed, subsequent reaction with
4
n i t r o s y l c h l o r i d e produces W(C0) (N0)C1 and W(C0)g as previously
4
described.
In support of t h i s reaction pathway i t was found that an
excess of n i t r o s y l c h l o r i d e simultaneously reduced the f i n a l y i e l d of
Et NW(C0) Cl and increased the y i e l d s of W(C0) (N0)C1 and W(C0) .
4
5
4
g
The
a n t i c i p a t e d product from the reaction between n i t r o s y l c h l o r i d e and
Et NW(C0) Me was the as yet unknown W(C0) (N0)Me complex.
4
5
4
This type of
conversion i s not without precedent since the anions [R M'M(C0) ]~
3
g
(M = Mo or W; M' = S i , Ge, Sn or Pb; R = Me or Ph) react with NOPFg
to give R M'M(C0) (N0) i n y i e l d s ranging from 7 to 74% ( 4 6 i ) .
3
4
Our
attempts to prepare W(C0) (N0)Me by the a c t i o n of methyl Grignard
4
reagents, methyl l i t h i u m
and trimethylaluminum on W(C0) (N0)X complexes
/l
- 30 -
f a i l e d , probably due to the great l a b i l i t y of the carbon monoxide ligand
in the halo-containing reactant.
2.3d
Reactions of t e t r a c a r b o n y l h a l o n i t r o s y l t u n g s t e n s , W(C0) (.N0)X
4
(X = Cl or B r ) , with tetrahydrofuran.
The f a c i l e s u b s t i t u t i o n of e i t h e r one or two carbonyl ligands
i n W(C0) (N0)X (X = C l , Br or I) has been amply demonstrated (57, 58).
4
For example, reaction with s t o i c h i o m e t r i c q u a n t i t i e s of e i t h e r PPh^ or
AsPhg i n r e f l u x i n g chloroform y i e l d s mer-W(C0),,(N0) (L)X, whereas excess
ligand produces c j ^ s - W t C O ^ N O H L ^ X .
If carbonyl s u b s t i t u t i o n
is
effected using bis(diphenylarsino)methane (dam) only the monodentate
mer_-W(C0) (N0)(dam)X and cis-W(C0) (NQ)(dam)pX complexes are formed.
3
2
However, when h a l f the s t o i c h i o m e t r i c amount of dam i s employed, the
product obtained i s [ W t C O ^ N O ^ ^ d a m , which i s believed to contain
both halide and dam bridges (58).
We discovered that s t i r r i n g a tetrahydrofuran s o l u t i o n of
W(C0) (N0)X (X = Cl or Br) for 24 hours r e s u l t e d i n the
4
quantitative
formation of the corresponding [W(C0) (NO.) (THF)X] complexes.
2
2
The
presence of tetrahydrofuran in these compounds was confirmed by
elemental analyses and proton NMR spectroscopy which displayed the
c h a r a c t e r i s t i c proton resonances at x5.60(m) and x7.85(m).
As expected,
these values are somewhat lower than the corresponding values of
uncomplexed tetrahydrofuran.
The e l e c t r o n donation from the THF oxygen
atom to the central metal i s expected to d e s h i e l d the protons on the
a-carbon more than those on the 3-carbon atoms.
In agreement with t h i s
- 31 -
e x p e c t a t i o n , the a-carbon proton resonances do indeed experience the
greater downfield
shift.
The [w^CO^NO) ( T H F j X ^ complexes are believed to be dimeric
with structures s i m i l a r to that proposed f o r [W^O^NCOXlgdam except
that the bridging dam i s replaced by two t e r m i n a l l y bonded tetrahydrofuran l i g a n d s , as shown by two p o s s i b l e structures A and B.
A
Consistent
B
with t h i s b e l i e f , both the tetrahydrofuran and dam containing compounds
d i s p l a y s i m i l a r i n f r a r e d spectra which are c h a r a c t e r i z e d by two
carbonyl and one n i t r o s y l
s t r e t c h i n g absorptions (58).
In compliance
with the "18-electron r u l e " , the dimers presumably are held together
by halide bridges without the aid of a metal-metal bond.
- 32 -
CHAPTER
III
REACTIONS OF NITROSYL CHLORIDE WITH NEUTRAL
METAL CARBONYL COMPLEXES
3.1
Introduction
As described i n the previous chapter the reactions of
nitrosyl
halides with metal carbonyl or n -cyc1opentadieny1metal carbonyl anions
5
had received v i r t u a l l y no a t t e n t i o n u n t i l t h i s study.
On the other
hand, t h e i r reactions with neutral complexes have been p r e v i o u s l y
i n v e s t i g a t e d , although by no means exhaustively (9).
n i t r o s y l halides are powerful
Because the
o x i d a n t s , s t r i c t a t t e n t i o n must be given
to the r e a c t i o n conditions which a r e , by and l a r g e , e m p i r i c a l l y
determined.
Thus the temperature, the s o l v e n t , the phase
and e s p e c i a l l y
the stoichiometry of the reaction are c r u c i a l i n determining the r e a c t i o n
products.
Reactive substrates include non-carbonyl as well as carbonyl
containing compounds.
In the former category, the n i t r o s y l
halides
u s u a l l y add o x i d a t i v e l y to the metal complex with or without ligand
displacement.
However, the majority of the published reports describe
the reactions of n i t r o s y l halides with t r a n s i t i o n metal complexes which
contain one or more carbonyl l i g a n d s , and only these are considered here.
33 -
At t h i s point a c l e a r d i s t i n c t i o n must be made between the
nitrosyl
halides and various nitrosonium s a l t s .
i n v a r i a b l y form c a t i o n i c n i t r o s y l
Cp M(C0) +
m
n
NOV
The l a t t e r reagents
complexes, as shown i n eq. 13,
[Cp M(C0) _ (N0)]V + CO
m
n
1
13.
(m = 0 or 1 and n = 2-6 depending on M)
since anions, A " , such as PFg" and BF^~ show a remarkable propensity to
remain uncoordinated.
Only one mole equivalent of carbon monoxide i s
displaced and the product formed i s a 1:1 e l e c t r o l y t e .
Any remaining
coordinated carbonyl ligands are u s u a l l y quite s u s c e p t i b l e to
displacement by even weakly n u c l e o p h i l i c neutral or anionic molecules
such as acetone or halide anions (59, 60).
The weakened metal-carbon
bond in c a t i o n i c carbonyl compounds i s c o n s i s t e n t with current bonding
t h e o r i e s , and the r e s u l t a n t enhanced l a b i l i t y of the CO ligand can be
advantageously e x p l o i t e d i n the synthesis of both c a t i o n i c and neutral
carbonyl compounds.
On the other hand, the n i t r o s y l
halides almost always react
with metal carbonyls to form products which contain a coordinated
halide l i g a n d .
However, two cases of c a t i o n i c products s i m i l a r to
those obtained with the nitrosonium s a l t s have been reported (61, 72a).
For i n s t a n c e , both ClNO and BrNO react with F e ( C 0 ) ( P P h )
3
3
2
in
a c e t o n i t r i l e to give [ F e ( C 0 ) ( N 0 ) ( P P h ) ] X ~ .
+
2
3
2
By f a r the most common mode of reaction i s that depicted in
- 34 -
equation 14
CpM(CO) + XNO
m
'n
r
x
^ C p M ( C 0 ) _ ( N 0 ) X + 2C0
m
n
14.
2
(m = 1 or 2 and n = 2-6 depending on M)
which involves the coordination of one mole of XNO f o r every two moles
of CO d i s p l a c e d .
Since NO i s a stronger Tr-acceptor than CO, i t
e f f e c t i v e l y competes with CO in the extent of u-backbonding with the
central m e t a l , thereby r e s u l t i n g i n a weaker metal-carbon bond.
Any remaining carbonyl ligand in the i n i t i a l
product i s now s u f f i c i e n t l y
l a b i l e to be displaced even by a c h l o r i n e atom which i s already
coordinated to a t r a n s i t i o n metal.
The r e s u l t i s the formation of
dimers, or polymers of indeterminate l e n g t h , which contain c h l o r i n e
bridges.
These general p r i n c i p l e s are exemplified i n the
following
syntheses.
Re(C0) (L)Cl + C1N0
* - Re(C0) (N0)LCl
4
2
+
2
2C0
(L = C H N , C H N0, C HgS, Bu P, P h P 0 , 4 - p i c o l i n e or
5
5
5
5
4
3
3
M(C0) + 2C1N0
[M(N0) Cl ]
6
2
2
n
(63)
3,4-1utidine)
+ 6C0
(64)
(M = Mo or W)
Mo(C0) (PPh )
4
3
2
+ 2BrN0—»-Mo(N0) Br (PPh )
2
2
3
2
LM(C0) (N Ph) + C1N0—*-LM(N0)(N Ph)Cl + 2C0
2
2
2
(M = Mo or W; L = H B ( p z ) :
3
M = Mo; L = Cp)
+ 4C0
(65)
(66)
- 35 -
RB(pz) M(C0) (N0) + C1N0
3
e»-RB(pz) M(N0) Cl + 2CO
2
3
(M = Mo; R = H or pz:
(10)
2
M = W; R = H)
HB(3,5-Me pz) Mo(C0) (N0) + C1N0 —*-HB(3,5-Me pz) Mo(N0) Cl
2
3
2
2
3
(10)
2
+ 2C0
The l a s t two of these r e a c t i o n s have a counterpart i n the
cyclopentadienyl d e r i v a t i v e s , CpM(C0) (N0) (M = C r , Mo or W), and the
2
reaction of C1N0 with these complexes has been developed i n t o a high
y i e l d synthetic route to the CpM(N0) Cl compounds, as described i n
2
Chapter IV.
In t h i s chapter the reactions of ClNO with F e ( C 0 ) ,
5
F e ( C 0 ) ( N 0 ) , [ C p F e ( C 0 ) ] , Co(C0) (N0) and CpCo(C0) are d e s c r i b e d .
2
3.2
2
2
2
3
2
Experimental
A l l experimental procedures described here were performed
under the same general conditions d e t a i l e d in s e c t i o n 2 . 2 .
Reactions of n i t r o s y l c h l o r i d e with neutral complexes of iron and c o b a l t .
To a vigorously s t i r r e d s o l u t i o n of Fe(C0)
g
(7.5 m l , 58 mmol)
in CHgCl2 (30 ml) was added at room temperature a CH C1
2
containing C1N0 (7.37 g , 112 mmol).
r e a c t i o n mixture became v i o l e t - b l a c k .
2
(50 ml)
solution
Gas was r a p i d l y evolved and the
The f i n a l mixture was taken to
dryness and the residue was extracted into hexanes (150 m l ) .
The e x t r a c t
was cooled to -78°C thereby p r e c i p i t a t i n g a dark red-brown s o l i d which
Table IV.
Reactions of N i t r o s y l Chloride with some Neutral Complexes of Iron and Cobalt.
T r a n s i t i o n metal compd.
(mmol)
Amt. of
C1N0
(mmol)
Solvent
(ml)
Temp.
Fe(C0) (N0) (28.8)
40
Pentane(20)
25°
29
2
Products(yields)
Fe(N0) Cl(31%)
3
I s o l a t i o n and
Identification
P r e c i p i t a t e d from pentane at
- 7 8 ° ; infrared spectrum >^,70
2
[CpFe(C0) ] (1.4)
2
2
4.0
CH C1 (30)
2
2
25°
CpFe(C0) Cl(51%)
2
Sublimation at 50-60°
(5xl0" mm);
J
infrared spectrum^
Co(C0) (N0) (4)
CH C1 (60)
29
3
2
2
-78°
[Co(N0) Cl]
2
2
I d e n t i f i c a t i o n in s o l u t i o n by
.
.
.
2,69,70
infrared spectroscopy
r
CpCo(C0) (10)
9
a
CH C1 (50)
2
2
-78°
[Co(N0) Cl]
2
2
Sublimation at 90-100° (5x10
~
j
.
2,69,70
infrared spectrum
,
elemental a n a l y s i s .
a
A CH^Cl2 s o l u t i o n of ClNO was added u n t i l the disappearance of the infrared absorptions due to the i n i t i a l
reactant.
rmi)
- 37 -
was c o l l e c t e d by suction f i l t r a t i o n .
In t h i s manner 3.0 g (17 mmol,
29% y i e l d ) of F e ^ O ^ C l , i d e n t i f i e d by i n f r a r e d spectroscopy ( 2 ) , was
isolated.
Fe(N0).jCl i s unstable at 25°C r e a d i l y l i b e r a t i n g n i t r i c o x i d e ,
e s p e c i a l l y under vacuum, so that an acceptable elemental a n a l y s i s f o r
t h i s compound could not be obtained.
The reactions of ClNO with other neutral complexes of i r o n and
cobalt were performed s i m i l a r l y and the experimental d e t a i l s are
summarized in Table IV.
3.3
3.3a
Results and Discussion
Reaction of n i t r o s y l c h l o r i d e with pentacarbonyliron, Fe(CO)^ .
When a dichloromethane s o l u t i o n of Fe(C0)g i s treated with
n i t r o s y l c h l o r i d e vigorous gas evolution occurs and the r e a c t i o n mixture
becomes v i o l e t - b l a c k .
The only n i t r o s y l containing product, Fe(N0).jCl,
i s subsequently i s o l a t e d i n 29% y i e l d and i d e n t i f i e d by i t s c h a r a c t e r i s t i c
i n f r a r e d spectrum (69, 70).
Optimum y i e l d s are obtained when the r a t i o
of n i t r o s y l c h l o r i d e to Fe(C0)g i s 2 : 1 , and r a t i o s l a r g e r than t h i s
r e s u l t in a r a p i d l y diminishing y i e l d of F e ^ O ^ C l .
In view of the
high o x i d i z i n g property of n i t r o s y l c h l o r i d e , FeCl^ i s l i k e l y a byproduct
of t h i s r e a c t i o n , although t h i s compound was not i s o l a t e d and i d e n t i f i e d .
No i n f r a r e d spectral evidence was obtained f o r the p o s s i b l e formation
of F e ( C 0 ) ( N 0 )
2
a s
2
a n
intermediate.
However, treatment of F e ( C 0 ) ( N 0 )
2
2
with n i t r o s y l c h l o r i d e under s i m i l a r experimental conditions did
produce F e ^ O ^ C l
in comparable y i e l d .
u t i l i z i n g Fe(C0)
i s much more convenient than the previously reported
procedure (71).
5
This preparative route to
Fe^O^d
- 38 -
Fe(N0) Cl i s a dark red-brown s o l i d which i s soluble in
3
common organic solvents i n c l u d i n g hexanes.
The complex loses n i t r i c
oxide at room temperature even under n i t r o g e n .
Sublimation under vacuum
i s p o s s i b l e but only with extensive attendant decomposition.
This
s u b s t a n t i a l thermal i n s t a b i l i t y thwarts a l l attempts to obtain an
acceptable elemental a n a l y s i s for t h i s compound.
