Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Thermal radiation wikipedia , lookup
Equipartition theorem wikipedia , lookup
Heat capacity wikipedia , lookup
Conservation of energy wikipedia , lookup
Thermal expansion wikipedia , lookup
Calorimetry wikipedia , lookup
Temperature wikipedia , lookup
Heat equation wikipedia , lookup
Van der Waals equation wikipedia , lookup
Countercurrent exchange wikipedia , lookup
Heat transfer wikipedia , lookup
First law of thermodynamics wikipedia , lookup
State of matter wikipedia , lookup
Internal energy wikipedia , lookup
Thermal conduction wikipedia , lookup
Equation of state wikipedia , lookup
Heat transfer physics wikipedia , lookup
Non-equilibrium thermodynamics wikipedia , lookup
Entropy in thermodynamics and information theory wikipedia , lookup
Maximum entropy thermodynamics wikipedia , lookup
Chemical thermodynamics wikipedia , lookup
Extremal principles in non-equilibrium thermodynamics wikipedia , lookup
History of thermodynamics wikipedia , lookup
Vapor–liquid equilibrium wikipedia , lookup
Gibbs free energy wikipedia , lookup
Adiabatic process wikipedia , lookup
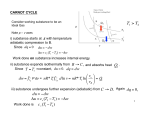
CARNOT CYCLE
T1 T2
Consider working substance to be an
Ideal Gas
Note p – v axes
i) substance starts at A with temperature T2,
T2
adiabatic compression to B.
Since dq 0 du dw
u cv (T1 T2 ) w
Work done on substance increases internal energy.
ii) substance expands isothermally from B C , and absorbs heat
Since T T1 =constant, du 0. dq dw
Q1 .
v
dw vvcB P dv = nR *T1 vvcB d ln v nR *T1 ln c Q1
vB
iii) substance undergoes further expansion (adiabatic) from C D. Again dq 0,
du dw
u cv (T2 T1 ) w
Work done is c v (T1 T2 )
1
iv) substance returns to original point A by undergoing an isothermal compression
from D A , and the heat exhausted is equal to Q2.
Since du=0 for this isothermal process, dq = dw:
dw
VA
VD
or
VD
pdv nR T2 d ln V nR T2 ln Q2
VA
VD
VA
V
Q2 nRT2 ln D
VA
Since the working substance returns to its original state, the work done by the
substance is equal to the net heat absorbed, Q1-Q2
Work done = Q1-Q2
(area inside the “rectangle”)
This can be readily verified by summing
the individual work terms.
?
wi wii wiii wiv Q1 Q2
This can be easily verified by adding up the individual terms.
Efficiency of the carnot cycle is
Q1 Q2
Q1
Q: How can
efficiency be
increased?
2
Only by transferring heat from a warm body to a cold body can heat be converted to work in a cyclic process.
For a CARNOT CYCLE
Q1 T1
Q2 T2
Proof: It can be shown that
pV
= constant for adiabatic process
Adiabatic legs
For A
B
PAVA PBVB
For C
D
PCVC PDVD
Isothermal legs
For B
C
PBVB PCVC
For D
A
PAVA PDVD
Combining 4 equations
We already know that
Vc VD
VB VA
V
Q1 RT1 ln C
VB
V
Q2 RT2 ln D
VA
3
Therefore
V
T1 ln C
Q1
VB T1
Q2 T ln VD T2
V
2
A
maximum (Carnot) efficiency of a power cycle
1
So
Q2
T
T
1 2 1 cold
Q1
T1
Twarm
• The maximum efficiency of an energy cycle is given by the Carnot efficiency,
which can be expressed as:
1
Tc
Th
If we consider the temperature difference between the equator and the poles:
Tpole (Tc)
Tequator (Th)
We note that Th varies little throughout
the year (estimate as ~301 K), compared
with Tc
Winter hemisphere: estimate Tc ~ 263 K
1
Tc
263
1
13%
Th
301
Summer hemisphere: estimate Tc ~ 283 K
1
Tc
283
1
6%
Th
301
--> More of the absorbed energy is converted into “work” in the winter hemisphere
4
The hurricane as an energy cycle
Consider a hurricane as a Carnot cycle:
http://apollo.lsc.vsc.edu/classes/met130/notes/chapter15/vertical_circ.html
3
Toutflow
4
2
1
Tsea surface
Analyze each leg of the cycle:
1: absorption of energy from ocean, ~isothermal expansion
2: convection in the eyewall, ~adiabatic expansion
3: upper level outflow, ~ isothermal compression (reject heat via radiation)
4: sinking branch, ~adiabatic compression
If we estimate SST~300 K, and outflow T~200 K,
1
Tc
200
1
30%
Th
300
U Hawaii class notes: Latent heat released when water vapor condenses in clouds is the key. Hurricanes are giant engines that
convert heat into wind energy. Consider a rain rate of 2 inches per day over an area of 300 mi radius: Over a 7 day lifecycle, the
energy released is equal to 50,000 1 MT nuclear explosions, equivalent to the total explosive yield of the nuclear arsenals of the
US and USSR at the height of the Cold War! www.soest.hawaii.edu/MET/Faculty/businger/.../18cHurricanes1.pdf
Our next state function: ENTROPY (consequence of Second Law)
There exists a function called entropy S, of the extensive variables of a system, defined for
all equilibrium states, such that the values assumed by the extensive variables are those
that maximize S (at equilibrium)
From the viewpoint of classical thermodynamics, entropy is defined as
ds
dqrev
T
where in this case heat is added to a substance undergoing a reversible transformation.
