Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Degenerate matter wikipedia , lookup
Marcus theory wikipedia , lookup
Eigenstate thermalization hypothesis wikipedia , lookup
George S. Hammond wikipedia , lookup
Thermal expansion wikipedia , lookup
Determination of equilibrium constants wikipedia , lookup
Stability constants of complexes wikipedia , lookup
Temperature wikipedia , lookup
Thermal conduction wikipedia , lookup
Heat transfer physics wikipedia , lookup
Maximum entropy thermodynamics wikipedia , lookup
Equilibrium chemistry wikipedia , lookup
Transition state theory wikipedia , lookup
Chemical equilibrium wikipedia , lookup
Gibbs paradox wikipedia , lookup
Thermodynamics wikipedia , lookup
Chemical thermodynamics wikipedia , lookup
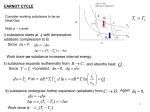
THERMODYNAMICS References Atkins, P.W., “Physical Chemistry”, Oxford University Press Castellan, G.W., “Physical Chemistry”, Addison Wesley Levine, I.R., “Physical Chemistry”, McGraw-Hill Laidler & Meiser, “Physical Chemistry”, Houghton Mifflin Co. Alberty, R.A. and Silbey, R., “Physical Chemistry”, Wiley thermo – heat dynamics – changes definitions Events,Experiments,Observations descriptions abstract generalize no proofs no violations Laws of Thermodynamics: 0th, 1st, 2nd, 3rd Applications, Verifications Note: does not worry about rate of changes (kinetics) but the states before and after the change not dealing with time Classical Thermodynamics Marcoscopic observables T, P, V, … Statistical Thermodynamics Microscopic details dipole moment, molecular size, shape Joule’s experiment Thermometer w adiabatic wall T mgh h (adiabatic process) U (energy change) = W (work) = mgh Page 59 w T time interval of heating U = q (heat) Conclusion: work and heat has the same effect to system (internal energy change) * FIRST LAW: U = q + W U: internal energy is a state function [ = Kinetic Energy (K.E.) + Potential Energy (P.E.) ] q: energy transfer by temp gradient W: force distance E-potential charge surface tension distance pressure volume First Law: The internal energy of an isolated system is constant Convention: Positive: heat flows into system work done onto system Negative: heat flows out of system work done by system Pressure-volume Work d work Fext dl M piston gdh weight Adh A Pext Adh Pext dV work Pext V2 V1 Mgh P1 = P2 M V1 M V2 If weight unknown, but only properties of system are measured, how can we evaluate work? P M V1 M V2 P Assume the process is slow and steady, Pint = Pext Free Expansion: Free expansion occurs when the external pressure is zero, i.e. there is no opposing force Reversible change: a change that can be reversed by an infinitesimal modification of a variable. Quasi equilibrium process: Pint = Pext + dP (takes a long time to complete) infinitesimal at any time W Pext dV Pint dV quasi equilibrium process Page 64-66 P 1 2 Example: P1 = 200kPa = P2 V1 = 0.04m3 V2 = 0.1m3 V * 2 W12 PdV P V2 V1 1 200kPa 01 . 0.04m 3 12kJ what we have consider was isobaric expansion (constant pressure) other types of reversible expansion of a gas: isothermal, adiabatic Page 65-66 Isothermal expansion remove sand slowly at the same time maintain temperature by heating slowly 2 Wrev PdV 21 nRT dV V 1 2 nRT 1 P * T dV V V2 V1 nRT ln V1 area under curve V2 V Ex. V1 = 0.04m3 P1 = 200kPa V2 = 0.1m3 Wrev V2 nRT1 ln V1 V2 PV 1 1 ln V1 0.1 200kPa 0.04m ln 0.04 3 7.33kJ PV 1 1 P2 80kPa V2 Adiabatic Reversible Expansion For this process PV = constant for ideal gas (proved later) Cp (slightly larger than 1) Cv PV 1 1 dV W PdV V 1 1 1 1 PV V V 1 1 2 1 1 2 2 P2V2 PV 1 1 1 V2 V1 P1 T1 P2 T2 Ex. V1 = 0.04m3, P1 = 200kPa, V2 = 0.1m3, = 1.3 0.04 P2 200 010 . W 1.3 60.77 kPa 60.77 01 . 