Previous i n v e s t i g a t i o n s of the reactions of ClNO with Fe(C0)
demonstrate that d i f f e r e n t
reaction c o n d i t i o n s .
5
products may be obtained depending on
In l i q u i d hydrogen c h l o r i d e the i s o l a b l e product
i s [ F e ( C 0 ) ( N 0 ) ] C l ~ which r a p i d l y d i s s o c i a t e s to C1N0 and Fe(C0)
at
+
5
5
room temperature (72a). The s u r p r i s i n g l y low N-0 s t r e t c h i n g frequency
of 1610 cm ^ displayed by t h i s compound i s i n d i c a t i v e of a n i t r o s y l
ligand coordinated as NO", ije. a bent M-N-0 geometry.
d i f f e r e n c e in the m e t a l - n i t r o s y l
ClNO with Fe(C0)
5
Apart from the
bonding modes, the e q u i l i b r i u m behaviour of
bears resemblance to the coordination reaction of ClNO
with a v a r i e t y of metal h a l i d e s , as exemplified by eq. 15 (15).
C1N0 + F e C l
,
3
ClN0-FeC1 -^-^N0
3
+
+ FeCl "
15.
4
In c o n t r a s t , other workers have found that n i t r o s y l c h l o r i d e
and Fe(C0)g,
i n a r a t i o of 1 . 5 : 1 , react at room temperature in a s t e e l bomb to give
a mixture of F e ( C 0 ) ( N 0 ) and Fe(C0)
2
2
5
(72b).
As the r a t i o i n c r e a s e s ,
lower y i e l d s of F e ( C 0 ) ( N 0 ) are obtained u n t i l , at the r a t i o of 2 . 7 : 1 ,
2
2
only a mixture of carbon monoxide and oxides of nitrogen i s found.
Unfortunately, the r e a c t i o n residue has not been examined for the p o s s i b l e
- 39 -
presence of F e ( N 0 ) C l .
If the r e a c t i o n i s repeated in pentane under
3
ambient temperature and pressure, only a mixture of F e ( C 0 ) ( N 0 ) and
2
2
Fe(C0)g i s obtained.
3.3b
Reaction of n i t r o s y l
dienyl)iron],
c h l o r i d e with b i s [ d i c a r b o n y l ( n - c y c l o p e n t a 5
[CpFe(C0) ] .
2
2
N i t r o s y l c h l o r i d e reacts with dimeric metal carbonyl complexes
to produce monomeric, dimeric or polymeric n i t r o s y l compounds as shown
i n eq. 16-18 (3, 63, 73).
[CpRu(C0) ]
2
+ C1N0
[Re(C0) Cl]
2
+ Cl NO
2
4
[M(C0) C1 ]
4
2
*— CpRu(N0)Cl
[Re(C0) (N0)Cl ]
2
+ C1N0
2
16.
2
*-[M(N0)Cl ]
3
n
2
17.
2
+ [M(N0) C1 ]
2
2
2
18.
(M = Mo or W)
In contrast to the r e a c t i o n depicted in eq. 16 we f i n d that
[CpFe(C0) ]
2
compound.
2
i s cleaved by C1N0 without the formation of a n i t r o s y l
The r e s u l t a n t CpFe(C0) Cl complex may be formed according to
2
the p l a u s i b l e r e a c t i o n scheme
[CpFe(C0) ]
2
2
+ C1N0
*»CpFe(C0) C1 + [CpFe(C0) ]~ + N0
2
2
followed by
[ C p F e ( C 0 ) ] " + N0
2
+
^ [ C p F e ( C 0 ) ] + NO
2
2
+
- 40 -
with the second step being i d e n t i c a l to the oxidation of [CpFe(C0) ]
2
by ClNO as described in Chapter II.
known a i r o x i d a t i o n of [ C p F e ( C 0 ) l
2
This transformation
p a r a l l e l s the
i n the presence of HC1 (68),
2
for
which the f o l l o w i n g r e a c t i o n sequence may be surmised.
[CpFe(C0) ]
2
2
+ HC1
«*~CpFe(C0) Cl + CpFe(C0) H
2
2CpFe(C0) H
2
[CpFe(C0) ] + H 0
2
2
2
2
The hydrido complex i s known to undergo a i r o x i d a t i o n to give
the i n i t i a l reactant dimer.
In a s i m i l a r manner, the M ( C 0 ) - J Q (M = Mn
2
or Re) compounds are cleaved by C1N0 to a f f o r d only the M(C0) Cl species
g
(74).
The formation of the metal halide complexes reported here does not
comprise a useful synthetic method since better y i e l d s are more
conveniently obtained according to e x i s t i n g procedures (68, 75).
3.3c
Reactions of n i t r o s y l c h l o r i d e with
tricarbonylnitrosylcobalt,
Co(C0) (N0), and d i c a r b o n y l ( n - c y c l o p e n t a d i e n y l ) c o b a l t ,
5
3
Both Co(C0) (N0) and CpCo(C0) react with n i t r o s y l
3
give [ C o ( N 0 ) C l ]
2
2
2
in moderate y i e l d s .
CpCo(C0) .
2
c h l o r i d e to
The r e a c t i o n i s rapid even at
-78°C and the use of excess n i t r o s y l c h l o r i d e r e s u l t s in extensive
decomposition.
The dimeric [ C o ( N 0 ) C l ]
2
2
was p r e v i o u s l y prepared in a
manner s i m i l a r to the preparation of Fe(N0) Cl (76).
3
of o l e f i n i c ligands by n i t r o s y l
The displacement
c h l o r i d e i s not unknown ( 3 ) , but the
displacement of the cyclopentadienyl ligand has not been p r e v i o u s l y
- 41 -
reported.
Thus, the formation of [ C o ( N 0 ) C l ]
2
i s unprecedented.
2
from CpCo(C0) and ClNO
S i m i l a r l y , the n -cyclopentadienyl
5
2
ligand in
CpRe(C0) has been shown to undergo a s i m i l a r displacement to y i e l d
3
the [ R e ( C 0 ) ( N 0 ) C l ]
2
reported
(63).
2
2
dimer (74), which has been previously
- 42 -
CHAPTER IV
CYCLOPENTADIENYLNITROSYL COMPLEXES OF CHROMIUM,
MOLYBDENUM, AND TUNGSTEN
4.1
Introduction
The number of c y c l o p e n t a d i e n y l n i t r o s y l complexes of the group
VI t r a n s i t i o n metals has grown s t e a d i l y i n the past two decades.
Both
mono- and d i n u c l e a r species are known, and most d i s p l a y a r i c h and
varied chemistry.
The CpMtCO^NO) (M = Mo or W) compounds have been
obtained in very low y i e l d s by the treatment of aqueous s o l u t i o n s of
NaCCpMCCO)^] with n i t r i c oxide (77).
Since these n u c l e o p h i l i c anions
react with p r o t i c solvents to form the hydrido complexes, C p M ^ O ^ H ,
the synthesis l i k e l y occurs by attack of n i t r i c oxide on these hydrido
intermediates.
The cleavage of the weak metal-metal bond of [ C p C r ( C O ) . ^
with n i t r i c oxide affords the chromium c a r b o n y l n i t r o s y l analog i n good
y i e l d s (78).
However, since the dimeric precursor can be obtained only
in low y i e l d s and with much expenditure of e f f o r t (79), t h i s
route i s not p r a c t i c a l .
synthetic
Although a number of polynuclear carbonyl
complexes react with n i t r i c oxide to produce mononuclear n i t r o s y l
d e r i v a t i v e s , the metal-metal bonds i n [ C p M t C O ) ^ (M = Mo or W) are too
strong to be cleaved by n i t r i c oxide.
A more general n i t r o s y l a t i n g agent
- 43 -
i s D i a z a l d , which converts CpM(C0) H (M = Mo or W) to CpM(C0) (N0) in
3
reasonable y i e l d s (27, 80).
2
However, i t was r e c e n t l y found that the
hydrido precursors are not necessary
and the synthesis can be performed
d i r e c t l y with the anions [CpM(C0) ]" (M = C r , Mo
or W) (81)
3
e l i m i n a t i n g an unnecessary s y n t h e t i c procedure.
thereby
A number of
substituted
complexes CpM(CO)(N0)L containing a v a r i e t y of donar ligands has been
reported
(28).
Another s e r i e s of r e l a t e d compounds can be represented by the
general formula CpM(N0) Cl, of which the chromium d e r i v a t i v e i s the
2
most s t u d i e d .
CrCl
3
It may be obtained in modest y i e l d when a mixture of
and NaCgH i s treated with n i t r i c oxide (27, 29, 82).
The
5
corresponding molybdenum compound i s obtained in poor y i e l d s by the
reaction of T1C H with [Mo(N0) Cl ]
5
5
2
2
(83), or by the r e a c t i o n of NaN0
with [CpMo(C0) NH ]Cl in h y d r o c h l o r i c ' a c i d (84).
3
3
The p r e v i o u s l y unknown
3
tungsten analog, obtained by the treatment of [CpW(N0) (C0)]PFg with
2
NaCl (59), was reported s h o r t l y a f t e r our synthesis of t h i s complex.
A number of compounds derived from C p C r ( N 0 ) C l , such as CpCr(N0) X
2
(X - F, Br, I,
2
CN, NCS, N0 and NCSe) (82, 8 5 ) , [CpCr(N0) L]C1,
2
2
[CpCr(N0)L ]Cl and CpCr(N0)(L)C1 (86) has already been obtained.
2
However,
no d e r i v a t i v e s of the molybdenum and tungsten chloro complexes were
known when t h i s research was undertaken.
P r i o r to t h i s work, f i v e dinuclear chromium s p e c i e s ,
[CpCr(N0)L]
2
(L = NMe , SPh
[CpCr(N0) ]
2
(90) had been c h a r a c t e r i z e d , and a l l these complexes
2
2
and SMe) (87, 88), C p C r ( N 0 ) N H
contain bridging NO and/or L ligands (91-94).
2
2
3
2
(89), and
The molybdenum dimers,
- 44 -
[CpMo(N0)L]
(L = I,
2
SPh and SCH Ph), [CpMo(NO)L J (L = I,
2
2
2
SPh, SCH Ph
2
and C 0 R ) and [CpMo(NO)(SCH Ph)I] have a l l been derived from
2
f
2
[CpMo(N0)I ]
2
[CpMo(N0)I ]
2
2
2
2
rather than from CpMo(N0) Cl (95-97).
The treatment of
2
with P P h , P(0Ph ) and CgHgN y i e l d s CpMo(N0)(L)I
3
3
2
(96).
U n t i l now, the only known a l k y l and a r y l compounds of the
type CpM(N0) R were the Me, E t , Ph
and a - C H
2
(27, 98).
5
5
d e r i v a t i v e s of chromium
The f i r s t three are obtained i n 60, 5, and 0.5 % y i e l d s ,
r e s p e c t i v e l y , by the r e a c t i o n of the corresponding Grignard reagent
with e i t h e r CpCr(N0) Br or CpCr(N0) I.
2
In g e n e r a l , the c h l o r o n i t r o s y l
2
complex gives the poorest y i e l d s , and when THF i s s u b s t i t u t e d f o r E t 0
2
as s o l v e n t , the CpCr(N0) Me complex i s obtained i n only 1% y i e l d .
2
Interestingly,
diazomethane i n s e r t s i n t o the Cr-Cl bond of CpCr(N0) Cl
2
to a f f o r d CpCr(N0) CH Cl i n 3% y i e l d , a r e a c t i o n s i m i l a r to the
2
2
i n s e r t i o n i n t o CpM(C0) H (M = Mo or W) and CpW(C0) CH to give the
3
3
3
corresponding methyl and ethyl d e r i v a t i v e s (27, 9 9 ) .
The ^-CgHg
d e r i v a t i v e i s obtained by the r e a c t i o n of NaC^Hg or TlC^Hg with the
h a l o n i t r o s y l compound (18, 98).
Recently, an i n t e r e s t i n g s e r i e s of complexes (C^Hg) Mo(N0)X
2
(X = I, Me and a-C^H^) has
[CpMo(N0)I ]
2
2
been prepared.
Thus, the treatment of
with 2T1C H and 4T1C H y i e l d s (C H ) Mo(N0)I (83) and
5
5
5
5
5
5
2
(C H ) Mo(N0) (0-CgHj.) (100), and the r e a c t i o n of the former product with
5
5
2
MeMgBr produces ( C g H ^ M o (NO)Me (83).
At room temperature, the NMR
spectrum of each of the three (C(-H ) Mo(N0)X complexes e x h i b i t s only one
5
2
sharp cyclopentadienyl proton resonance.
"18-electron r u l e " , an instantaneous
In order to s a t i s f y the
s t r u c t u r e containing r a p i d l y
- 45 -
interchanging n - and n - C H
5
3
g
g
rings was proposed (83).
However, the
x-ray s t r u c t u r e s of ( C H ) M o ( N 0 ) ( n ^ H g ) (101) and (C H ) Mo(N0)Me (102)
5
5
2
5
5
2
show that the n o n - n ' - C ^ r i n g s , though magnetically non-equivalent, are
equivalent with respect to the c e n t r a l metal atom.
Assuming s i m i l a r
structures i n s o l u t i o n , the NMR behavior over a wide temperature range
then does not require the n - C H =^==n -C H
5
fluxionality.
3
5
5
5
5
Moreover,
the complexes (C H ) Mo(N0)S CNMe and [ P P h ] [(C^H ) Mo(N0)S CC(CN) ],
5
5
2
2
2
4
which have the c h e l a t i n g S CNMe and S C ( C N )
2
2
2
2
2
2
2
l i g a n d s , contain both
2
n^-CgHg and n - C H g rings which become equivalent at 70°C due to
5
g
rapid r ^ - C ^ : ^ ^ n - C H
interchange
5
5
5
(103).
This chapter describes the chain of events which subsequently
leads to the a c q u i s i t i o n of the CpM(N0) R complexes, where M = C r , Mo
2
or W; R = a l k y l or a r y l .
The s y n t h e t i c r o u t e , which i s g e n e r a l l y
a p p l i c a b l e to a l l three m e t a l s , begins with the commercially a v a i l a b l e
metal hexacarbonyl compounds, as in the scheme:
M(C0) — N a [ C p M ( C 0 ) ]
6
^-CpM(C0) (N0)
3
2
»-CpM(N0) Cl—*-CpM(N0) R
2
2
The success of t h i s preparative method i s l a r g e l y due to the high y i e l d
synthesis of each intermediate compound, as described h e r e i n .
Although
the CpM(C0) (N0) species have been p r e v i o u s l y c h a r a c t e r i z e d , the
2
CpM(N0) Cl and CpM(N0) R compounds have not been thoroughly
2
studied
2
e i t h e r because of the d i f f i c u l t y i n t h e i r preparation or simply because
they were p r e v i o u s l y unknown.
of CpMo(C0) (N0), CpM(N0) X
2
2
Therefore, not only are the syntheses
and CpM(N0) R d e s c r i b e d , but the chemical and
2
physical properties of some f u r t h e r d e r i v a r i v e s of CpM(N0) X are reported.