Clausius (1822-1888) stated that the cyclic integral of δQ/T is always less than or equal to zero:
Q
T
0
The cyclic integrals of work and of heat are not zero (engines).
Entropy is a STATE VARIABLE. (We can make up an internally reversible path from A to B to
compute change in entropy from the integral of δQ/T.)
Since dq= Tds, the First Law can be written as,
Tds du pdv
Even though we imagined a reversible
process to substitute for dq, this equation
applies to both reversible and irreversible
transitions because they are state variables
Generalized Statement of the Second Law
•
•
We need to generalize the definition of entropy since real systems are typically spontaneous
and irreversible, moving from a state of non-equilibrium to a state of equilibrium.
Second law can be formulated as 4 postulates:
1. There exists a STATE VARIABLE for any substance called the ENTROPY.
2. Entropy (S) may change by one of two ways; the substance may contact a thermal reservoir (heat
source), or S may change due to “internal” changes in the substance.
dse (externally-forced changes in S)
dsi (internally-forced changes in S)
Examples: dse Heat flow into a substance causing molecules to rearrange
dsi Mixing a gas, forcing molecules to change position.
Total entropy change ds = dse + dsi
3. Changes in S due to external influences are given by
dse
•
dq
T
where dq = heat imported or exported, while substance is in
contact with a thermal reservoir at temperature T
dsi 0
4. For reversible transformations,
For irreversible transformations, dsi 0
So reversible and irreversible processes are distinguished by entropy changes. For reversible
processes, there are no entropy changes associated with internal processes, or they have ceased as the
system undergoes only reversible changes, say via contact with a heat reservoir.
Now consider the system + its surroundings ( = “universe”)
•
In a reversible process any heat flow between the system and surroundings
must occur with no finite temperature difference between the system and
surroundings. Otherwise the process would be irreversible
dqrev
Tsystem
Tsurroundings
UNIVERSE
dqrev
dsuniverse dssystem dssurroundings
dqrev
dqrev
(
)0
Tsystem Tsurroundings
Since Tsystem Tsurroundings
•
•
Therefore Suniverse 0 for reversible process.
As expected and can be shown via a similar argument,
Suniverse 0
In general
Irreversible process
Suniverse 0
Statement of 2nd Law!
Calculations of Entropy Changes
•
•
•
2
For reversible processes, s s2 s1 dqrev
T
1
dqirrev
For irreversible processes, s is not necessarily equal to
T
However since S is a state function, S depends only upon endpoints.
Hence Sirrev can be found by devising a series of reversible transformations
that are equivalent to the irreversible transformations.
1. Reversible adiabatic process
Hence dqrev 0 , hence s 0
2. Reversible phase change at constant T, p
since T constant
2
s
1
dqrev
T1
q
L*m
rev
T
T
3. Reversible, isothermal process
where m=mass, L= latent heat
s
qrev
T
4. Reversible change of state for an Ideal Gas
From First Law:
dqrev du dw cv dT pdv
then
ds
dqrev
cv d ln T nR * d ln V
T
2
2
1
1
S cv d ln(T ) nR * d ln V
T2
V2
cv ln( ) nR * ln( )
T1
V1
Heating term
Volume term
5. Constant-pressure heating (irreversible)
For constant pressure:
dp 0
dqrev c p dT dp
T2
Hence
S c p d ln T
T1
T
S c p ln( 2
T1
)
for
c p f (T )
6. Similarly, for constant-volume heating (irreversible)
dqrev cv dT pdv
pdv 0
T2
S cv ln dT
T1
T2
S cv ln
T1
More entropy calculations at end of notes
“Thermodynamic Potentials”
•
The internal energy, when expressed as a function of S and V, is one of the
so-called “thermodynamic potentials”. The differential of a thermodynamic
potential gives the first and second laws of thermodynamics combined:
du dq dw
du Tds Pdv
•
•
S and V are the “native variables” for U (although we can express U as a
function of any two variables)
Remember
enthalpy is:
H U PV
dh Tds vdP
•
•
So S and P are the “native variables” for H.
