Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Discovery and development of proton pump inhibitors wikipedia , lookup
Gastrointestinal tract wikipedia , lookup
Drug interaction wikipedia , lookup
Psychopharmacology wikipedia , lookup
Neuropharmacology wikipedia , lookup
Neuropsychopharmacology wikipedia , lookup
Discovery and development of cephalosporins wikipedia , lookup
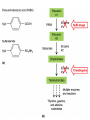
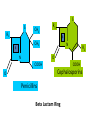
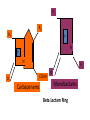
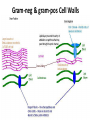
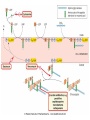
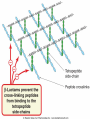
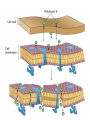
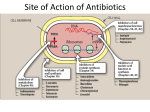
ANTI-BACTERIAL CHEMOTHERAPY..(1) By Dr Israa Omar ANTIMICROBIAL AGENTS THAT INTERFERE WITH THE SYNTHESIS OR ACTION OF FOLATE sulfonamides • In a landmark discovery in the 1930s, Domagk demonstrated that it was possible for a drug to influence the course of a bacterial infection. The agent was prontosil, a dye that proved to be an inactive prodrug but which is metabolised in vivo to give the active product, sulfanilamide ( Many sulfonamides have been developed since, and some are still useful drugs although their importance has declined in the face of increasing resistance. • Examples include sulfadiazine , sulfadimidine , sulfamethoxazole (intermediate acting) • Sulfametopyrazine (long acting) • Sulfasalazine (poorly absorbed in the gastrointestinal tract) • Sulfamethoxazole (in combination with trimethoprim as co-trimoxazole). Mechanism of action • Sulfanilamide is a structural analogue of paminobenzoic acid (PABA), which is an essential precursor in the synthesis of folic acid in bacteria. • folate is required for the synthesis of the precursors of DNA and RNA both in bacteria and in mammals, but whereas bacteria need to synthesise folic acid, mammals can obtain it from dietary sources. • Sulfonamides compete with PABA for the enzyme dihydropteroate synthetase, and the effect of the sulfonamide may be overcome by adding excess PABA. Mechanism of action • The action of a sulfonamide is to inhibit growth of the bacteria, not to kill them; that is to say, it is bacteriostatic rather than bactericidal. • The action is vitiated in the presence of pus or products of tissue breakdown, because these contain thymidine and purines, which bacteria utilise directly, bypassing the requirement for folic acid. • Resistance to the drugs, which is common, is plasmid-mediated and results from the synthesis of a bacterial enzyme insensitive to the drug. Pharmacokinetics • Most sulfonamides are readily absorbed in the gastrointestinal tract and reach maximum concentrations in the plasma in 4-6 hours. They are usually not given topically because of the risk of sensitisation or allergic reactions. • The drugs pass into inflammatory exudates and cross both placental and blood-brain barriers. They are metabolised mainly in the liver, the major product being an acetylated derivative that lacks antibacterial action. Side effects • Mild to moderate side effects include nausea and vomiting, headache and mental depression. • Cyanosis caused by methaemoglobinaemia may occur but is a lot less alarming than it looks. • Serious adverse effects include hepatitis, hypersensitivity reactions (rashes, fever, anaphylactic reactions), bone marrow depression • Crystalluria; this effect results from the precipitation of acetylated metabolites in the urine. Clinical uses of sulfonamides • Combined with trimethoprim (co-trimoxazole) for Pneumocystis carinii. • Combined with pyrimethamine for drug-resistant malaria , and for toxoplasmosis. • In inflammatory bowel disease: sulfasalazine (sulfapyridine-aminosalicylate combination. • For infected burns (silver sulfadiazine given topically). • For some sexually transmitted infections (e.g. trachoma, chlamydia, chancroid). • For respiratory infections: use now confined to a few special problems (e.g. infection with Nocardia). • For acute urinary tract infection (now seldom used). TRIMETHOPRIM • Trimethoprim is chemically related to the antimalarial drug pyrimethamine , both being folate antagonists. Structurally, it resembles the pteridine moiety of folate and the similarity is close enough to fool the bacterial dihydrofolate reductase, which is many times more sensitive to trimethoprim than the equivalent enzyme in humans . Pharmacokinetic aspects • Trimethoprim