9
- 46 -
4.2
Experimental
A l l experimental procedures described here were performed
under the same general conditions d e t a i l e d in section 2 . 2 .
4.2a
Preparation of d i c a r b o n y l ( n - c y c 1 o p e n t a d i e n y l ) n i t r o s y l complexes
5
of chromium, molybdenum
and tungsten, CpM(C0) (N0) (M = C r , Mo or W).
2
The success of the high y i e l d syntheses of the C p M ^ O ^ N O )
complexes depends on the a v a i l a b i l i t y of the Na[CpM(C0) ] anions
3
uncontaminated by any NaC^Hg.
The molybdenum and tungsten s a l t s were
obtained according to a published procedure (27), although a 5% mole
excess of the M(C0)g compounds was used to improve the y i e l d s of the
desired anions.
A l s o , the tungsten r e a c t i o n was allowed to continue f o r
70 h i n order to ensure more complete conversion.
The chromium s a l t
was e f f i c i e n t l y prepared in the f o l l o w i n g manner.
A THF s o l u t i o n
containing NaCgHg (27) (4.18 g , 47.5 mmol) was concentrated in vacuo j u s t
u n t i l a s l u r r y formed (approx. 20 m l ) . n-Butyl ether (100 m l ; Eastman
Kodak p r a c t i c a l grade) and Cr(C0)g (11.0 g, 50.0 mmol; Pressure Chemical
Co.) were added and the mixture was heated under gentle r e f l u x
12 h with vigorous s t i r r i n g .
room temperature and f i l t e r e d .
for
The f i n a l mixture was allowed to cool to
The pale yellow s o l i d thus c o l l e c t e d
was washed with n-butyl ether (3 x 30 ml) and once with hexanes (30 m l ) ,
and dried at 25°C (5 x 1 0 " mm) f o r 18 h.
3
The Na[CpM(C0) ] complexes
3
of molybdenum and tungsten were freed of any unreacted M(C0)g by
simply taking the f i n a l r e a c t i o n mixture to dryness in vacuo and heating
- 47 -
the residue to 60° (5 x 10" mm) f o r 12 h.
without f u r t h e r
A l l three s a l t s were used
purification.
The three CpM(C0) (N0) complexes were prepared s i m i l a r l y and
2
the experimental procedure using the molybdenum complex as a t y p i c a l
example, was as f o l l o w s .
To a r a p i d l y s t i r r e d THF s o l u t i o n (120 ml)
NaCCpMo^O^]
containing
(13.0 g , 48.7 mmol) was added dropwise at room temperature
a s o l u t i o n of Diazald (10.4 g , 48.6 mmol; Eastman Kodak reagent grade)
i n THF (50 m l ) .
Gas was evolved and an orange s o l i d p r e c i p i t a t e d .
The f i n a l r e a c t i o n mixture was taken to dryness in vacuo and
sublimation of the residue at 60°C (5 x 10
mm) onto a water-cooled
probe for 18 h produced 11.2 g (45.3 mmol, 93% y i e l d ) of
pure CpMo(C0) (N0).
analytically
The chromium and tungsten compounds were obtained
2
i n y i e l d s of 82 and 84% r e s p e c t i v e l y .
A n a l . Calcd. f o r C H C r ( C 0 ) ( N 0 ) :
5
Found:
1945(s).-
5
2
C, 41.40; H, 2.60; N, 6.70. v(C0) cm"
v(N0) cm"
1
(in C H C 1 ) :
2
5
1937(s).
5
2
C, 34.28; H, 2.24; N, 5.54.
v(N0) cm"
1
(in CHpClpj:
C, 25.29; H, 1.70; N, 4 . 1 3 .
v(N0) cm"
1
(in-CH^C"^):
1
(in C H C 1 ) :
2
2
2020(s);
1663(s).
2
1925(s).
2020(s);
C, 34.03; H, 2.04; N, 5.67.
v(C0) cm"
Calcd. f o r C,-HgW(C0) (NO):
Found:
(in CHpClp):
1
1680(s).
2
C a l c d . f o r C H Mo(C0) (N0):
Found:
C, 41.39; H, 2.48; N, 6.90.
C, 25.10; H, 1.50; N, 4.18.
v(C0) c m
1655(s).
-1
(in C H C 1 ) :
2
2
2010(s);
- 48 -
4.2b
Preparation of chloro(n -c,yclopentadienyl )dinitrosy1 complexes of
5
chromium, molybdenum
All
manner.
and tungsten,
CpM(N0) Cl (M = C r , Mo or W) .
2
three CpM(N0) Cl compounds were prepared i n a s i m i l a r
2
The y i e l d s of the chromium and tungsten complexes were optimized
by performing the reactions at -78°C.
The molybdenum compound was
obtained in e x c e l l e n t y i e l d s even at room temperature and a t y p i c a l
synthesis was as f o l l o w s .
A CH C1
2
2
s o l u t i o n (100 ml) containing CpMo(C0) (N0) (6.5 g ,
2
26 mmol) was treated dropwise with r a p i d s t i r r i n g at room temperature
with a s o l u t i o n of ClNO i n C H C 1 .
2
2
Gas e v o l u t i o n occurred and the
i n i t i a l orange s o l u t i o n became dark green.
The course of the r e a c t i o n
was followed by i n f r a r e d spectroscopy and C1N0 was added j u s t u n t i l the
carbonyl absorptions due to the reactant disappeared.
(Great care was
taken not to add excessive amounts of ClNO since t h i s r e s u l t e d i n a
s i g n i f i c a n t reduction i n the y i e l d of the desired product.
Conversely
i f i n s u f f i c i e n t C1N0 was employed, then some d i f f i c u l t y
was encountered i n the separation of the unreacted CpM(C0) (N0) from
2
the desired product.)
The f i n a l r e a c t i o n mixture was concentrated
in vacuo to ca. 30 ml and f i l t e r e d through a short ( 3 x 5 cm) F l o r i s i 1
column.
The column was washed with CH C1
2
2
u n t i l the washings were
c o l o r l e s s , and the combined f i l t r a t e s were taken to dryness to give
6.0 g (2.3 mmol, 90% y i e l d ) of a n a l y t i c a l l y pure CpMo(N0) Cl.
2
The
chromium and tungsten complexes were obtained i n y i e l d s of 77 and 72%
respectively.
Table V. Elemental Analyses and Physical Properties of CpM(N0) X (M = C r , Mo or W; X = Cl or
9
Color
Compound
Mp, °C
(under N )
Analyses, %
Proton NMR,
x(in C D )
2
CpCr(N0) Cl
9
CpMo(N0) Cl
2
CpW(N0) Cl
2
CpCr(N0) I
2
CpMo(N0) I
2
CpW(N0) I
9
gold
green
green
dark gold
o l i v e green
o l i v e green
144 (dec)
116
127 (dec)
146 (dec)
114 (dec)
129 (dec)
I).
g
6
v(N0) c m
( i n CH C1 )
-1
2
2
H
N
Cl
^ " 5 5
5.22(s)
1816(s), 1711(s)
4.93(s)
1759(s), 1665(s)
5.02(s)
1733(s), 1650(s)
5.28(s)
1808(s), 1718(s)
5.00(s)
1764(s), 1677(s)
4.94(s)
1740(s), 1657(s)
calcd:
28.26
2.37
13.18
16.68
found:
28.29
2.55
12.86
16.99
calcd:
23.42
1.96
10.92
found:
23.53
1.90
10.70
calcd:
17.44
1.46
8.13
10.29
found:
17.68
1.62
8.10
10.26
calcd:
19.75
1.66
9.22
found:
19.71
1.76
9.00
calcd:
17.26
1.45
8.05
found:
17.33
1.40
7.88
calcd:
13.78
1.16
6.43
found:
13.78
1.17
6.36
5
C
H
- 50 -
The complexes, along with t h e i r elemental analyses and
physical p r o p e r t i e s , are l i s t e d in Table V.
4.2c
Preparation of ( n - c y c l o p e n t a d i e n y l ) i o d o d i n i t r o s y l
5
chromium, molybdenum
complexes of
and tungsten, CpM(N0),,I (M = C r , Mo or W).
A l l the CpM(N0) I complexes were prepared i n a s i m i l a r manner
2
and the customary procedure f o r the synthesis of the CpCr(N0) I
2
compound was as f o l l o w s .
A THF s o l u t i o n (150 ml) containing CpCr(N0) Cl (2.54g, 12.0
2
mmol) and Nal (9.0 g , 60 mmol; M a l l i n c k r o d t reagent grade) was s t i r r e d
at room temperature f o r 18 h.
The r e a c t i o n mixture became a very
dark yellow-brown in c o l o r and a white s o l i d formed.
The f i n a l
mixture
was taken to dryness in vacuo and the r e s u l t a n t residue extracted i n t o
CH C1
2
2
(150 ml).
The e x t r a c t
was f i l t e r e d through a short ( 3 x 5 cm)
F l o r i s i 1 column and taken to dryness i n vacuo (5 x 10
mm) thereby
a f f o r d i n g a v i r t u a l l y q u a n t i t a t i v e y i e l d of the a n a l y t i c a l l y pure
CpCr(N0) I complex.
2
The elemental analyses and physical properties of
a l l three CpM(N0) I compounds are given i n Table V.
2
4.2d
Preparation of b i s [ ( n - c y c l o p e n t a d i e n y 1 ) e t h o x o n i t r o s y l c h r o m i u m ] ,
5
[CpCr(N0)(0Et)] .
2
A s o l u t i o n of CpCr(N0) Cl (2.13 g , 10.0 mmol) in EtOH (120 ml)
2
was treated with NaOEt (0.80 g , 12.0 mmol) at room temperature.
Immediately, the mixture became y e l l o w - r e d and a f i n e white s o l i d formed.
- 51 -
The mixture was s t i r r e d f o r 0.5 h and the i n f r a r e d spectrum of the
supernatant l i q u i d displayed absorptions at 1792 c m ( s ) and 1585 c m ( s )
-1
i n d i c a t i v e of the CpCr(N0) (0Et) complex (cf.
i n f r a r e d spectrum of
2
C p C r ( N 0 ) C l , v(N0) cm"
(in EtOH): 1812(s); 1708(s)).
1
2
removed i n vacuo y i e l d i n g a red o i l .
(5 x 10
-1
The solvent was
During f i n a l drying at 25°C
mm) f o r 0.5 h, the red o i l was transformed to a green s o l i d .
This product was extracted i n t o CH^Cl^ (25 ml) and chromatographed
through a F l o r i s i 1 column (2.5 x 8 cm) with CH C1
2
eluate was taken to dryness in vacuo (5 x 10
2
as e l u e n t .
mm) y i e l d i n g 0.64 g
(3.3 mmol, 33% y i e l d ) of the o l i v e - g r e e n [CpCr(N0)(0Et)]
A n a l . Calcd. f o r [C H Cr(N0)(0Et)] :
5
N, 7.40.
1660.
Found:
5
compound.
2
C, 43.58; H, 5 . 2 1 ;
2
C, 43.78; H, 5.26; N, 7.29.
Mp. (under N ) :
The
v(N0) cm"
1
(in C H C 1 ) :
2
2
233°C (dec).
2
The CpM(N0) Cl (M = M or W) complexes reacted with LiOEt i n
2
THF producing y e l l o w - r e d s o l u t i o n s which, when taken to dryness, y i e l d e d
yellow-red o i l s .
95°C (5 x 10
These o i l s remained unchanged even a f t e r heating at
mm) f o r 2 h.
Their i n f r a r e d spectra suggested that the
products were CpMo(N0) Et (v(N0) cm"
(in C H C 1 ) :
1
2
and CpW(N0) Et (v(N0) cm"
2
4.2e
Preparation of
1
2
(in C H C 1 ) :
2
2
2
1740(s); 1630(s))
1710(s); 1610(s)).
bis[ch1oro(n -cyclopentadieny1)nitrosylchromium],
5
[CpCr(N0)Cl] ,
2
A sample of [CpCr(N0) (0Et)] (0.19 g , 1.0 mmol) was d i s s o l v e d i n
2
2
benzene (30 ml) and dry HCl(g) was bubbled through the s o l u t i o n .
Immediately, the off-green reaction mixture became a very intense green
- 52 -
in color.
Enough HCI(g) was added to j u s t completely consume the
i n i t i a l reactant (monitored by i n f r a r e d spectroscopy).
The mixture
was taken to dryness in vacuo and the product c r y s t a l l i z e d from CH C1
2
(20 ml) by the a d d i t i o n of hexanes (50 m l ) .
2
The s o l i d was c o l l e c t e d
by f i l t r a t i o n and d r i e d at 25°C (5 x 10~ mm) to give 0.16 g (0.88 mmol,
3
88% y i e l d ) of the green [CpCr(N0)C1] complex.
g
A n a l . Calcd. f o r [ C ^ C r (N0)C1 ] :
2
Found:
C, 32.50; H, 2 . 6 1 ; N, 7.66.
C, 32.90; H, 2.76; N, 7.67.
v(N0) cm"
1
(in C H g C l ) :
2
1678.
Mp. (under N ) : 140 (dec).
2
4.2f
Preparation of a l k y l - and a r y l - ( n - c y c ! o p e n t a d i e n y l ) d i n i t r o s y l
5
complexes of chromium, molybdenum
and tungsten, CpM(N0) R
2
(M = C r , Mo or W; R = a l k y l or a r y l ) .
Since the various a l k y l and a r y l d e r i v a t i v e s were s i m i l a r l y
prepared, two representative syntheses are described below.
Table VI
summarizes the p a r t i c u l a r experimental d e t a i l s .
4.2fl
Preparation of ( n - c y c l o p e n t a d i e n y l ) m e t h y l d i n i t r o s y l t u n g s t e n ,
5
CpW(N0) Me .
2
A benzene s o l u t i o n (12 ml) containing jte^Al
(0.22 g , 3.1 mmol;
Texas A l k y l s ) was added dropwise at room temperature to a s t i r r e d
of CpW(N0) Cl (1.0 g , 2.9 mmol) in benzene (25 ml).
2
solution
The green s o l u t i o n
was allowed to s t i r f o r 48 h during which time a red-brown o i l y s o l i d
deposited on the w a l l s of the r e a c t i o n f l a s k .
(The r e a c t i o n was followed
- 53 -
Table VI. Reaction Conditions and P u r i f i c a t i o n Methods for CpM(N0) R .