We define two more thermodynamic potentials that have “native variables” ,
respectively, of T and V and T and P.
Very useful for equilibrium calcs!
Helmholtz & Gibbs Functions
•
Consider the following system/reservoir arrangement,
Q
system
•
-Q
Total work done by the system, whether it be in a reversible or irreversible
process is,
First Law
•
reservoir
w (u1 u2 ) Q
Q is positive if it flows into the
system
(1)
w is positive if done by the
system
dq=du+dw
From principle of entropy,
w=Q-(u2-u1)
( s2 s1 ) sR 0
s2 s1 is entropy change of system
sR is entropy change of reservoir
(2)
A general expression
that entropy must stay
constant or increase
sR
Q
T
•
For the reservoir,
•
Therefore we have ( S 2 S1 )
•
•
Note T= constant; reservoir has infinite heat capacity.
From (3) we have the following inequality, T ( s2 s1 ) Q
heat flow out of reservoir
Q
0
T
> applies for irreversible processes (ssystem
(3)
Q
)
T
= applies for reversible processes
•
•
From First Law or Eq (1) above (substitute for Q)
WT (u1 u2 ) T ( s1 s2 )
Reversible process
WT (u1 u2 ) T ( s1 s2 )
Irreversible process
For the irreversible case the work done is less than u T s since some of the
heat added to the system can go into changing the ‘internal’ entropy of the system.
Combining the two equations above
WT (u1 u2 ) T ( s1 s2 )
Now define the Helmholtz energy as
F u Ts
(4)
•
The difference in this energy between 2 equilibrium states at the same
temperature is
F ( F1 F2 ) (u1 u2 ) T ( s1 s2 )
From Eq n (4) we now have
WT ( F1 F2 )
•
(5)
Hence change in F sets an upper limit to the work that can be done in
any process between 2 equilibrium states at constant T.
For a reversible process WT F1 F2
For an irreversible process WT F1 F2
•
F is often referred to as the ‘free energy’ since ∆F represents the maximum
amount of work that can be done in a transformation at constant
temperature.
• Returning to the equation
WT (u1 u2 ) T ( s1 s2 ) (6)
• This equation is general and can be applied to any system, change
of state, phase, or even a chemical reaction.
• In general, work in processes we are considering consist of pdV
work and other possible forms of work, e.g. work required to form a
curve interface between 2 phases, frictional dissipation, etc.
• Let WT represent pdV type work and
work, so we now have
AT
WT AT F1 F2
represent other forms of
(7)
• If we consider a constant volume process, WT 0,
AT F1 F2
• Hence at constant T and V, Helmholtz energy sets an upper limit
to the non- pdV work that can be done in such a transition.
•
For a reversible constant volume process
AT ( F1 F2 )
•
Now consider a situation where no pdV and no
0 ( F1 F2 )
AT work is done, then
F2 F1
• Hence for a process at constant volume, for which AT 0 (and
T=constant), F can only decrease or remain constant. A transition
will not occur if F F .
2
1
Wikipedia definition:
du Tds Pdv
The Helmholtz free energy is a thermodynamic
potential which measures the “useful” work
dh Tds vdP
obtainable from a closed thermodynamic system at a
f u Ts (by definition)
constant temperature and volume. For such a
system, the negative of the difference in the
df du Tds sdT
Helmholtz energy is equal to the maximum amount
of work extractable from a thermodynamic process
sdT Pdv
in which temperature and volume are held constant.
Under these conditions (constant T and v), it is
Constant v constraint is not
minimized at equilibrium.