200 0.04 1 13 . d: pressure drop without volume change 6.41kJ 200kPa const P isothermal W PdV 0 |Wa|>|Wb|>|Wc|>|Wd| a b d adibatic V State Function Vs Path Function State function: depends only on position in the x,y plane e.g.: height (elevation) 300 1 B 2 200 100m Y X A Path Function: depends on which path is taken to reach destination from 1 2, difference of 300m (state function) but path A will require more effort. Internal energy is a state function, heat and work are path functions P 200kPa 1 2 481.13K 3 0.04 0.1 192.45K V/m3 5 moles of monoatomic gas 3 CV R 2 U nCV T 3 2 R 289 5 18kJ W1 2 12 kJ Q1 2 U 12 kJ 30kJ W1 3 2 7.33kJ 0 7.33kJ Q1 3 2 7.33kJ 18kJ 25.33kJ Multivariable Calculus f x, y, z, w, Consider a 2 variable function f x, y or z f x, y a surface in three dimensional plot z or C’ C xc x yc y At point C, there are two slopes orthogonal to each other in constant x or constant y direction z C’ C’ z C C x=xC y=yc y x the change in can be calculated as a sum of two parts: change in x direction and change in y direction zC zC d dx dy x y y c partial derivative w.r.t. x xc partial derivative w.r.t. y Joule’s second experiment Energy UU(V,T) ??? thermometer V1 V2 adiabatic wall At time zero, open valve thermometer No temperature change adiabatic wall After time zero, V1 V1+V2 T=0 Q=0 W=0 no Pext U=0 Page 99 U=U(T) Energy is only a function of temperature for ideal gas * U 0 V T U U dU dT dV CV dT T V V T 0 (for ideal gas) CV is constant volume heat capacity Kinetic Model for Gases Qualitatively: • Gases consists of spheres of negligible size, far apart from one other. • Particles in ceaseless random motion; no interactions except collisions U V Constant Temperature U V T U U CV T V T * Energy is a function of Volume and temperature for real gases Interaction among molecules *Enthalpy Define Page 101 H = U + PV state function intensive variables locating the state Enthalpy is also a state function H = U + PV + VP At constant pressure H = U+PV = U - W =Q H = QP constant pressure heating H H T , P H is expressed as a functional of T and P H H dH dT dP CP 0 CP T P P T C P CV R CP CV for ideal gases (proved later) Thermochemistry Heat transferred at constant volume qV = U Heat transferred at constant pessure qP = H Exothermic H = -ve Endothermic H = +ve Standard states, standard conditions do not measure energies and enthalpies absolutely but only the differences, U or H The choice of standard state is purely a matter of convenience Analogy – differences in altitudes between 100 points and their elevation with respect to sea level * What is the standard state ? The standard states of a substance at a specified temperature is its pure form at 1 bar 25oC, 1 bar: the most stable forms of elements assign “zero enthalpy” H o 298 = 0 used for chemical reactions Standard enthalpy of formation Standard enthalpy change for the formation of the compound from its elements in their reference states. Reference state of an element is its most stable state at the specified temperature & 1 bar C (s) + 2H2 (g) CH4 (g) 289K, 1 atm o Hf = -75 kJ From the definition, Hfo for elements 0 Hess’s Law The standard enthalpy of an overall reaction is the sum of the standard enthalpies of the individual reactions into which a reaction may be divided. Standard reaction enthalpy is the change in enthalpy when the reactants in their standard states change to products in their standard states. Hess’ Law H2 H3 H4 R X Y P H1 H1 = H2 + H3 + H4 function state Hess’s law is a simple application of the first law of thermodynamics e.g. C (s) + 2H2 (g) CH4 (g) 298K, 1 atm H1o = ? C (s,graphite) + O2 (g) CO2 (g) Ho = -393.7 kJ H2 (g) + ½O2(g) H2O (l) Ho = -285.8 kJ CH4 (g) + 2O2 (g) CO2 (g) + 2H2O (l) Ho = -890.4 kJ H1o = -393.7 + 2(-285.8) - (-890.4) = -75 kJ/mole Heat of Reaction (Enthalpy of Reaction) Enthalpy change