9
Compound
CpCr(N0) C H
2
g
CpCr(N0) CH
2
2
2
4
g
CpMo(N0) CH
2
2
CpW(N0) CH
2
3
5
(CH ) A1
0.5
AB
(C H ) A1
0.5
A
(i-C H ) A!H
0.5
A
(C H ) A1
1 .5
AA
(CH ) A1
1
A
(C H ) A1
1
A
g
0.5
A
1
AB
48
AB
3
3
5
3
4
g
5
2
3
3
2
5
4
g
AB
3
CpMo(N0) i-C H
2
0.5
5
6
3
2
CpW(N0) C H
g
5
CpMo(N0) C H
2
(C H ) A1
2
5
2
2
Purification
methods
3
CpCr(N0) i-C H
CpMo(N0) C H
Reaction
time (hours)
6
5
3
CpCr(N0) C H
A l k y l a t i n g or
A r y l a t i n g Agent
5
3
(i-C H ) A!H
4
W
g
2
A1
3
(CH ) A1
3
3
+ A - chromatographic separation on alumina using benzene as the e l u a n t .
B - sublimation i n vacuum
- 54 -
by i n f r a r e d spectroscopy and such monitoring of several experiments
showed these to be the optimum stoichiometry and reaction time.)
Judging from the r e l a t i v e v(N0) band i n t e n s i t i e s of the f i n a l
reaction
mixture, the CpW(N0) Me product and unreacted CpW(N0) Cl e r e present
2
2
in ca_. equimolar q u a n t i t i e s .
W
The r e a c t i o n mixture was concentrated
in vacuo to c a . 15 ml and the supernatant l i q u i d chromatographed i n a
short ( 3 x 7
as eluent.
cm) column of alumina (Woelm neutral grade 1) with benzene
The eluate was taken to dryness in vacuo and the r e s u l t a n t
_q
s o l i d sublimed at 30°C (5 x 10
mm) onto a water-cooled probe a f f o r d i n g
0.20 g (0.62 mmol, 20% y i e l d ) of a n a l y t i c a l l y pure CpW(N0) Me.
2
4.2f2
Preparation of
[n -cyclopentadieny1)dinitrosylphenylmo1ybdenum,
5
CpMo(N0) Ph .
2
A s o l u t i o n of Ph Al (28) (0.35 g , 1.4 mmol) i n benzene (70 ml)
3
was added dropwise to a s o l u t i o n of CpMo(N0) Cl (1.0 g , 3.9 mmol) i n the
2
same solvent (30 ml).
A red-brown s o l i d formed and the mixture was
f u r t h e r s t i r r e d for 1 h at which point the i n f r a r e d spectrum of the
supernatant l i q u i d confirmed the depletion of the i n i t i a l
reactant.
The f i n a l reaction mixture was concentrated i n vacuo to c a . 20 ml and
the supernatant l i q u i d was chromatographed on an alumina column.
The
product was eluted with benzene and only the main portion of the green
product band was c o l l e c t e d .
The solvent was removed from the eluate and
the residue was d r i e d at room temperature i n vacuo (5 x 10
mm) f o r 4 h
thus providing 0.65 g (2.2 mmol, 56% y i e l d ) of CpMo(N0) Ph as an
2
a n a l y t i c a l l y pure o l i v e - g r e e n o i l .
- 55 -
A l l the other complexes i n Table VII were obtained in y i e l d s
of 30-50%.
Their elemental analyses and physical properties are given
in Tables VII and V I I I .
A l l the complexes are stable in a i r f o r
short
periods of time but are best stored under nitrogen and below 0°C.
They
are very soluble i n common organic solvents i n c l u d i n g hexanes.
4.2g
Preparation of
bis[(n -cyclopentadienyl)dinitrosylchromium],
5
[CpO(N0) ] .
2
2
To an amalgam made from sodium (0.5 g , 20 mmol) and mercury
metal (20 ml) was added a s o l u t i o n of CpCr(N0) Cl (1.06 g , 5.00 mmol) i n
2
benzene (120 ml).
This reaction mixture was s t i r r e d vigorously at room
temperature u n t i l the i n f r a r e d spectrum of the supernatant
liquid
contained no absorption bands due to the i n i t i a l reactant (ca_. 1.5 h).
[Reaction beyond t h i s point led to decomposition of the desired product.]
During the course of the reaction a grey s o l i d was deposited and the
s o l u t i o n became red-purple in c o l o r .
The supernatant l i q u i d was syringed
from the amalgam and concentrated in vacuo to ca_. 20 ml.
This s o l u t i o n
was t r a n s f e r r e d to a short (2 x 5 cm) column of alumina (Woelm neutral
grade 1) and the column was eluted with benzene u n t i l the washings were
c o l o r l e s s (ca_. 120 m l ) .
Solvent was removed from the eluate in vacuo and
the remaining purple-black s o l i d was dried at room temperature under high
vacuum'(5 x 10
mm) f o r 2 h.
In t h i s manner 0.68 g (1.9 mmol, 77% y i e l d )
of the a n a l y t i c a l l y pure [ C p C r ( N 0 ) l
2
2
complex was obtained.
Anal. Calcd. for [ ( C H ) C r ( N 0 ) ] :
5
Found:
1512(m).
5
C, 33.78; H, 2.75; N, 15.82.
Mp. (in a i r ) :
147°C (dec).
2
2
C, 33.91; H, 2.85; N, 15.82.
v(N0) c m
-1
(in C H C 1 ) :
2
2
1667(s);
- 56 -
Table VII.
Elemental Analyses and Physical Properties of CpM(N0) R.
9
Compound
Color
Mp,°C
(
CpCr(N0) C H
2
g
CpCr(N0) CH
2
3
CpCr(N0) C H
2
5
2
5
CpCr(N0) i-C H
2
4
CpMo(N0) C H
2
6
CpMo(N0) CH
2
5
2
5
CpMo(N0) i-C H
2
4
CpW(N0) CgH
2
CpW(N0) CH
2
3
5
g
a i r
Analyses,%
)
C
calculated
H
N
C
found
H
dark green
48-48.5
51.97 3 .97 11 .02 52 .05 4 .10 10 .97
dark green
80.5-81.0
37.51 4 .20 14 .58 37 .42 4,.19 14 .49
green
oil
40.79 4 .89 13 .59 40 .74 4,.90 13 .46
green
oil
46.15 6 .03 11 .96 46 .38 6,.20 11 .62
o l i v e green
oil
44.31 3 .38
green
3
CpMo(N0) C H
2
g
i n
61.5-62.0
green
oil
green
oil
9 .40 44 .28 3,.46
9 .13
30.53 3 .42 11 .87 30 .69 3 .33 11 .82
33.52 4 .03 11 .20 34 .05 4 .38 10 .90
.
38.86 5 .07 10 .07 39 .33 5 .23 10 .06
green
109-110
34.22 2 .61
7 .26 34 .38 2 .74
7 .03
pale green
75-76
22.24 2 .49
8 .65 22 .54 2 .42
8 .60
- 57 -
Table VIII.
IR and 'h! NMR Data f o r CpM(N0) R.
2
Compound
IR
a
v(N0) cm"
CpCr(N0) Cl
2
CpCr(N0) C H
2
6
CpCr(N0) CH
2
3
CpCr(N0) C H
2
5
2
5
CpCr(N0) i-C H
2
4
g
CpMo(N0) Cl
2
CpMo(N0) C H
2
g
CpMo(N0) CH
2
3
CpMo(N0) C H
2
5
2
5
CpMo(N0) i-C H
2
4
CpW(N0) Cl
2
CpW(N0) C H
2
g
CpW(N0) CH
2
3
5
g
]
n -C,-rL
1
5
H NMR x ( i n CgDg)
others
5.22
-
1690
5.27
2.7(m)
1780
1675
5.63
9.55 ( s i n g l e t CH )
1772
1670
5.45
8.6(m)
1775
1675
5.33
8-9(m)
1758
1665
1745
1658
. 4.90
2.7(m)
1728
1640
4.92
9.20 ( s i n g l e t CH )
1735
1643
4.90
8.2(m)
1725
1638
4.93
7.6-8.8(m)
1733
1650
5.02
-
1713
1630
4.95
2.6(m)
1705
1620
4.98
8.82 ( s i n g l e t CH )
1815(s)
1710(vs)
1792
b
b
b
3
-
4.93
3
3
Hexane s o l u t i o n unless indicated otherwise.
Dichloromethane s o l u t i o n .
Approximate centroid of complex m u l t i p l e t
C
(m).
- 58 -
4.3
4.3a
Results and Discussion
The d i c a r b o n y l ( n - c y c l o p e n t a d i e n y l ) n i t r o s y l
5
complexes of chromium,
molybdenum and tungsten, CpM(C0)p(N0) (M = C r , Mo or W) .
A l l three CpM(C0) (N0) compounds were obtained according to the
2
f o l l o w i n g sequence of r e a c t i o n s :
NaC H + M(C0)
5
5
* - Na[CpM(C0) ] + 3C0
6
3
Na[CpM(C0) ] + p-CH C H S0 N(N0)CH
3
3
6
4
2
——»~
3
CpM(C0) (N0) + C0 + p-CH C H S0 N(CH )Na
2
3
6
4
2
3
The success of t h i s synthetic route depends c r i t i c a l l y on the a v a i l a b i l i t y
of the corresponding Na[CpM(C0) ] s a l t s which are free of any unreacted
3
NaCgHg.
The presence of t h i s contaminant i s immediately evident upon
subsequent reaction with Diazald since a brown-black, rather than the
expected orange-red, reaction mixture i s obtained.
Furthermore, the
y i e l d of the desired c a r b o n y l n i t r o s y l i s d r a s t i c a l l y reduced.
However,
once the s u f f i c i e n t l y pure anions are obtained, t h e i r r e a c t i o n with
Diazald in THF produces the CpM(C0) (N0) complexes i n e x c e l l e n t ,
2
reproducible y i e l d s ranging from 82-93%, depending on M.
A l s o , these
syntheses can be e a s i l y scaled to produce the desired products in
q u a n t i t i e s as large as 30 g while s t i l l maintaining the same high y i e l d s .
Undoubtedly, the experimental procedures described f o r the preparation of
these complexes are the most convenient thus f a r developed (27, 77, 78, 80, 81 ).
- 59 -
The Na[CpM(C0) ] (M = Mo or W) s a l t s can be r e a d i l y o b t a i n e d ,
3
i n THF under r e f l u x , according to a published procedure (27).
However,
to ensure complete conversion important modifications have been n e c e s s a r i l y
made.
Thus, the syntheses are performed with NaCp as the l i m i t i n g
reagent and the r e a c t i o n i n v o l v i n g the tungsten compound i s allowed to
continue f o r the extended period of 72 hours.
Unfortunately, under the
same c o n d i t i o n s , Cr(C0)g and NaC^Hg react very slowly and the conversion
a f t e r seven days of reaction i s s t i l l
r e l a t i v e l y low.
Although diglyme
has been previously employed in the preparation of Na[CpCr(C0) ]
(79),
3
i t s strong s o l v a t i n g c h a r a c t e r i s t i c s renders i t s use i m p r a c t i c a l .
However, i f Cr(C0)g and NaCgHg are allowed to react i n n-BupO under
r e f l u x f o r 18 hours, a v i r t u a l l y q u a n t i t a t i v e y i e l d of the desired
product i s obtained.
The Na[CpCr(C0) ], i n s o l u b l e i n n-BupO, p r e c i p i t a t e s
3
as a c r y s t a l l i n e pale yellow s o l i d during the course of the r e a c t i o n .
Subsequent p u r i f i c a t i o n , to remove excess M(C0)g, of the Na[CpM(C0) ]
3
compounds i s e a s i l y performed as described in the Experimental s e c t i o n .
The properties of the C p M ^ O ^ N O ) compounds have been
previously described (2, 3, 28).
The complexes are orange to orange-red
s o l i d s r e a d i l y soluble i n organic solvent and stable in a i r f o r
periods of time.
However, they may be stored i n d e f i n i t e l y
short
under nitrogen
at room temperature.
4.3b
The c h l o r o ( n - c y c 1 o p e n t a d i e n y l ) d i n i t r o s y l
5
complexes of chromium,
molybdenum and tungsten, CpM(N0) Cl (M = C r , Mo or W).
9
The high y i e l d syntheses of a l l three complexes are e f f e c t e d by
- 60 -
the treatment of CpM(C0) (N0) with C1N0 in C H C 1 , according to eq. 19.
2
2
CpM(C0) (N0) + C1N0
2
e-CpM(N0) Cl + 2C0
2
2
19.
(M = C r , Mo or W)
In order to maximize y i e l d s , the transformations i n v o l v i n g the chromium
and tungsten containing compounds must be performed at -78°C, while the
molybdenum complex can be obtained i n e x c e l l e n t y i e l d s at room temperature.
The l a t t e r r e a c t i o n i s accompanied by the formation of a small amount
of [ C p M o ( N 0 ) C l ] , i d e n t i f i e d by elemental a n a l y s i s and i t s c h a r a c t e r i s t i c
2
2
i n f r a r e d spectrum (104).
Since t h i s byproduct can a l s o be formed by
the d i r e c t treatment of CpM(C0) (N0) with C l
2
2
(104), i t s appearance
simply r e f l e c t s the f a c t that ClNO e x i s t s in s o l u t i o n as part of the
equilibrium
2C1N0-^
— 2N0 + C l
2
As in most reactions between the n i t r o s y l halides and metal c a r b o n y l s ,
special a t t e n t i o n must be taken to avoid excess n i t r o s y l h a l i d e .
T y p i c a l l y , the above conversions are followed by i n f r a r e d spectroscopy
and enough C1N0 i s added u n t i l the carbonyl absorptions of the reactant
have j u s t disappeared.
In t h i s manner, the f u r t h e r r e a c t i o n of the
desired products i s e f f e c t i v e l y prevented.
In any case, the general
a p p l i c a b i l i t y of the reaction given by eq. 19, u t i l i z i n g the now r e a d i l y
a v a i l a b l e s t a r t i n g m a t e r i a l s , makes t h i s high y i e l d preparative route
superior to a l l others thus f a r reported (27, 29, 59, 83, 84, 105).
- 61 -
The CpM(N0) Cl complexes, along with some of t h e i r physical
2
p r o p e r t i e s , are presented in Table V.
They are c r y s t a l l i n e s o l i d s which
have low s o l u b i l i t y in hexanes but are very soluble in most other
organic s o l v e n t s .
Contrary to a previous report (83), CpMo(N0) Cl, as
2
well as the chromium and tungsten d e r i v a t i v e s , are stable
indefinitely
under nitrogen at room temperature, and a l l three can be exposed to
a i r f o r short periods of time without noticeable decomposition.
Furthermore, the molybdenum complex melts at 116°C without
decomposition and a l l three compounds may be sublimed at 40-50°C
(5 x 10
mm) although some decomposition occurs with the chromium and
tungsten s p e c i e s .
Chemically, the CpM(N0) Cl compounds are useful
2
precursors f o r the synthesis of a wide v a r i e t y of stable a l k y l and a r y l
complexes, CpM(N0) R, as discussed subsequently.