is given orally. It is fully absorbed from the gastrointestinal tract and widely distributed throughout the tissues and body fluids. It reaches high concentrations in the lungs and kidneys, and fairly high concentrations in the cerebrospinal fluid (CSF). When given with sulfamethoxazole, about half the dose of each is excreted within 24 hours. Because trimethoprim is a weak base, its elimination by the kidney increases with decreasing urinary pH. Antimicrobial agents that interfere with the synthesis or action of folate • Sulfonamides are bacteriostatic; they act by interfering with folate synthesis and thus with nucleotide synthesis. Unwanted effects include crystalluria and hypersensitivities. • Trimethoprim is bacteriostatic. It acts by antagonising folate. • Co-trimoxazole is a mixture of trimethoprim with sulfamethoxazole, which affects bacterial nucleotide synthesis at two points in the pathway Clinical uses of trimethoprim/cotrimoxazole • For urinary tract and respiratory infections: trimethoprim, used on its own, is usually preferred. • For infection with Pneumocystis carinii, which causes pneumonia in patients with AIDS: cotrimoxazole is used in high dose. Cell Wall Inhibitors Cell Wall Inhibitors • b-Lactams – Penicillins – Cephalosporins – Monobactams (Aztreonam) – Carbapenems (Imipenem) • Non b-Lactams – Vancomycin (glycopeptide) – Cycloserine – Fosfomycin – Bacitracin (cyclic peptide) S S R1 CH3 R1 β CH3 β N N R2 O COOH COOH Cephalosporins O Penicillins Beta Lactam Ring R1 R2 R1 β N β N O R2 COOH Carbapenems O Monobactams Beta Lactam Ring Dye Binds to Exposed Cell Wall Peptidoglycan, Gram positive (Rt.) Cell coverings of bacteria • • • • Inner: Cell / plasma membrane Outer: Cell wall Peptidoglycan layer consists of glycan chains Glycan chains: linear strands of two alternating amino sugars N-acetylglucosamine (Nag) N-acetylmuramic acid (Nam) • Cross linking of these strands by peptide chains Gram-neg & gram-pos Cell Walls β-LACTAM ANTIBIOTICS Penicillins and cephalosporins • Penicillins and cephalosporins – Part of group of drugs called β –lactams • Have shared chemical structure called β-lactam ring – Competitively inhibits function of penicillinbinding proteins • Inhibits peptide bridge formation between glycan molecules • This causes the cell wall to develop weak points at the growth sites and become fragile. PENICILLIN • In 1928, Alexander Fleming, working at St Mary's Hospital in London, observed that a culture plate on which staphylococci were being grown had become contaminated with a mould of the genus Penicillium, and that bacterial growth in the vicinity of the mould had been inhibited. He isolated the mould in pure culture and demonstrated that it produced an antibacterial substance, which he called penicillin. This substance was subsequently extracted and its antibacterial effects analysed by Florey, Chain and their colleagues at Oxford in 1940. They showed that it had powerful chemotherapeutic properties in infected mice, and that it was non-toxic Mechanism of action • All β-lactam antibiotics interfere with the synthesis of the bacterial cell wall peptidoglycan . • After attachment to penicillin-binding proteins on bacteria (there may be seven or more types in different organisms), they inhibit the transpeptidation enzyme that cross-links the peptide chains attached to the backbone of the peptidoglycan. Mechanism of action Mechanism of action • The final bactericidal event is the inactivation of an inhibitor of autolytic enzymes in the cell wall, leading to lysis of the bacterium. Some organisms, referred to as 'tolerant', have defective autolytic enzymes and are inhibited but not lysed in the presence of the drug. Types of penicillin and their antimicrobial activity • The first penicillins were the naturally occurring benzylpenicillin and its congeners, including phenoxymethylpenicillin. Benzylpenicillin is active against a wide range of organisms and is the drug of first choice for many infections. Its main drawbacks are poor absorption in the gastrointestinal tract (which means it must be given by injection) and its susceptibility to bacterial β-lactamases Types of penicillin and their antimicrobial activity • Various semisynthetic penicillins have been prepared by adding different side-chains to the penicillin nucleus . • β-lactamase-resistant penicillins (e.g. flucloxacillin) • broad-spectrum penicillins (e.g. ampicillin, pivampicillin