in a reaction, which may be o obtained from Hf of products and reactants Reactants Products H r o H n p H f , prod o products i nR H f ,reaction o reaactan ts o f i products reac tan ts I stoichiometric coefficient, + ve products, - ve reactants E.g. CH4 (g) + Cl2 (g) CH3Cl (g) + HCl (g) Hfo/ kJ CH3Cl -83.7 HCl -92.0 Cl2 0 CH4 -75.3 Hro = (-83.7-92.0) - (-75.3+0) = -100.4 kJ n Reactants R H f , R o elements Products n H elements p o f ,p Page 83 2A + B 3C + D 0 = 3C + D - 2A - B Generally, 0 = J J J J denotes substances, J are the stoichiometric numbers * H r j H o o f ,j Bond energy (enthalpy) Assumption – the strength of the bond is independent of the molecular environment in which the atom pair may occur. o C (s,graphite) + 2H2 (g) CH4(g)H = -75.4 kJ H2 (g) 2H (g) Ho = 435.3 kJ o C (s,graphite) C (g) H = 715.8 kJ C (s,graphite) + 2H2 (g) C (g) + 4H (g) Ho = 2(435.3)+715.8 = 1586.5 kJ C (g) + 4H (g) CH4 (g) H = -75.4-1586.5 = -1661.9 kJ CH Bond enthalpy = 1661.9/4 kJ = 415.5 kJ Temperature dependence of Hr T H r H r C p dT T o To C P C p i ,productsn p products reactants P HrT CP,P Hro R CP,R 298 C p i ,reaction nR T C i reactants products pi What is the enthalpy change for vaporization (enthalpy of vaporization) of water at 0oC? H2O (l) H2O (g) Ho = -241.93 - (-286.1) = 44.01 kJmol-1 o H2 = H + CP(T2-T1) assume CP,i constant wrt T H (273) = Ho(298) + CP(H2O,g) - CP (H2O,l)(273-298) = 44.10 - (33.59-75.33)(-25) = 43.0 kJ/mole For ideal Gases: H H 0, CP CV R P T T P U U dU dV dT V T T V H H dH dP dT P T T P dH dU PdV VdP V U U V dV dT VdP P dP dT , V T T V T P P T U U V V dV dP dT V T V T P T T P U U V V V U V dH P dT V P dP T P P T V T P T T V V T T P U V H U P CV R T P T V V T T P 0 = R/P for ideal gas H U V RT V P V P 2 V V 0 P T V T P T P 0 for ideal gas Prove PV = constant for adiabatic reversible expansion of an ideal gas U dU CV dT dV V T 0 for ideal gas Adiabatic expansion dU Q W , Q 0 RT CV dT PdV dV V dT dV CV R T V CV d ln T Rd ln V T2 R V2 ln ln T1 CV V1 T2 V2 T1 V1 R CV P2V2 V2 P1V1 V1 1 P2 V1 P1 V2 R R CV CV V1 V2 P2V2 P1V1 Reversible vs Irreversible Non-spontaneous changes vs Spontaneous changes Reversibility vs Spontaneity First law does not predict direction of changes, cannot tell which process is spontaneous. Only U = Q + W Second Law of Thermodynamics •Origin of the driving force of physical and chemical change •The driving force: Entropy •Application of Entropy: • Heat Engines & Refrigerators • Spontaneous Chemical Reactions •Free Energy * Page 120 Second Law of Thermodynamics No process is possible in which the sole result is the absorption of heat from a reservoir and its complete conversion into work Hot Reservoir q w Engine Direction of Spontaneous Change More Chaotic !!! Spontaneous change is usually accompanied by a dispersal of energy into a disorder form, and its consequence is equivalent to heating Entropy (S) is a measurement of the randomness of the system, and is a state function! SQ S 1/T Expansion into a vacuum irreversible reversible Vac V2 V2 V1 V1 Wirr Pext dV 0 U Qirr 0 V2 V1 ,Wmin V2 nRT ln V1 Wirr = 0 = qirr = U Wrev Pext dV V2 nRT ln V1 q rev Wrev qrev > qirr -Wrev > -Wirr Page 122 Entropy S For a reversible process, the change of entropy is defined as dqrev dS T (thermodynamic definition of the entropy) * Another expression of the Second Law: The entropy of an isolated systems increases in the course of a spontaneous change: Stot > 0 where Stot is the total entropy of the isolated system Entropy S The entropy of an isolated systems increases in the course of a reversible change: Stot = 0 where Stot is the total entropy of the isolated system Entropy Change for an isothermal expansion of a perfect gas V2 V1 S 1 T dq rev qrev V2 nR ln T V1 W rev Pext dV Depend only on