2
A l l three CpM(N0) I complexes are obtained in
2
y i e l d s by the d i s p r o p o r t i o n a t e
quantitative
r e a c t i o n between Nal and the
corresponding chloro complexes i n THF.
Their physical (Table V) and
chemical properties are very s i m i l a r to those of the chloro compounds.
The molybdenum and tungsten complexes have not been p r e v i o u s l y reported.
The x-ray c r y s t a l structure of CpCr(N0) Cl i n d i c a t e s a pseudo2
tetrahedral
("piano s t o o l " ) arrangement of ligands about the chromium
atom (106) as in C.
The Cj-H^ r i n g randomly occupies one of two coplanar
O
- 62 -
o r i e n t a t i o n s i n which a l l the Cr-C distances are approximately e q u a l , the
o
average distance being 2.20 A.
The Cr-NObond distances of 1.717 (0.012) and 1.704 (0.013) A ,
which are v i r t u a l l y
i d e n t i c a l to the value of 1.716 (0.003) found in
CpCr(N0) (NC0) (107), are about 0.4 A shorter than the expected Cr-N(sp)
2
bond length.
In f a c t , the Cr-N bond order i s estimated to be 1.7,
i n d i c a t i n g the n i t r o s y l i s bonded as N0 with appreciable e l e c t r o n
+
donation from the Cr(d)
to the N(pir*) o r b i t a l s .
The Cr-N-0 fragments
i n both the chloro and isocyanato compounds depart from l i n e a r i t y by
about 10°.
This i s ascribed to e l e c t r o s t a t i c r e p u l s i o n between the
oxygen atom of the n i t r o s y l ligand and the c h l o r i d e or isocyanate l i g a n d
rather than to c r y s t a l packing f o r c e s .
The mass spectral data for the CpM(N0) Cl complexes, given i n
2
Tables IX and X, e x h i b i t the expected fragmentation p a t t e r n s .
The
presence of MCp and MCpCl and the concurrent absence of M(N0) and
+
+
+
M(N0)C1 fragments can be a t t r i b u t e d to the f a c t that the M-Cp linkage
+
i s stronger than the M-N0 bond.
The mass spectrum of CpMo(C0) (N0)
d i s p l a y s a s i m i l a r behavior (108).
2
Furthermore, as i s found in the
cases of CpFe(C0) Cl (108) and CpMo(C0) Cl (109), the M-Cp and M-Cl bonds
2
3
in the CpM(N0),,Cl complexes are ruptured with about equal
difficulty.
The [ M C H ] fragments, which occur frequently i n the mass spectra of
+
3
3
Cp-containing complexes, are not observed f o r the chromium compound but
become i n c r e a s i n g l y abundant for the molybdenum and tungsten analogs.
a d d i t i o n , the r e l a t i v e abundance of fragments containing the n i t r o s y l
group suggest a M-N0 bond strength which increases as Mo < Cr < W,
In
- 63 -
Table IX. High Resolution Mass Spectral Data f o r C p C r ( N 0 ) C l .
9
measured
m/e
' calculated
relative
abundance
assignment
213.9423
213.9414
11.9
C H Cr(N0)
211.9440
211.9444
32.6
C H Cr(N0)
183.9447
183.9434
13.8
C H Cr(N0) Cl
+
181.9476
181.9464
36.6
C H Cr(N0) Cl
+
153.9459
153.9455
36.6
151.9481
151.9484
100
176.9796
176.9756
5.3
C H Cr(N0)
146.9818
146.9776
3.4
C H Cr(N0)
116.9784
116.9796
18.0
C H Cr
88.9050
88.9063
10.6
Cr Cl
+
86.9089
86.9093
23.9
Cr
+
65.0396
65.0391
13.3
C
51.9412
51.9404
13.8
Cr
5
37
5
5
5
2
Cl"
Cl
2
3 7
5
5
3 5
5
5
37
5 5
+
35 +
C H C r °C1
5
5
5
5
c
c
5
+
5
3 7
3 5
5 5
H
+
Cl
+
+
+
2
- 64 -
Table X. Low Resolution Mass Spectral
Data f o r
CpM(N0) Cl .
a
2
(M = Mo or W)
CpMo(N0) Cl
CpW(N0) Cl
2
2
m/e
m/e
Relative
Abundance
Assignment
258
43.7
C H Mo(N0) Cl
228
33.5
C H Mo(N0)Cl
198
100
C H MoCl
+
172
30.4
C H MoCl
+
163
7.5
C H Mo
+
137
12.4
C H Mo
+
133
24.2
MoCl
98
10.4
Mo
65
3.7
a
5
c
9 8
Mo,
5
3
3
5
5
3
1 8 4
+
+
344
100
C H W(N0) C1
314
81.0
C H W(N0)C1
284
53.7
C H WC1
258
71.4
C
249
28.1
CHW
223
13.0
C
219
16.0
WC1
184
2.1
w
65
6.8
C
5
5
5
+
2
+
5
0
5
C
2
c
O
The assignments
i.e.
5
Assignment
Relative
Abundance
3
+
+
5 5
H
+
5
3 3
H
5
W C 1 +
+
5
3 3
H
W +
+
+
5 5
H
+
involve the most abundant n a t u r a l l y occurring
W and
3 5
C 1 , in each fragment.
+
5
isotopes,
- 65 -
s i m i l a r to the ordering of the M-CO bond strengths i n the CpM(C0) Cl
3
compounds (28).
A wide v a r i e t y of d e r i v a t i v e s of the type CpCr(N0) X (X = F, Br,
2
I,
CN, NCS, N0 (82), and NCSe (85)) have been obtained by the
2
treatment
of an aqueous s o l u t i o n of [ C p C r ( N 0 ) ] , generated from CpCr(N0) Cl and
+
2
AgNO-j, with NaX or KX.
2
The isoselenocyanato complex decomposes r e a d i l y
i n s o l u t i o n to give CpCr(N0) CN and elemental selenium.
Also,
2
CpCr(N0) SCF i s formed when the chloro compound i s allowed to react with
2
3
AgSCF i n acetone (110).
3
In the above r e a c t i o n s , the intermediary
s a l t has not been previously i s o l a t e d .
However, we f i n d that
treatment
of CpCr(N0) Cl with AgNO^ or AgBF^ in water produces a p r e c i p i t a t e
2
AgCl and a dark green supernatant s o l u t i o n .
of the dried reaction mixture i n t o CH^Cl
2
2
2
of
Subsequent e x t r a c t i o n
followed by c r y s t a l l i z a t i o n
from CH Cl /hexanes y i e l d s the CpCr(N0) N0 and CpCr(N0) BF
2
nitrate
3
2
4
compounds as stable green s o l i d s which are soluble i n most
organic solvents except hydrocarbons.
CH C12 d i s p l a y n i t r o s y l
2
Their i n f r a r e d spectra i n
s t r e t c h i n g absorptions at 1841 and 1738 c m " , and
1
at 1848 and 1744 cm" f o r the N 0 " and BF^" s a l t s r e s p e c t i v e l y .
These
1
3
absorptions are s i m i l a r to those displayed by [ C p C r ( N 0 ) ] [ A l C l ] "
+
2
and are consistent with reduced Cr(diO to N(pir*) donation.
4
(111)
However,
present attempts to perform the same reaction with the molybdenum complex
proceeded with gas evolution and attendant decomposition.
- 66 -
4.3c
Bis[(n -cyclopentadienyl)ethoxonitrosylchromium],
[CpCr(NO)(0Et)] ,
5
2
and bis[ch1oro(n -cyclopentadieny1)nitrosylchromium,
[CpCr(NQ)C1] .
5
2
When CpCr(N0) C1 i s treated with NaOEt in EtOH or with LiOEt
2
in THF, the CpCr(N0) 0Et can be i s o l a t e d as an unstable red o i l .
Its
2
i n f r a r e d spectrum in EtOH d i s p l a y s absorptions at 1792 and 1685 c m ,
- 1
which are s i g n i f i c a n t l y lower than those of the chloro complex
( i . e . 1812 and 1708 c m
-1
i n EtOH) due to the greater e l e c t r o n donating
a b i l i t y of the alkoxide l i g a n d .
Upon exposure to high vacuum at room
temperature, CpCr(N0) (0Et) spontaneously loses n i t r i c oxide to form
2
the new o l i v e green s o l i d , [CpCr(NO)(0Et)] » according to eq. 20.
This
2
2CpCr(N0) (0Et)
2
[CpCr(NO) ( 0 E t ) ]
2
+ 2N0
20.
same transformation can be effected by the e l u t i o n of a CH C1
2
the d i n i t r o s y l compound through an alumina column.
2
s o l u t i o n of
The dimeric s p e c i e s ,
i d e n t i f i e d by elemental a n a l y s i s and mass spectrometry, contains in
i n f r a r e d spectrum a s i n g l e terminal n i t r o s y l absorption at 1660 c m
C H C 1 , s i m i l a r to the p r e v i o u s l y reported [CpCr(N0)L]
2
2
and SMe) compounds (87, 88).
2
its
-1
in
(L = NMe , SPh
2
The [CpCr(NO)(OEt)] complex i s soluble in
most organic solvents and can be handled i n a i r f o r short periods of
time without noticeable decomposition.
It decomposes without melting at
233°C, under n i t r o g e n .
On the other hand, the s i m i l a r reactions of the molybdenum and
tungsten chloro compounds with the above reagents y i e l d only reddish o i l s
s i m i l a r in appearance to CpCr(N0) (0Et).
9
A comparison of t h e i r
infrared
- 67 -
spectra with those of the s t a r t i n g chloro complexes, given in the
Experimental section and in Table V, suggests that these compounds can
be formulated as CpM(N0) (0Et).
2
They possess a remarkable thermal
s t a b i l i t y since heating to 95°C under vacuum f o r two hours e f f e c t s no
change.
A l s o , the attempted dimerization on an alumina column resulted
only in complete decomposition, and a n a l y t i c a l l y pure samples of the
d i n i t r o s y l s cannot be obtained.
The CpM(N0) L (L = SBu, NPh and NH )
2
2
2
complexes are prepared s i m i l a r l y and they e x h i b i t a behavior s i m i l a r to
the ethoxide d e r i v a t i v e s .
(L = I,
A v a r i e t y of [CpMo(N0)L] and [CpMo(N0)L 1
2
2
2
SPh, SCH Ph and C0 R.p) have been reported but they are obtained
2
from [CpMo(N0)I ]
2
2
2
rather than CpMo(N0) Cl (95-97).
2
The r e a c t i o n of [CpCr(NO)(0Et)]
2
with gaseous HC1 in benzene
affords the novel green [CpCr(N0)C1] complex i n high y i e l d , according to
2
eq. 21.
Without making any i m p l i c a t i o n s as to the mechanism, t h i s conversion
[CpCr(N0)(0Et)]
2
+ 2HC1
>- [CpCr(N0)C1] + 2EtOH
21.
2
can be considered as an acid-base reaction between H and O E t " , and i n
+
p r i n c i p l e , a s i m i l a r transformation should occur with any acid stronger
than EtOH.
As expected, the s u b s t i t u t i o n of the ethoxide group by the
c h l o r i d e anion causes the terminal n i t r o s y l
from 1660 to 1678 c m " .
1
s t r e t c h i n g frequency to increase
Like [CpCr(NO)(0Et)] , the chloro d e r i v a t i v e
2
is
very soluble i n common organic solvents except hexanes and, though stable
in a i r for short periods of time, i s best stored under n i t r o g e n .
The structures of [CpCr(NO)(0Et)]
2
and [CpCr(N0)C1]
expected to be s i m i l a r to those of [CpCr(NO)(SPh)]
0
(91) and
2
are
- 68 -
[CpCr(MO)(NMe )]
2
2
(92), which are characterized by symmetrically bridging
SPh and NMe ligands and by a d i s t i n c t metal-metal bond.
Both the trans
2
and c i s forms, D and E, can be i s o l a t e d .
A l s o , the s t r u c t u r e of
D
Cp Cr (N0) (NH ) shows a trans arrangement containing both n i t r o s y l and
2
2
2
2
amido bridges (94).
The Cr-N linkages of the t e r m i n a l l y coordinated
n i t r o s y l groups have considerable double bond c h a r a c t e r , while the Cr-N-0
bond angles are approximately 170°, as they are a l s o in CpCr(N0) Cl and
2
CpCr(N0) (NC0) (106, 107).
2
The dimeric nature of [CpCr(NO)(0Et)]
2
and [CpCr(N0)Cl]
confirmed by t h e i r mass spectral d a t a , shown in Table X I .
2
is
The fragmen-
t a t i o n p a t t e r n s , i n d i c a t i n g the l o s s of NO, e t h y l , ethoxo, chloro
and
Cj-H fragments, are s i m i l a r to those found i n the mass spectra of
5
[CpMo(N0)I]
2
and [CpMo(NO)(SCH Ph)] , i n which the parent ions are a l s o
2
r e l a t i v e l y abundant (95).
2
In the mass spectra of both the chloro and ethoxo
complexes the i n t e n s i t y pattern at m/e = 182 i s i n d i c a t i v e of [ C p C r ]
2
rather than the [ ( C H ) C r ]
3
3
2
2
+
or [CpCr(N0)C1] fragments.
The same
+
assignment has been made i n the cases of [CpCr(NO)(SMe)] (88) and
2
Cp Cr (N0)^(NH ) (89),
9
9
9
where [ C p C r ]
9
+
i s thought to a r i s e from the
+
- 69 -
Table XI. Low Resolution Mass Spectral Data f o r
[CpCr(N0)(0Et)]
[CpCr(N0)Cl]
2
0.4
Cp Cr Cl (N0)
2
334
3.3
Cp Cr Cl (N0)
2
304
3.4
Cp Cr Cl
182
10.0
152
2.9
CpCrCl
117
3.1
CpCr
52
2.3
Cr
384
0.6
Cp Cr (0Et) (N0)
354
6.8
Cp Cr (0Et) (N0)
324
10.0
Cp Cr (0Et)
295
1.5
Cp Cr (0Et)0
279
1.7
Cp Cr (0Et)
250
2.1
Cp Cr 0
182
6.4
Cp Cr
162
1.4
CpCr(0Et)
117
0.9
CpCr
52
1.4
Cr
a
2
2
2
2
2
2
2
2
2
+
2
+
2
Assignment
364
Assignment
2
2
Relative
Abundance
Relative
Abundance
2
a
m/e
m/e
2
[CpCr(N0)X]
+
+
2
+
2
2
2
2
2
2
^ C r and
J 0
2
+
2
+
+
+
+
+
+
The assignments involve the most common n a t u r a l l y occurring
i.e.
+
2
2
Cp Cr
2
C1.
isotopes,
- 70
fragmentation of the metastable [Cp2Cr2$2] and [Cp2Cr2(NH2)]
+
+
species
respectively.