and amoxicillin) • ( ticarcilin) with antipseudomonal activity against P. aeruginosa. • Amoxicillin is sometimes combined with β-lactamase inhibitor clavulanic acid as co-amoxiclav Pharmacokinetic aspects • When given orally, different penicillins are absorbed to differing degrees depending on their stability in acid and their adsorption to foodstuffs in the gut. • Penicillins can be given IM or IV , but intrathecal administration is inadvisable, particularly in the case of benzylpenicillin, as it can cause convulsions. Pharmacokinetic aspects • The penicillins are widely distributed in body fluids, passing into joints; into pleural and pericardial cavities; into bile, saliva and milk; and across the placenta. • Being lipid-insoluble, they do not enter mammalian cells and do not, therefore, cross the blood-brain barrier unless the meninges are inflamed, in which case they readily reach therapeutically effective concentrations in the CSF as well. Pharmacokinetic aspects • Elimination of most penicillins occurs rapidly and is mainly renal, 90% being through tubular secretion. • The relatively short plasma half-life is a potential problem in the clinical use of benzylpenicillin Unwanted effects • Penicillins are relatively free from direct toxic effects (other than their proconvulsant effect when given intrathecally). • The main unwanted effects are hypersensitivity reactions caused by the degradation products of penicillin, which combine with host protein and become antigenic. Skin rashes and fever are common; a delayed type of serum sickness occurs infrequently. Unwanted effects • Much more serious is acute anaphylactic shock, which although fortunately very rare, may in some cases be fatal. • When given orally, penicillins, particularly the broad-spectrum type, alter the bacterial flora in the gut. This can be associated with gastrointestinal disturbances and in some cases with suprainfection by other, penicillininsensitive, micro-organisms. Clinical uses of the penicillins – bacterial meningitis (e.g. by N. meningitidis, S.pneumoniae): benzylpenicillin, high doses IV – bone and joint infections (e.g. with Staph. aureus): flucloxacillin – skin and soft tissue infections (e.g. with Strep. pyogenes or Staph. aureus): benzylpenicillin, flucloxacillin; animal bites: co-amoxiclav – pharyngitis (from Strep. pyogenes): phenoxylmethylpenicillin – otitis media (organisms commonly include Strep. pyogenes, Haemophilus influenzae): amoxicillin – bronchitis (mixed infections common): amoxicillin – pneumonia: amoxicillin – urinary tract infections (e.g. with Escherichia coli): amoxicillin – gonorrhea: amoxicillin (plus probenecid) – syphilis: procaine benzylpenicillin – endocarditis (e.g. with Strep. viridans or Enterococcus faecalis) – serious infections with Pseudomonas aeruginosa: ticarcillin, piperacillin. CEPHALOSPORINS AND CEPHAMYCINS • They are usually classified inarbitrary in terms of chronological order in which they were produced: • First generation : cephalexin,cefadroxil • Second generation: cefuroxime, cephamandole and cefoxitin • Third generation: cefotaxime,ceftazidine, cefixime and ceftrixone,more active against g-ve • Fourth generation: cefepime highly resistant to beta lactamase enzyme Mechanism of action • The mechanism of action of these agents is similar to that of the penicillins: interference with bacterial peptidoglycan synthesis after binding to the β-lactam-binding proteins. Resistance • Resistance to this group of drugs has increased because of plasmid-encoded or chromosomal β-lactamase. • Nearly all Gram-negative bacteria have a chromosomal gene coding for a β-lactamase that is more active in hydrolysing cephalosporins than penicillins, and in several organisms a single mutation can result in highlevel constitutive production of this enzyme. Pharmacokinetic aspects • Some cephalosporins may be given orally , but most are given parenterally, IM or IV. • After absorption, they are widely distributed in the body and some, such as cefotaxime, cefuroxime and ceftriaxone, cross the bloodbrain barrier. • Excretion is mostly via the kidney, largely by tubular secretion, but 40% of ceftriaxone is eliminated in the bile Adverse effects • Hypersensitivity reactions, very similar to those seen with penicillin, may occur, and there may be some cross-sensitivity; about 10% of penicillin-sensitive individuals will have allergic reactions to