(8)
always helpful for us …
•
Next, consider a process that occurs at constant pressure. In this case the
pdV work is
WT p(V2 V1 )
•
Then returning to the equation:
•
We have:
WT AT F1 F2
AT ( F1 F2 ) p(V1 V2 ) or AT (u1 u2 ) T (s1 s2 ) p(V1 V2 )
Now define the Gibbs Free Energy as,
G F pV U TS pV
(dG = -SdT + VdP)
Then for a transition between 2 states at the same T ,p:
G G1 G2 (U1 U 2 ) T (S1 S2 ) p(V1 V2 )
and substituting into (8) we have
AT G1 G2 G
∆G represents the total non- pdV work for a constant T,P process. This
is of fundamental importance for cloud physics.
(8)
quick look ahead to one application we’ll be discussing:
•
During nucleation, some energy is released when molecules move from the
vapor to the “liquid” phase
• A is the work (energy required) necessary to form the surface of the small
T
liquid (or ice) embryo from the vapor phase:
AT Aembryo
For reversible process AT , p G
For irreversible process AT , p G
•
surface tension ( Jm 2 )
Aembryo area of embryo
AT , p bounded by G
If WT AT 0 then 0 G1 G2
So G can only decrease in a transition occurring at constant T, P.
•
If the hypothetical nucleus is too small (known as an unstable nucleus or "embryo"),
the energy that would be released by forming its volume is not enough to create its
surface
Thermodynamic Potential
•
•
•
Given F U TS ; G U TS pV
Consider a closed system which is one where no mass is allowed to cross
the boundaries of the system.
Taking total differentials,
(1)
dF dU TdS SdT
dG dU TdS SdT Vdp pdV
•
(2)
Using First Law in the form: dq dU pdV
TdS dU pdV
•
We can substitute for (1) and (2) the dU term
dF TdS pdV TdS SdT
pdV SdT
For dG,
dG TdS pdV TdS SdT pdV Vdp
dG SdT Vdp
•
•
Consider the following series of equations,
dU TdS pdV
U ( S ,V )
dF SdT pdV
dG SdT Vdp
F (T , V )
G (T , p )
dH TdS Vdp
H (S , p)
It is useful to identify the coefficients of the equations on the left hand side
with partial derivatives of the variables on the right hand side.
•
U
U
For: U ( S ,V ); dU
dS
dV
S V
V S
•
Therefore we have
•
U
T;
S V
Since F F (T , V )
dF
•
U
p
V S
F
F
dT
dV
T V
V T
Therefore F S ;
T V
F
p
V S
•
Since G G (T , p)
G
G
dG
dp
dT
T p
p T
•
And
•
Since H H ( S , p )
G
S
T p
G
V
p T
H
H
dH
dp
ds
S p
p S
H
T
S p
H
V
p S
•
And
•
Since P, V, T and S can be expressed as partial derivatives of U, F, G and
H, the latter variables are referred to as Thermodynamic Potentials.
Maxwell Relations
• The “Gibbs relations” are the following equations that we have already seen:
du Tds Pdv
dh Tds vdP
da sdT Pdv
dg sdT vdP
• We recognize that they are exact differentials and have the form
dz Mdx Ndy
M N
y x x y
• We can deduce the “Maxwell Relations”:
T
P
v s
s v
T v
P s s P
s P
v T T v
s
v
P T
T P
Can get
unmeasurables in
terms of measurables
Stable and Unstable Equilibrium
•
•
Stable Equilibrium: a state in which all irreversible processes have ceased,
and the system is in a state of maximum entropy. Reversible
transformations are possible (for which Stotal 0 ).
For state equilibrium, S 0
where S S Seq MAX
ENTROPY
•
•
a)
Seq S
This equilibrium condition is for an isolated system (no heat transfer, no
mass transfer)
A typical system we will deal with is NOT in thermal isolation, but rather in
contact with a heat reservoir (e.g., as approximated by the atmosphere).