4.3d
The a l k y l - and ary1-(n -c,yclopentadienyl ) d i n i t r o s y l
5
complexes of
chromium, molybdenum and tungsten, CpM(N0)pR (M = C r , Mo or W;
R = a.l kyl or a r y l ) .
These complexes are obtained in reasonable y i e l d s by the reaction
of the corresponding C p M ^ O ^ C l compounds with the appropriate organoaluminum reagent according to the general eq. 2 1 ,
CpM(N0) Cl + R - A l ^
2
b
- £ ! l ^ j £ ^ CpM(N0) R + C l - A l ^
r
H 5
where M = C r , Mo or W and R = a l k y l or a r y l .
2
21.
During the course of the
r e a c t i o n an o i l y red-brown s o l i d of undetermined composition i s d e p o s i t e d ,
and the exact nature of the aluminum byproduct remains to be a s c e r t a i n e d .
This synthetic procedure appears to be quite general f o r the chromium
and molybdenum compounds and i s l i m i t e d only by the a v a i l a b i l i t y of the
appropriate A l r e a g e n t .
However, i n the case of tungsten, only the
phenyl and methyl d e r i v a t i v e s are o b t a i n e d , the l a t t e r only a f t e r a
prolonged r e a c t i o n time.
Treatment of C p l ^ N O ^ C l with Et^Al or
AlH(i-Bu)2 under various reaction conditions f a i l s to give the expected
a l k y l a t e d products.
Interestingly,
in the reactions i n v o l v i n g A l H ( i - B u )
2
only the i-Bu group, rather than the hydride, i s t r a n s f e r r e d to the
metal.
The Me, Et and Ph d e r i v a t i v e s of chromium were p r e v i o u s l y
reported (27) but are obtained here in better y i e l d s .
The remaining a l k y l
- 71 -
and a r y l complexes were unknown p r i o r to t h i s work.
The physical
properties of these complexes are summarized i n Tables VII and V I I I .
A l l the complexes are very soluble i n organic sol v e n t s , i n c l u d i n g
hexanes, and may be exposed to a i r f o r short periods of time without
decomposition.
Those compounds which are s o l i d s at room temperature
_q
sublime r e a d i l y at 30-40°C (5 x 10 mm) without decomposition and a r e ,
indeed, best p u r i f i e d i n t h i s manner.
However, the ones which are
l i q u i d s a t room temperature decompose slowly under nitrogen but may be
stored f o r many months a t -5°C or lower.
The methyl and phenyl d e r i v a -
t i v e s , e s p e c i a l l y those of chromium, are the most thermally stable and
melt without decomposition.
The i n f r a r e d spectra (Table V I I I ) of a l l the complexes d i s p l a y
two strong a b s o r p t i o n s , s i m i l a r to those in CpM(N0) Cl, due to terminal
2
n i t r o s y l N-0 s t r e t c h i n g s .
As expected, these s t r e t c h i n g frequencies
decrease i n the order Cl > Ph > a l k y l which p a r a l l e l s the a b i l i t y of these
ligands to withdraw e l e c t r o n i c charge from the c e n t r a l metal.
The proton NMR spectra (Table V I I I ) e x h i b i t a sharp s i n g l e t f o r
the C^Hg protons as well as the expected patterns and i n t e g r a t i o n
f o r the a l k y l and a r y l protons.
ratios
Not too s u r p r i s i n g l y , the p o s i t i o n of
the Cp resonance remains nearly independent of the c h l o r o , a l k y l or a r y l
substituent present f o r any given metal.
Any e l e c t r o n i c
perturbation
about the central metal i s buffered by the presence of the strong T T accepting n i t r o s y l l i g a n d s .
In f a c t , the N-0 s t r e t c h i n g frequencies do
vary s i g n i f i c a n t l y , as mentioned, and hence provide a more s e n s i t i v e
measure f o r detecting changes i n e l e c t r o n i c d i s t r i b u t i o n around the central
Figure2.
*H
N M R of
(T7 -C H )MO(N0) CH CH
5
5
5
2
2
3
Experimental spectrum
T
I
1
1
1
1
1
1
1
1
1
1
1
1
1
1
1
"
1
1
1
1
1
1
1
1
1
1
1
1
1
1
r
1
1
1
r
- 73 -
metal.
The presence of the a-bonded ethyl group in CpMo(N0) Et i s
2
unequivocally demonstrated by the computer f i t t i n g of the A B
2
3
multiplet
due to the a l k y l proton resonance, from which the f o l l o w i n g parameters
can be extracted (Figure 2 ) :
x(CH )
2
8.10,
T(CH )
3
8.26,
J(CH ,
2
CH )
3
7.54 Hz^.
The mass spectral data for each complex in Table XII
the molecular ion as the highest-mass fragment.
exhibit
The fragmentation
patterns f o r the Et and i-Bu d e r i v a t i v e s of molybdenum are s i m i l a r , but
assignments are complicated by concomitant fragmentation of the a l k y l
groups and by the large number of molybdenum i s o t o p e s .
The most s t r i k i n g
d i f f e r e n c e between the phenyl and methyl complexes i s the systematic
absence of the [CpM(N0)] and [CpM(N0) ]
+
+
2
fragments in the phenyl compounds,
and the absence of [ ( C H ) M R ] and [CpMR] i n the methyl d e r i v a t i v e s .
+
3
+
3
These data strongly suggest that the metal-carbon bond i s stronger in the
phenyl than in the methyl compounds, and that the trend i s independent of
the metal.
The major d i f f e r e n c e among the metals i s that the chromium-
containing complexes show an abundance of C r
+
and [ C p C r ]
+
fragments
whereas, f o r the molybdenum- and tungsten-containing analogs, the c o r r e s ponding fragments are much l e s s abundant.
The two most general preparative routes to a-bonded a l k y l and a r y l
complexes involve the n u c l e o p h i l i c displacement of the halide from e i t h e r
the t r a n s i t i o n metal complex, eq. 22, or from an a l k y l or a r y l h a l i d e ,
We thank Ms. V. Gibb f o r assistance in obtaining these s p e c t r a .
Table XII. Mass Spectral Fragmentation Data f o r CpM(N0) R.
9
. b
ion
Compound
M
+
C
5 5
H
M +
C H M(N0)
5
5
+
C H M(N0)
5
5
+
2
C
3 3
H
M R +
C H MR
5
5
+
C H M(N0)R
5
5
+
C H M(N0) R
5
5
10.0
8.3
-
-
-
2.3
7.5
0.7
-
-
-
-
2.7
3.6
8.2
3.6
CpW(N0) C H -
-
.1-7
-
-
4.2
7.2
10.0
5.6
CpCr(N0) CH
3
10.0
8.4
2.7
1.2
-
2.1
7.2
1.6
CpMo(N0) CH
3
5.7
3.6
10.0
6.2
-
-
6.2
6.0
3
0.4
-
2.9
3.4
-
-
4.6
6.2
CpCr(N0) Et
10.0
2.7
1.9
2.0
-
1.9
5.6
0.4
CpCr(NO) i-Bu
10.0
9.9
3.5
4.1
7.2
-
9.9
2.9
CpCr(N0) C H
5
CpMo(N0) C H
5
2
g
2
2
6
g
5
2
2
CpW(N0) CH
2
2
2
+
2
Entries i n t h i s t a b l e are r e l a t i v e ion i n t e n s i t i e s with the most intense metal containing ion assigned an
a r b i t r a r y value of 10.0
R e l a t i v e i n t e n s i t i e s were evaluated by measuring peak heights corresponding to the
most abundant isotope of each metal:
5 2
Cr,
98
M o and
1 8 4
W.
Only unambiguously assignable ions are g i v e n ; f o r the p o l y i s o t o p i c molybdenum and tungsten overlapping of
some medium to strong i n t e n s i t y peaks i n the lower mass range made assignments d i f f i c u l t .
Selected compounds
were run on the MS902 high r e s o l u t i o n instrument which measured exact masses of a l l major peaks; c a l c u l a t e d
and measured values were in good agreement.
- 75 -
eq.
23, (112).
The synthetic route given by eq. 23 cannot be e x p l o i t e d
L M-X + R-M
is— L MR + M'X
1
n
(M
1
22.
n
= Li or MgX)
[ L M ] " + R-X
» - L MR + X"
n
23.
n
in t h i s instance since a l l attempts to obtain the [ C p M ^ O ^ ] " anions
have thus f a r f a i l e d .
Indeed, the only well c h a r a c t e r i z e d organometallic
n i t r o s y l anion which has been s u c c e s s f u l l y used as a synthetic precursor
i s [Fe(C0) (N0)]" (53, 113, 114).
3
While eq. 22 has been applied with
modest success in the p r e p a r a t i o n ' o f C p C r ^ O ^ R (R = Me, Et or Ph) (27),
the s i m i l a r reactions of C p M ^ O ^ C l
(M = Mo or W) with various
alkyllithium
and Grignard reagents f a i l to y i e l d even s p e c t r o s c o p i c a l l y detectable
q u a n t i t i e s of the corresponding a l k y l or a r y l d e r i v a t i v e s .
These reagents
are not s u f f i c i e n t l y s e l e c t i v e in the n u c l e o p h i l i c displacement of the
halide and they appear to attack other f u n c t i o n a l groups.
In c o n t r a s t ,
we f i n d the organoaluminum reagents to be p a r t i c u l a r l y mild and s e l e c t i v e
in t h e i r reactions with C p M ^ O ^ C l complexes.
This feature i s a l s o
i l l u s t r a t e d by the e x c l u s i v e monoalkylation of some d i c h l o r o compounds
of ruthenium even when an excess of the organoaluminum reagent i s used at
elevated temperatures
(115).
Organoaluminum compounds are r e l a t i v e l y strong Lewis acids which
are known to coordinate to the oxygen end of carbonyl ligands i n a
v a r i e t y of neutral
complexes.
(116) and anionic (117) n -cyclopentadienylmetal carbonyl
5
Non-bonding e l e c t r o n p a i r s l o c a l i z e d on the metal atom may also
- 76 -
serve as a Lewis base s i t e and a number of 1:1 adducts containing the M-Al
bond have been i s o l a t e d (118).
However, no i n f r a r e d spectroscopic
evidence was obtained in support of any s i m i l a r Lewis acid-base
adducts during our preparation of the C p M ^ O ^ R complexes.
Although useful in the syntheses of main group metal a l k y l and
a r y l compounds (119), the organoaluminum reagents have received very
l i t t l e a t t e n t i o n i n the formation of t r a n s i t i o n metal-carbon bonds (112).
The general r e a c t i o n of organoaluminum compounds with metal halides (eq. 24)
R-Al
+ M-X
*~
A l - X + M-R
24.
i s favored when M i s s u b s t a n t i a l l y e l e c t r o p o s i t i v e and X i s e l e c t r o n e g a t i v e .
The conversion does not l i k e l y take place by a d i s s o c i a t i v e and
r e a s s o c i a t i v e i o n i c pathway since the organoaluminum compounds are l a r g e l y
covalent.
A l s o , the reactions are t y p i c a l l y performed i n solvents which
do not support i o n i z a t i o n and, indeed, the use of coordinating solvents
or the presence of Lewis bases retard the r e a c t i o n .
Consistent with the
above f a c t s , the present a l k y l a t i o n reactions can be considered to occur
v i a a Lewis acid-base intermediate as in III
or IV, where M i s an organo-
metallic residue.
R
M
X
X
EZ
+
- 77 -
If M i s s u f f i c i e n t l y e l e c t r o p o s i t i v e , as i s the case with the
a l k a l i metals, III
and IV are formed as stable products rather than
intermediates (119).
Since the CpM(N0) Cl complexes d i s p l a y a c e r t a i n
2
i n c l i n a t i o n to form the c a t i o n i c [CpM(N0) ]
+
2
s p e c i e s , the benzene
i n s o l u b l e byproduct deposited during the syntheses of CpM(N0) R may
2
reasonably be formulated as [CpM(N0) ]
2
+
[AlR^Cl]".
However, t h i s
p o s s i b i l i t y was not i n v e s t i g a t e d .
In contrast to the numerous CpCr(N0) R complexes, the only known
2
i s o e l e c t r o n i c t r i c a r b o n y l analog i s the thermally unstable CpCr(C0) Me'
3
compound.
However, the CpM(C0) R (M = Mo or W) compounds are more
3
abundant and have been the subject of considerable study.
Notably,
these complexes undergo a thermal transformation to y i e l d [CpM(C0) ]
3
(120, 121) or, when R = E t , Ph or CHpPh, [RC H M(C0) 1
5
4
3
2
(121, 122).
2
Our
attempts to obtain s i m i l a r l y the analogous, but as yet unknown, [CpM(N0) ]
2
2
(M = Mo or W) dimers by the thermolysis of the a l k y l - or a r y l d i n i t r o s y l
complexes have been u n s u c c e s s f u l .
Moreover, the n i t r o s y l complexes are
understandably more i n e r t toward s u b s t i t u t i o n than the i s o e l e c t r o n i c
CpM(C0) R analogs, and no r e a c t i o n p a r a l l e l to carbonyl " i n s e r t i o n " has
3
yet been found.
On the other hand, both CpCr(N0) R and CpM(C0) R undergo
2
3
i n s e r t i o n of S 0 a f f o r d i n g d e r i v a t i v e s which contain the S - s u l f i n a t e
2
l i n k a g e , M-'s'-R (123-125).
8
The rates of i n s e r t i o n into the n i t r o s y l
containing complexes a r e , by f a r , the greatest among the v a r i e t y of
compounds studied (126).
Infrared spectroscopic evidence (127) and k i n e t i c
studies (126) i n d i c a t e that S 0 i n s e r t i o n occurs with the
9
initial
- 78 -
II
formation of the O - s u l f i n a t e linkage M-OSR followed by rearrangement to
the S - s u l f i n a t e compound.
S i m i l a r l y , (CN) C=C(CN) reacts with the
2
above carbonyl and n i t r o s y l
2
complexes to give the d i r e c t
insertion
product, MC(CN) C(CN) R, and the keteniminato d e r i v a t i v e , MN=C=C(CN)C(CN) R
2
2
2
(124, 128).
Stannous c h l o r i d e and bromide i n s e r t i n t o the metal-carbon bond
a l k y l c a r b o n y l ( n - c y c l o p e n t a d i e n y l ) m e t a l complexes (M-R)
of a v a r i e t y of
5
to y i e l d MSnRX , MSnX^ and MX, depending on r e a c t i o n conditions
(129).
2
We f i n d that reaction of S n C l
of CpMo(N0) Cl e x c l u s i v e l y .
2
2
with CpMo(N0) i-Bu r e s u l t s i n the formation
2
However, S n C l
2
does i n s e r t i n t o the metal-
c h l o r i n e bond of CpCr(N0) Cl according to the solvent dependent e q u i l i b r i u m ,
2
eq. 25.