cephalosporins. • Nephrotoxicity has been reported (especially with cefradine), as has drug-induced alcohol intolerance. • Diarrhoea can occur with oral cephalosporins Clinical uses of the cephalosporins • • • • • septicaemia (e.g. cefuroxime, cefotaxime) pneumonia caused by susceptible organisms meningitis (e.g. ceftriaxone, cefotaxime) biliary tract infection urinary tract infection (especially in pregnancy or in patients unresponsive to other drugs) • sinusitis (e.g. cefadroxil). OTHER β-LACTAM ANTIBIOTICS CARBAPENEMS AND MONOBACTAMS • Carbapenems and monobactams were developed to deal with β-lactamase-producing Gram-negative organisms resistant to penicillins carbapenem • Imipenem, an example of a carbapenem, acts in the same way as the other β-lactams . • It has a very broad spectrum of antimicrobial activity, being active against many aerobic and anaerobic Gram-positive and Gram-negative organisms. • Imipenem was originally resistant to all βlactamases, but some organisms now have chromosomal genes that code for imipenemhydrolysing β-lactamases. It is sometimes given together with cilastatin, which inhibits its inactivation by renal enzymes.. • Meropenem is similar but is not metabolised by the kidney. • Ertapenem has a broad spectrum of antibacterial actions but is licensed only for a limited range of indications Unwanted effects • Unwanted effects are generally similar to those seen with other β-lactams, nausea and vomiting being the most frequently seen. Neurotoxicity can occur with high plasma concentrations MONOBACTAMS • The main monobactam is aztreonam, a simple monocyclic β-lactam with a complex substituent which is resistant to most βlactamases. • It is given parenterally and has a plasma halflife of 2 hours. • Aztreonam has an unusual spectrum of activity and is effective only against Gramnegative aerobic rods such as pseudomonads, N.meningitidis and H.influenzae. It has no action against Gram-positive organisms or anaerobes Unwanted effects • Unwanted effects are, in general, similar to those of other β-lactam antibiotics, but this agent does not necessarily cross-react immunologically with penicillin and its products, and so does not usually cause allergic reactions in penicillin-sensitive individuals. Non b-Lactams Vancomycin • Non Beta-Lactam • teicoplanin is similar but longer lasting • Inhibits transglycosylase enzyme from binding to D-ala-D-ala; stops elongation of peptidoglycan chian; Bactericidal • IV only; Renal elimination • Used to treat Serious infections (sepsis) due to Staph,Methicillin-resistant strains, Alt. to βlactams in endocarditis caused by Staphylococci and Streptococci • C.difficile enterocolitis is treated orally with vancomycin, metronidazole is preferred. Adverse effect of vancomycin • Red neck syndrome • Phlebitis @inj. site • Ototoxicity / nephrotoxicity with aminoglycoside Bacitracin • Cyclic peptide mixture – not β-Lactam • Nephrotoxic; Topical use only • Blocks dephosphorylation of lipid deliver to peptidoglycan subunits of the growing cell wall • Often combine with other antibiotics Fosfomycin • Inhibits synthesis of N-acetylmuramic acid by inhibiting enolpyruvyl transferase(MurA) • One single oral dose • Uncomplicated urinary tract infection esp. in women • Unchanged drug present in high conc. in urine up to 46 hrs CYCLOSERINE • Structural analog of D-alanine • Exclusively use in strains of Mycobecteria resistant to first line anti tuberculous drugs • Serious dose related nervous toxicity ie psychosis, tremors, convulsions Interference with cell membrane integrity Drugs that interfere with cell membrane integrity • • • • • • • • • Few damage cell membrane Polymixn B (e.g. colistin) most common Common ingredient in first-aid skin ointments Binds membrane of Gram –ve cells Alters permeability Leads to leakage of cell and cell death Also bind eukaryotic cells but to lesser extent Limits use to topical application Can cause severe neurotoxicity and nephrotoxic Daptomycin • Cyclic lipopeptide • Treat infections by resistant gram positive organism MRSA and vancomycin-resistant enterococci • Work by rapidly depolarizing bacterial membrane, thus disrupting multiple aspects of membrane function and inhibit synthesis of DNA and RNA • It is bacteriocidal antibiotics and can be inactivated by lung surfactant;thus is not indicated in treatment of pneumonia • Can cause potential muscle toxicity and elevation of liver transaminases THANK YOU