For a transition at constant volume, in thermal contact with a reservoir at
temperature T, the stable state is defined by,
F 0
F Feqm Fother
b)
Helmholtz free energy is a minimum
For a transition at constant pressure and temperature, (in contact with a
thermal reservoir), stable state is,
G 0
G Geqm Gother
Gibbs free energy is a minimum
Applying equilibrium criteria
• We can now add moisture to the air parcel we’ve computed changes
for
– We could have added water to the “dry air mixture” and computed a
new average molecular weight, etc., BUT we know that simple
approach, assuming water stays as a vapor during the adiabatic ascent,
is only valid over small regions
– That is, we’ll need to consider phase changes for the water
• First, we’ll figure out where vapor, liquid and ice are in equilibrium,
and the energy changes associated with the transitions
– Can calculate changes such as heat released so we can treat the
energy changes in our parcel
– Can also figure out if the change is favored (e.g., if the transition is
allowed to occur thermodynamically, given specified conditions)
LATENT HEATS:
During a phase change, the pressure is constant and equal to the saturation pressure
(which is a function of T only). Adding heat to a substance at constant pressure and
resulting in a change in physical state, i.e. change in phase, is
described by,
Starting with First Law in the
dq dh Ldm
form dq = dh when dp = 0
where L is the latent heat (J kg-1) for the particular transition and m is mass
transferred between phases. The heat absorbed goes into changing the
molecular configuration/structure. For water substance:
L f 3.34 105 Jkg 1
Heat of fusion
Heat of condensation
Lc 2.25 106 Jkg 1
Heat of sublimation Ls 2.6 106 Jkg 1
SOLID
Lf
LIQUID
Lc
L is an energy change to
go from one phase to
another
VAPOR
26
LS
Thermodynamics of Moist Air
•
Phase transitions: what are the equilibrium conditions for a mixed-phase
system, e.g. a mixture of water and water vapor?
•
Consider a system consisting of liquid water and H2O(v) undergoing a
change in state,
{
V1
vapor
liquid
{
V2
″ denotes liquid phase
″′ denotes vapor phase
vapor
liquid
•
Define V1 total volume of state 1
V2 total volume of state 2
n1 # of moles of liquid, state 1
n1 # of moles of vapor, state 1
n2 # of moles of liquid, state 2
n2 # of moles of vapor, state 2
g specific Gibbs free energy of liquid phase
g specific Gibbs free energy of vapor phase
G1 Gibbs energy, state 1
•
Hence
G2 Gibbs energy, state 2
G1 n1 " g " n1 "' g "'
G2 n2 " g " n2 "' g "'
•
•
•
Since both states are stable equilibrium states, G1=G2,
and therefore
g
n1g n1g n
g n
2
2
But since the system is closed ntotal 0
(n
n1
( n1 n
)g
2) g
2
Therefore
g g
n1 n1 n
2 n
2
n1 n
n
2 n1
2
•
•
•
At equilibrium, the specific Gibbs free energy is the same for both phases
At the triple point, g S g L gV
Lets look at g between 2 phases in more detail:
vapor
c
liquid
g
b,e
f
d
a
T
g
s
T p
•
From earlier discussions,
•
Where s′″ is the specific entropy of the vapor phase. Hence the slope of g vs. T
gives the specific entropy.
Likewise g
for the liquid phase
•
•
s
T p
Note the slopes are <0, since larger values of g correspond to lower T’s. The
difference between entropies is
s s
•
Since
23
23
0, s s 0 s s
vapor
T
liquid
( ds dq )
T
Clausius-Clapeyron Equation
•
This equation is very important since it describes the variation in pressure
with temperature, for a system consisting of two phases in
equilibrium. For the case of a liquid/vapor or vapor/solid system, this
“pressure” is the saturation vapor pressure.
vapor
liqui d
•
•
•
es ,w sat vapor pressure for water
es ,i sat vapor pressure for ice
For a system in equilibrium, g g
Suppose the system undergoes a small change to a new pressure and
temperature, p+dp, T+dT such that the new state is also a stable state.
Each phase changes by dp, dT amounts.
Then for each phase:
dg sdT vdp
dg sdT vdp
•
Since the new state is also an equilibrium state,
dg dg
•
Or,
sdT vdp sdT vdp
( s s)dT (v v)dp
dp s s s
dT v v v
•
Since s s
CLAUSIUS-CLAPEYRON
EQUATION
23
23 =latent
T
dp
23
dT T (v v)
•
•
•
s, v are specific
quantities!
heat for vapor-liquid interface
For vapor-liquid system
For vapor-liquid system dp de
s ,w
For vapor-ice system
dp des ,i
Geometrical Interpretation
SOLID
LIQUID
S-L
P
L-V
Triple point
Critical point
S-V
VAPOR
T
dp
dT gives the slope of the
equilibrium line between
the 2 phases involved.
(on P-V-T diagram.)
•
Looking again at the Clausius-Clapeyron Equation we have three interfaces,
dp
23
dT T (v v)
dp
13
dT T (v v)
dp
12
dT T (v v)
•
1.
vaporliquid
solidvapor
liquidsolid
Since xy 0 always, the slope of the equilibrium line between the 2
phases involved is determined by the difference in specific volumes.
dp
is >0 for
dT
v v, v
vaporliquid
vaporsolid
] ALWAYS
For all materials!