In donor solvents such as THF the l e f t hand side of eq. 25 i s
CpCr(N0) Cl + SnCl =^===^== CpCr(N0) SnCl
2
2
2
25.
3
favored probably due to Lewis acid-base i n t e r a c t i o n between S n C l
s o l v e n t , whereas CH C1
2
CpCr(N0) SnCl 2
1825 c m
- 1
and benzene s h i f t the e q u i l i b r i u m to the side of
2
The i n f r a r e d n i t r o s y l
3
i n CH C1 ) of CpCr(N0) SnCl
2
and the
2
2
2
s t r e t c h i n g frequencies (1733 and
are approximately 20 c m
3
-1
higher
than those of C p C r ( N 0 ) C l , which we a t t r i b u t e to the p r e v i o u s l y demonstrated
2
poorer o-donating and stronger ^-accepting properties of the SnCl^
entity
4.3e
(26a).
Bis[(n -cyclopentadienyl)dinitrosylchromium], [CpCr(N0) ]Q,
5
2
When a benzene s o l u t i o n containing CpCr(N0) Cl i s allowed to
2
s t i r over a sodium amalgam, [ C p C r ( N 0 ) ]
9
o
i s obtained i n over 60% y i e l d .
- 79 -
This synthetic route i s f a r superior to the one previously reported in
which the desired product i s extracted in only 5% y i e l d (90).
The same
conversion can be effected by using a zinc amalgam and, to a l e s s e r
e x t e n t , with f i n e l y divided magnesium or ytterbium metal although a
much longer reaction time i s necessary.
In these l a t t e r reactions no
evidence i s obtained f o r the existence of a c t i v a t e d species (105) such
as "CpCr(N0) MgCl".
When f i n e l y divided z i n c metal i s employed, no
2
reaction occurs and the use o f a magnesium amalgam in THF causes
immediate decomposition of the s t a r t i n g chloro complex.
Correspondingly,
the attempted reduction of the CpM(N0) Cl (M = Mo or W) compounds using
2
a wide v a r i e t y of reaction conditions f a i l s to y i e l d the s t i l l
[CpM(N0) ]
2
2
unknown
(M = Mo or W) complexes.
The o r i g i n a l synthesis of [ C p C r ( N 0 ) J from CpCr(N0) Cl and
2
2
2
NaBH^ i s believed to occur through the formation of CpCr(N0) H although
2
no evidence f o r t h i s hydrido intermediate was obtained (90).
However, we
f i n d that CpMo(N0) Cl reacts with NaBH^ i n MeOH or EtOH at -30°C to y i e l d
2
a v o l a t i l e pale-green s o l i d which decomposes r a p i d l y at room temperature.
Its i n f r a r e d spectrum (1740 and 1645 cm" i n EtOH) bears a s t r i k i n g
1
resemblance to those of the a l k y l and a r y l d e r i v a t i v e s , and on t h i s basis
the complex i s formulated as CpMo(N0) H.
The thermal or o x i d a t i v e
2
degradation of t h i s s p e c i e s , e i t h e r in s o l u t i o n or i n the s o l i d , f a i l s
to provide the [CpMo(N0) J compound.
2
2
The i n f r a r e d spectrum of [ C p C r ( N 0 ) ]
2
2
e x h i b i t s two strong
n i t r o s y l absorptions at 1667 and 1512 cm" i n CH^Cl > i n d i c a t i v e of both
1
2
terminal and bridging n i t r o s y l
ligands.
In CDCl^ s o l u t i o n s the dimer
e x i s t s i n both the c i s and trans forms, F and G, as evidenced by i t s
- 80 -
F
G
room temperature proton NMR which contains two sharp s i n g l e t s at T5.20
and T5.00 i n the r a t i o of 1:20 (93).
(93),
In the s o l i d s t a t e , [ C p C r ( N 0 ) l
2
i s i s o s t r u c t u r a l with t r a n s - [ C p F e ( C 0 ) o ]
[CpCr(N0)(SPh)]
2
(91)
2
2
(130), t r a n s -
and trans-[CpCr (NO) ( N H j ] , , (92).
The metal-metal
o
distance of 2.615 A in the d i n i t r o s y l dimer i s s i g n i f i c a n t l y shorter than
those of the SPh (2.90 A) and NH (2.67 A) containing s p e c i e s .
While
2
the terminal Cr-N-0 bond angles in the SPh and NH compounds deviate
2
from l i n e a r i t y by about 1 0 ° , ' t h e corresponding angle in the d i n i t r o s y l
species i s v i r t u a l l y
l i n e a r ( 1 7 6 . 6 ° ) , p o s s i b l y as a r e s u l t of reduced
e l e c t r o n density on the metal atoms of the n i t r o s y l
bridged compound.
The
terminal Cr-N bond lengths f o r a l l three dimers f a l l w i t h i n the range of
o
o
1.64-1.69 A and are not much d i f f e r e n t from the value of 1.71 A in the
CpCr(N0) Cl compound (106).
2
As expected, the Cr-N distance (1.960 A) in
the symmetrically-bridging n i t r o s y l
ligands i s considerably longer than
the terminal Cr-N bonds.
The mass spectrum of [ C p C r ( N 0 ) l
2
2
(Table X I I I ) d i s p l a y s the
molecular ion peak and i s c o n s i s t e n t with the dimeric f o r m u l a t i o n .
no [CpCr(N0) ]
n
or [C^H^Cr(N0) ]
n
(n = 1 or 2) fragments are formed,
Since
it
- 81 -
Table X I I I .
Low Resolution Mass Spectral Data f o r
[CpCr(N0) ]
9
9
m/e
Relative
Abundance
354
3.1
Cp Cr (N0)
4
324
2.2
Cp Cr (N0)
3
294
0.3
Cp Cr (N0)
2
264
10.0
Cp Cr (N0)
182
6.1
(C H ) Cr
117
2.3
CpCr
52
4.2
Cr
The assignments involve the most common n a t u r a l l y
52
i.e.
Cr.
r
Assignment
2
2
2
2
2
2
2
+
+
+
+
2
3
3
2
+
2
+
+
occurring
isotope,
- 82 -
appears that fragmentation prefers to occur without rupture of the
metal-metal bond.
In c o n t r a s t , the i s o e l e c t r o n i c non-bridged [CpCrv^CO)^]
dimer does not d i s p l a y a molecular ion peak and, indeed, only mononuclear
fragments are found (131).
Unlike [ C p C r ( N 0 ) ( 0 E t ) ]
contrary to a previous report
[CpCr(N0) ]
2
[Cp Cr]
2
+
2
2
or [CpCr(N0)C1] , and
2
(89), we f i n d that the mass spectrum of
does not contain a peak which may be assigned to the
fragment.
Instead we assign the peak at m/e = 182 to the
d i n u c l e a r [ ( C ^ H ^ ^ C r ^ * fragment.
Furthermore, the greater abundance
of dinuclear species i n the mass spectrum of [ C p C r ( N 0 ) l suggests that
2
the n i t r o s y l
2
ligand forms a stronger bridge than do the OEt or Cl
ligands in r e l a t e d compounds.
- 83 -
CHAPTER V
Conclusion
It should be stressed that while m e t a l - n i t r o s y l
bonds may be
formed by a v a r i e t y of synthetic methods,' none o f these preparative
routes has general a p p l i c a b i l i t y .
In c o n t r a s t , t h i s work has shown
n i t r o s y l c h l o r i d e to be the s i n g l e most v e r s a t i l e s y n t h e t i c reagent
a v a i l a b l e for the formation of M-NO l i n k a g e s .
The d i r e c t formation of
c h l o r o n i t r o s y l complexes from neutral carbonyl compounds i s of p a r t i c u l a r
interest.
As discussed in t h i s t h e s i s , the former compounds, which
contain a very r e a c t i v e m e t a l - c h l o r i n e bond, serve as useful precursors
in the preparation of f u r t h e r n i t r o s y l d e r i v a t i v e s .
For example, the
metal-halide bond may be converted to a large v a r i e t y of metal-heteroatom
a-bonds containing elements from Groups IVA, VA and VIA.
Obviously, the behavior of n i t r o s y l c h l o r i d e with many more
neutral and anionic metal carbonyl complexes remains to be i n v e s t i g a t e d .
Most importantly, such studies should be undertaken with the aim of
developing convenient synthetic routes to n i t r o s y l complexes, and not
simply as a study of the chemistry o f n i t r o s y l
chloride.
The s c a r c i t y of anionic n i t r o s y l complexes i s s u r p r i s i n g ,
e s p e c i a l l y in view of the multitude of anionic carbonyl compounds which
- 84 -
has been c h a r a c t e r i z e d .
of various n i t r o s y l
While attempts to e f f e c t the chemical reduction
compounds have so f a r been u n s u c c e s s f u l , n i t r o s y l -
containing anions might be obtained by electrochemical reduction at a
controlled potential.
Further work in t h i s d i r e c t i o n i s c u r r e n t l y
in
progress.
While numerous n i t r o s y l compounds are known, many, which are
expected to be stable under ambient c o n d i t i o n s , are simply nonexistent at
present.
For i n s t a n c e , i n the "pseudo i r o n - c a r b o n y l " s e r i e s , Fe(C0)g,
Mn(C0) (N0), C r ( C 0 ) ( N 0 )
4
3
yet to be reported.
2
and V ( C 0 ) ( N 0 )
2
3>
the l a s t two compounds have
The synthesis of such unknown species should be
endeavored i n order to enhance our knowledge of p o t e n t i a l l y useful
complexes.
nitrosyl
S p e c i f i c a l l y , the preparation of these compounds should not
be viewed as an end i n i t s e l f .
Rather, future studies should be d i r e c t e d
at e s t a b l i s h i n g the comparative chemistry between s i m i l a r n i t r o s y l and
carbonyl compounds and the p r a c t i c a l a p p l i c a t i o n of n i t r o s y l complexes
in various organic syntheses.
- 85 -
REFERENCES
1.
W.P. G r i f f i t h , Adv. Organomet. Chem., 7, 211 (1968).
2.
B.F.G. Johnson and J . A . McCleverty, Progr. Inorg. Chem., 7_, 277 (1966).
3.
N.G. Connelly, Inorg. Chem. Acta R e v . , 6, 47 (1972).
4.
(a) J . A . McGinnety,
229 (1972).
HTP Int.
Rev. S c i . , Inorg. Chem., Ser One, 5_,
(b) B.A. Frenz and J . A . I b e r s , HTP Int.
One, 11, 33 (1972).
5.
Rev. S c i . , Phys. Chem., Ser.
(a) J . B . Raynor, Inorg. Chim. A c t a , 6_, 347 (1972).
(b) R.D. Harcourt and J . A . Bowden, J . Inorq. N u d . Chem., 36_, 1115
(1974).
(c) Chan-Cheng Su and J.W. F a l l e r , J . Organomet. Chem., 84, 53 (1975).
6.
J . P . Candlin and W.H. Janes, J . Chem. Soc. C, 1856 (1968).
7.
J . P . C o l l man, N.W. Hoffman and D.E. M o r r i s , J . Am. Chem. S o c , 91_,
5659 (1969).
8.
W.B. Hughes, Adv. Chem. S e r . , 132_, 192 (1974).
9.
K.G. Caulton, Coord. Chem. R e v . , T4, 317 (1975).
10.
S. Trofimenko, Inorg. Chem., 8, 2675 (1969).
11.
(a) R.B. K i n g , Adv. Organomet. Chem., 2, 157 (1964).
(b) J . E . E l l i s , J . Organomet. Chem., 86, 1 (1975).
12.
D.D. P e r r i n , W.L.F. Armarego and D.R. P e r r i n , " P u r i f i c a t i o n
Laboratory Chemicals," Pergamon P r e s s , Oxford, 1966.
of
13.
William J . J o l l y , "The Synthesis and C h a r a c t e r i z a t i o n of
Compounds", P r e n t i c e - H a l l I n c . , New J e r s e y , 1970.
14.
G. Pass and H. S u t c l i f f e , " P r a c t i c a l Inorganic Chemistry", Chapman and
H a l l , London, 1968, pp. 145-146.
15.
J . Jander and U. Engelhardt, "Developments in Inorganic Nitrogen
Chemistry", V o l . 2, C.B. Colburn E d . , E l s e v i e r S c i e n t i f i c Publishing
Co. New York, N . Y . , 1973, p. 141-155.
16.
J . K . Ruff and W.J. S c h l i e n t z , Inorg. Synth. 1_5, 84 (1974).
Inorganic
- 86 -
17.
J . K . Ruff, Inorg. Chem., 7, 1818 (1968).
18.
P.M. T r e i c h e l , E. P i t c h e r , R.B. King and F.G.A. Stone, J . Am. Chem.
S o c , 83, 2593 (1961 ).
19.
W. Hieber and G. Wagner, Z. N a t u r f o r s c h . , 12b 478 (1957); 13b, 339
(1958).
20.
A . N . Nesmeyanov, K.N. Anisimov, N.E. Kolobova and V . N . Khandozhko,
Dokl. Chem. (Eng. T r a n s l . ) , 156, 502 (1964).
21.
H. Behrens and R. Weber, Z. Anorg. A l l g . Chem., 28J_, 190 (1955).
22.
E.O. Fischer and R. Bottcher, Z. N a t u r f o r s c h . , 10b, 600 (1955).
23.
C.G. Barraclough and J . Lewis, J . Chem. S o c , 4842 (1960).
24.
R.D. G o r s i c h , J . Am. Chem. S o c , 8_4, 2480 (1962).
25.
D.S. F i e l d s and M . J . Newlands, J . Organomet. Chem., 27, 213 (1971).
26.
(a) W. J e t z , P. Simons, J . Thompson and W.A.G. Graham, Inorg. Chem.,
5, 2217 (1966).
(b) J . A . J . Thompson and W.A.G. Graham, i b i d . , 6, 1875 (1967).
27.
T . S . Piper and G. W i l k i n s o n , J . Org. N u c l . Chem., _3, 104 (1956).
28.
K.W. Barnett and D.W. Slocum, J . Organomet. Chem., 44_, 1 (1972).
29.
R.B. King, "Organometallic Syntheses", V o l . 1, Academic P r e s s , New
York, N . Y . , 1965, pp. 167-168.
30.
C.G. Barraclough, J . A . Bowden, R. Colton and C . J . Commons, Aust. J .
Chem., 26, 241 (1973).
31.
E.W. A b e l , I.S.
32.
C P . Casey, S.W. Polichnowski and R.L. Anderson, J . Am. Chem. S o c ,
97, 7375 (1975).
33.
R.D. Wilson and R. Bau, J . Am. Chem. S o c , 96, 7601 (1974) and
references t h e r e i n .
34.
W.E. Swartz, J r . , J . K . Ruff and D.M. Hercules, J . Am. Chem. S o c , 94,
5227 (1972).
35.
E.W. Abel and G. W i l k i n s o n , J . Chem. S o c , 1501 (1959).
36.