2.
dp
can be of either sign for liquid solid interface.
dT
a) for v v 0
v v
For substance that expands upon freezing (like water). Hence
dp
0
dT
P
dp
0
dT
L
S
Triple
V
T
b) for v v 0
v v
substance contracts upon freezing.
MOST SUBSTANCES!
dp
0
dT
•
Integrate the Clausius-Clapeyron Eqn/ to get es as a function of T. For a
vapor/liquid system,
dp
23
dT T (v v)
1) need 23 (T )
2)v v as a function of T .
•
•
constant
RvT
v v v
p
Assume
23
Therefore dp
dT
GAS LAW
L23
RvT 2 P
L23
ln p
constant
RvT
p es
es 6.11mb @ T 0C
after integration
More entropy calculations
35
Rapid expansion
7. Irreversible change of state of an Ideal Gas
Let n moles of an ideal gas of P1,V1, T1 irreversibly change state to P2 ,V2 , T2
This irreversible process can be broken into 2 reversible processes:
1.) A slow expansion to V2 at constant T1
-put gas in a frictionless piston-cylinder, place in constant temperature
bath; slowly move piston out
2.) Hold gas at V2 , heat gas to temperature
T2 and pressure P2
S S1 S2
Hence
2
2
1
1
Sirrev cv (T )d ln T nR * d ln V
Constant-volume heating
Isothermal expansion
8. Irreversible phase change
Let 10g of supercooled water at -10°C change to ice at -10°C. Consider
constant pressure.
This process is IRREVERSIBLE e.g. if the ice is slowly heated, it will not
become water at -10°C.
Water at -10°C
IRREVERSIBLE
Ice at -10°C
1
3
2
Water at 0°C
Ice at 0°C
Find equivalent reversible path and compute entropy change
2
1.) Isobaric heating of water
2.) Phase change
S2
T
dqrev 2
S1
mc p , w d ln T
T
1
T1
Lf m
T
H m
T
2
3.) Isobaric cooling of ice S3 m c p ,i d ln T
1
STotal S1 S2 S3
STotal
Lf m
T2
T
mc p , w ln( )
mc p ,i ln( 1 )
T2
T1
T
273
J
) 1.57
263
deg
2
J
10
kg
5 J
S2 3.34 10
12.23
kg
273 K
deg
263
J
S3 102 kg (2106 Jkg 1 deg 1 ) ln(
) 0.8
273
deg
J
STotal S 11.46
deg
S1 102 kg (4218 Jkg 1 deg 1 ) ln(
T2 0 C
T1 10 C
m 10 g
c p , w 4218 Jkg 1 K 1
c p ,i 2106 Jkg 1 K 1
More entropy calculations
•
Calculate the change in entropy when 5g of water at 0°C are raised to
100°C and converted into vapor (steam) at that temperature.
Lv 2.25 106 Jkg 1
1. Raise temperature of H2O(l) from 0°C to 100°C
S S373 S273
T2
dqrev
T1
Let
dq mcwdT
T
Specific heat of water
cw 4.18 103 J deg 1 kg 1
T2
T
S1 mcw d ln T mcw ln( 2 ) 6.58 J deg 1
T1
T1
2. Convert to vapor (steam) at 100°C
Lv m (2.25 106 Jkg 1 )(5 103 kg )
S2
30.2 J deg 1
T
373 K
Stotal S1 S 2 36.8 J deg 1
•
Find S for the melting of 5g of ice at 0°C at 1 atm. ( L f 79.7cal g 1 )
This process is reversible since both ice and liquid states are possible at
0°C. (Equilibrium states!)
S
•
Lf m
T
79.7cal ( g )(5 g )
1.46 cal 6.1 J
K
K
273K
For freezing of liquid water,
qrev 0
and
Why the specific signs for melting vs. freezing?
S 6.1 J
K
•
Let n moles of a perfect gas undergo an adiabatic free expansion into a
vacuum (this is Joule’s experiment).
– Express ∆S in terms of the initial and final temperature and volume.
– Calculate ∆S if V2 2V1
•
The initial state is T1 ,V1 and the final state is T2 ,V2
Although the process is adiabatic, (dq=0), entropy change is non-zero
because this process is irreversible. Entropy of course changes (and
increases) since the molecules changed their configuration expanding into
the vacuum.
Hence
V
S nR* ln( 2
V1
)
nR ln 2
*
This is the so-called Entropy of Mixing.
With
V2 2V1