R . J . A n g e l i c i and M.D. Malone, J . Inorg. Chem., 9, 1731
Butler and J . G . R e i d , J . Chem. S o c , 2068 (1963).
(1967).
- 87 -
37.
J . K . Ruff,
Inorg. Chem., 8, 86 (1969).
38.
W.J. S c h l i e n t z , Y. Lavender, N. Welcman, R.B. King and J . K . R u f f ,
J . Organomet. Chem., 33, 357 (1971).
39.
W. Schlientz-and J . K . Ruff, Syn. Inorg. Metal-Org. Chem., 1_, 215
(1971) and references t h e r e i n .
40.
J . K . Ruff, Inorg. Chem., 7_, 1821
41.
W.J. S c h l i e n t z and J . K . Ruff,
42.
R.B. King, Acc. Chem. R e s . , 3_, 417
43.
D . J . M i l l e n and J . P a n n e l l , J . Chem. S o c , 1322 (1961 ).
44.
F.A. Cotton and G. W i l k i n s o n , "Advanced Inorganic Chemistry", 3rd e d . ,
John Wiley and Sons, I n c . , 1972, p. 356.
45.
A.M. Bond and R. C o l t o n , Inorg. Chem., 1_5, 446
46.
(a) B . J . Hathaway and A . E . U n d e r h i l l , J . Chem. S o c , 2705
(b) B . J . Hathaway, D.G. Holah
(1968).
Inorg. Chem., 11_, 2265 (1972).
(1970).
(1976).
(1960).
and A . E . U n d e r h i l l , i b i d . , 2444
(c) R.F. Schramm and B.B. Wayland, Inorg. Chem., 8, 971
(1962).
(1969).
(d) E.K. B a r e f i e l d and D.H. Busch, J . Chem. S o c , Chem. Comm., 522
(1970).
(e) J . J . Fowler and J . K l e i n b e r g , Inorg. Chem., 9, 1005
(1970).
(f) R.H. Reimann and E. S i n g l e t o n , J . Organomet. Chem., 32_, C44 (1971 )
(g) N.G. Connelly and J . D . D a v i s , i b i d . , 38, 385 (1972).
(h) R.H. Reimann and E. S i n g l e t o n , J . Chem. S o c , Dalton T r a n s . ,
2658 (1973).
(i)
E.E. Isaacs and W.A.G. Graham, J . Organomet. Chem., 99_, 119 (1975)
47.
W. Hieber, Z. Anorg. A l l g . Chem., 204, 165 (1932).
48.
R.B. King and F.G.A. Stone, Inorg. S y n t h . , 7_, 193 (1963).
49.
J . R . C h i p p e r f i e l d , J . Ford and D.E. Webster, J . Oraanomet. Chem., 102,
417 (1975).
50.
A . N . Nesmeyanov, K.N. Anisimov, N.E. Kolobova and V . N . Khandozhko,
Dokl. Akad. Nauk. S . S . S . R . , 156, 383 (1964).
51.
R . E . J . B i c h l e r , H.C. C l a r k , B.K. Hunter and A . T . Rake, J . Organomet.
Chem., 69, 367 (1974).
- 88 -
52.
W. Hieber and H. Beutner, Z. Anorg. A l l g . Chem., 320_, 101 (1963).
53.
F. S t e e l , i b i d . , 269, 40 (1952).
54.
R.B. King, M.S. Saran and S . P . Anand, Inorg. Chem., 1_3, 3038 (1974).
55.
M. Andrew, D.L. Lipton and S.W. K i r t l e y , J . Chem. S o c , Chem. Comm.,
181 (1973).
56.
J . P . Olsen, T . F . K o e t z l e , S.W. K i r t l e y , M. Andrews, D.L. Tipton and
R. Bau, J . Am. Chem. S o c , 96_, 6621 (1974).
57.
R. Colton and C . J . Commons, Aust. J . Chem., 1487 (1973).
58.
R. Colton and C . J . Commons, i b i d . , 1493 (1973).
59.
R.P. Stewart, J r . and G.T. Moore, Inorg. Chem., T4, 2699 (1975).
60.
(a) B.F.G. Johnson and J . A . S e g a l , J . Organomet. Chem., 31_, C79 (1971 ).
(b) B.F.G. Johnson, S. Bhaduri and M.G. Connelly, i b i d . , 40, C36 (1972)
(c) B.F.G. Johnson and J . A . S e g a l , J . Chem. S o c , Dalton T r a n s . ,
1268 (1972).
(d) M. Green and S.H. T a y l e r , i b i d . , 2629 (1972).
(e) N.G. C o n n e l l y , i b i d . , 2183 (1973).
(f) F. Faraone, R. P i e t r o p a o l o , G.G. T r o i l o and P. P i r a i n o , Inorg.
Chim. A c t a , 7, 729 (1973).
61.
G.R. Crooks and B.F.G. Johnson, J . Chem. S o c A , 1238 (1968).
62.
Z . Iqbal and T . C . Waddington, i b i d . , 2958 (1968).
63.
F. Z i n g a l e s , A. T r o v a t i , F. C a r i a t i and P. U g u a g l i a t i , Inorg. Chem.,
10, 507 (1971).
64.
F.A. Cotton and B.F.G. Johnson, Inorg. Chem., 3 , 1609 (1964).
65.
B.F.G. Johnson, J . Chem. S o c A, 475 (1967).
66.
M. Deane and F . J . L a l o r , J . Organomet. Chem., 5_7, C61 (1973).
67.
W. Hieber and W. Beck, Z. N a t u r f o r s c h . , Ub_, 194 (1958).
68.
(a) T . S . Piper and G. W i l k i n s o n , J . Inorg. Nucl. Chem., 1_, 165 (1955).
(b) E.O. Fischer and E. Moser, Inorg. S y n t h . , 1_2, 36 (1970).
69.
A. Jahn, Z. Anorg. A l l g . Chem., 301_, 301 (1959).
- 89 -
70.
W. Hieber and A. Jahn, Z. N a t u r f o r s c h . , 13b, 195 (1958).
71.
W. Hieber and R. Nast, Z. Anorg. A l l g . Chem., 244, 23 (1940).
72.
(a) Z. Iqbal and T . C . Haddington, J . Chem. Soc. A , 1238 (1968).
(b) D.W. McBride, S . L . Stafford and F.G.A. Stone, Inorg. Chem., 1_,
386 (1962).
73.
R. D a v i s , B . F . G . Johnson and K.H. A l - O b a i d i , J . Chem. S o c ,
Dalton T r a n s . , 508 (1972).
74.
J . K . Hoyano, unpublished r e s u l t s .
75.
W. Hieber, R. Schuh and H. Fuchs, Z. Anorg. A l l g . Chem., 28_4, 243
(1941).
76.
W. Hieber and R. M a r i n , Z. Anorg. A l l g . Chem., 240, 241 (1939).
77.
E.O. F i s c h e r , 0. Beckert, W. Hafner and H.O. S t a h l , Z. N a t u r f o r s c h . ,
10b, 598 (1955).
78.
E.O. Fischer and K. P l e s s k e , Chem. B e r . , 94, 93 (1961).
79.
R.B. King and F.G.A.
80.
R.B. King and M.B. B i s n e t t e , Inorg. Chem., 6, 469 (1967).
81.
A . E . Crease and P. Legzdins, J . Chem. S o c , Dalton T r a n s . , 1501
(1973).
82.
T . S . Piper and G. W i l k i n s o n , Inorg. N u c l . Chem., 2, 38 (1956).
83.
R.B. K i n g , Inorg. Chem., 7, 90 (1968).
84.
M.L.H. Green, T.R. Sanders and R.N. Whiteley, Z. N a t u r f o r s c h . ,
23b, 106 (1968).
85.
M.A. Jennings and A. W o j c i c k i , J . Organomet. Chem., j_4, 231 (1968).
86.
E.O. Fischer and H. Strametz, J . Organomet. Chem., j_0, 323 (1967).
87.
M. Ahmad, R. Bruce and G. Knox, Z. N a t u r f o r s c h . , 2fb, 289 (1966).
88.
J . F . Preston and R.L. Reed, J . Chem. S o c , Chem. Comm., 51 (1966).
89.
N. F l i t c r o f t ,
90.
R.B. King and M.B. B i s n e t t e , Inorg. Chem., 4, 791 (1964).
Stone, Inorg. S y n t h . , 7_, 99 (1963).
J . Organomet. Chem., 1_5, 254 (1968).
- 90 -
91.
A.T. McPhail and G.A. Sim, J . Chem. Soc. A, 1858 (1968).
92.
M.A. Bush and G.A. Sim, J . Chem. Soc. A,
93.
J . L . Calderon, S. Fontana, E. Frauendorfer
Organomet. Chem., 64, CIO (1974).
94.
L . Y . Y . Chan and F.W. B. E i n s t e i n , Acta C r y s t . , 26b, 1899 (1970).
95.
T.A. James and J . A . McCleverty, J . Chem. Soc. A, 1068 (1971).
96.
R.B. King, Inorg. Chem., 6, 30 (1967).
97.
R.B. King and R.N. Kapoor, J . Organomet. Chem., ]_5, 457 (1968).
98.
F.A. Cotton, A. Musco and G. Yagupsky, J . Am. Chem. S o c , 89,
6136 (1967).
99.
E.O. F i s c h e r , Angew. Chem., 67, 475 (1955).
611 (1970).
and V.W. Day, J .
100.
F.A. Cotton and P. Legzdins, J . Am. Chem. S o c , 90, 6232 (1968).
101.
J . L . Calderon, F.A. Cotton and P. Legzdins, i b i d . , 91_, 2528
(1969).
102.
F.A. Cotton and G.A. Rusholme, i b i d . , 94, 402 (1972).
103.
W.G. K i t a , M.K. Lloyd and J . A . McCleverty, J . Chem. S o c , Chem.
Comm., 420 (1971).
104.
J . A . McCleverty and D. Seddon, J . Chem. S o c , Dalton T r a n s . ,
(1972).
105.
(a) J . M . B u r l i t c h and S.W. Ulmer, J . Organomet. Chem., 1_9, P21
(1969)
2526
b) A . E . Crease and P. Legzdins, J . Chem. S o c , Chem. Comm., 775
1973).
(c)
A . E . Crease, Ph.D. Thesis (1973).
(d) A . E . Crease, P. Legzdins and E.R. Sigurdson, Ann. New York
Acad. S c i . , 239, 129 (1974).
106.
O.L. C a r t e r , A.T. McPhail and G.A. Sim, J . Chem. Soc. A, 1095
(1966).
107.
M.A. Bush and G.A. Sim, i b i d . , 605 (1970).
108.
R.E. Winters and R.W. K i s e r , J . Phys. Chem., 69, 3198 (1965).
- 91 -
109.
E. Schumacher and R. Taubenest, Helv. Chim. A c t a , 49, 1447 (1966).
110.
R.B. King and N.A. Welcman, Inorg. Chem., 8, 2540 (1969).
111.
B.V. Lokshin, E.B. Rusach, N.E. Kolobova, Yu. V. Makarova, N.A.
Ustynyuk, V . I . Zdanovich, A . Z h . Zhakaeva and V . N . S e t k i n a , J .
Organomet. Chem. 108, 353 (1976).
112.
(a) G.W. P a r s h a l l and J . J . Mrowca, Adv. Organomet. Chem., 7_, 157
(1968).
(b) P . S . Braterman and R . J . Cross, Chem. Soc. R e v . , 2, 271
(1973) .
(c)
F. Calderazzo, Pure A p p l . Chem., 33, 453 (1973).
(d) P . J . Davidson, M.F. LaDpert and R. Pearce, Acc. Chem. R e s . ,
7, 209 (1974).
113.
M. Casey and A.R. Manning, J . Chem. Soc. A, 256 (1971).
114.
A . J . C l e l a n d , S.A. Fieldhouse, B.H. Freeland, C.D.M. Mann and R . J .
O ' B r i e n , i b i d . , 376 (1971).
115.
J . Chatt and R.G. Hayter, J . Chem. S o c , 6017 (1963).
115.
(a) N . J . Nelson, N.E. Kime and D.F. S h r i v e r , J . Am. Chem. S o c ,
91_, 5173 (1969).
(b) J . C . Kotz and C D . Turnipseed, J . Chem. S o c , Chem. Comm.,
41 (1970).
(c) A. A l i c h , N . J . Nelson, D. Strope and D.F. S h r i v e r , Inorg.
Chem., n_, 2976 (1972).
117.
J . M . B u r l i t c h and R.B. Peterson, J . Organomet. Chem., 24, C65
(1970).
118.
(a) H. Brunner, P . C Wailes and H.D. Kaesz, Inorg. N u c l . Chem.
L e t . , 1, 125 (1965).
(b)
A. S t o r r and B . S . Thomas, Can. J . Chem., 49, 2504 (1971).
119.
T. Mole and E.A. J e f f r e y , "Organoaluminum Compounds", E l s e v i e r
P u b l . C o . , New York, N . Y . , 1972.
120.
J . A . McCleverty and G. W i l k i n s o n , J . Chem. S o c , 4096 (1963).
121.
C U . Pittman J r . and R.F. F e l i s , J . Organomet. Chem., 1_2, 399
(1974) .
- 92 -
122.
A . N . Nesmeyanov, Y.A. C h a p o s v s k i i , B.V. Lockshin, A . V . K i s i n and
E.G. Makarova, Dokl. Acad. Nauk. S . S . S . R . , 171,
(1966).
6 7 3
123.
M. G r a z i a n i , J . P . B i b l e r , R.M. Montesano and A. W o j c i c k i , J .
Organomet. Chem., 1_6, 507 (1969).
124.
J . A . Hanna and A. W o j c i c k i , Inorg. Chim. A c t a , 9, 55 (1974).
125.
R.G. Severson and A. W o j c i c k i , i b i d . , U,
126.
S . E . Jacobson and A. W o j c i c k i , J . Organomet. Chem., 72> 113 (1974).
127.
S . E . Jacobson, P. Reich-Rohrwig and A. W o j c i c k i , Inorg. Chem., 1_2,
717 (1973).
128.
S.R. Su and A. W o j c i c k i , J . Organomet. Chem.., 3]_, C34 (1971).
129.
F. Bonati and G W i l k i n s o n , J . Chem. S o c , 179 (1964).
130.
O.S. M i l l s , Acta C r y s t . ,
131.
R.B. K i n g , J . Am. Chem. S o c , 88, 2075 (1966)..
132.
James K. Hoyano, Peter Legzdins and John T. M a l i t o , Inorg. S y n t h . ,
17. (in press)
133.
a. James K. Hoyano, Peter Legzdins and John T. M a l i t o , J . Chem. S o c ,
Dalton Trans. 1022 (1975).
17 (1975).
Y\_, 620 (1958).
b. Brian B. W. S. Kolthammer, Peter Legzdins and John T.
Inorg. S y n t h . , 29 ( i n p r e s s ) .
Malito,