V V2 nRT ln V1 qrev W rev Entropy is a state function Carnot’s Theoretical Heat Engine Heat flows from a high temperature reservoir to a low temperature body. The heat can be utilized to generate work. e.g. steam engine. Consider the sequence of reversible processes For the cycle, -Wnet = (W1+W2+W3+W4) = qnet = q1+q3 Ucycle=0 q1 positive q3 negative qnet positive |q3| < |q1| Net effect of the gas going through a cycle |q1| qH |Wnet| W |q3| qL Efficiency of theoretical heat engine Wnet Wnet Wnet available heat qH q1 q3 q1 q3 q3 1 1 q1 q1 q1 Carnot theorem: Engines operating between two temperature TH, TL have the same efficiency VB W1 nRT1 ln q1 , U 0 VA W3 nRT3 ln 1 VD q3 , U 0 VC T3 VC T3 VD , T1 VB T1 V A VB VC V A VD 1 VB VD nRT1 ln nRT3 ln q1 q3 VA VC T3 1 V q1 T1 nRT1 ln B VA th TL 1 TH limiting thermal efficiency of a heat engine In actual cases, heat engine have much lower efficient. irreversible processes, friction losses, etc.. Kelvin: It is impossible, by means of a cycle to take heat from q reservoir and convent it to work without at the same time transferring heat from a hot to cold reservoir. (We cannot have a 100% efficient heat engine) Show that 2nd law and Kelvin’s principle are equivalent Examine an adiabatic irreversible process. We want to evaluate the entropy change for the process by an reversible path BCDA D C Wnet A B Q=0 irreversible B C adiabatic compression C D isothermal compression D A adiabatic expansion 1st law for the cycle, U = 0 -(WAB + WBA) = QBA + QAB -Wnet = QBA 0 By Kelvin’s principle, -Wnet must be negative, otherwise a 100% efficient heat engine QBA = -Wnet must be negative QBA 0 S A B Q A B rev T QB A TH 0 Evaluation of Entropy Changes isothermal expansion: TdS dq rev dU PdV dq rev PdV S T T V2 P1 nRdV nR ln nR ln V V1 P2 isobaric heating: S Tm T1 C P , solid T H m b C P ,liq H v 2 C p , gas dT dT dT Tm Tm T Tb Tb T T T Tm: melting pt., Tb: boiling pt., If CP = constant, T2 S C P ln T1 H/S V L S T1 Tm Tb T2 change of temperature and volume (pressure) U TdS dU PdV dT PdV nCV dT PdV T V 2 2 CV P S n dT dV T T 1 1 2 P T2 V2 nCV ln nR ln T1 V1 T2 P1 nC P ln nR ln T1 P2 1 V Page 142 The efficiencies of heat engines Hot Reservoir q w Engine * S = - |q|/Th < 0 not possible! contrary to the second law Page 130 The efficiencies of heat engines Sh = - |qh|/Th Hot Reservoir S = - |qh|/Th + |qc|/Tc 0 qh w qc Cold Reservoir |wmax| = |qh|- |qc,min| = (1- Tc /Th) |qh| Maximum efficiency: erev = |wmax|/|qh|= 1- Tc /Th Sc = + |qc|/Tc erev 1 as Tc 0 or Th Page 144 Energetics of Refrigeration Hot Reservoir Sh = + |qc|/Th S = - |qc|/Tc + |qc|/Th < 0 |qc| Cold Reservoir not possible! Sc = - |qc|/Tc Page 144 The energetics of refrigeration Sh = + |qh|/Th S = |qh|/Th - |qc|/Tc 0 Hot Reservoir qh qh w qc Cold Reservoir |wmin| = |qh,min|- |qc,| = (Th /Tc-1) |qc| Maximum efficiency of performance: crev = |qc|/ |wmin| = Tc /(Th- Tc) Sc = - |qc|/Tc Page 145 The energetics of refrigeration Sh = + |qh|/Th Hot Reservoir How to keep it cool? dqc/dt = A(Th -Tc) qh qh w d|w|/dt = (1/ crev) dqc/dt = A (Th -Tc)2 / Tc qc Cold Reservoir Sc = - |qc|/Tc The Nernst Theorem The entropy change accompanying any physical or chemical transformation approaches zero as the temperature approaches zero. S 0 as T0 * Third Law of Thermodynamics Page 147 If the entropy of each element in its most state is taken as zero at the absolute zero of temperature, every substance has a positive entropy. But at 0K, the entropy of substance may equals to 0, and does become zero in perfect crystalline solids. Implication: all perfect materials have the same entropy (S=0) at absolute zero temperature Crystalline form: complete ordered, minimum entropy Statistical Interpretation of S S=0 at 0K for perfect crystals S = k ln Boltzmann postulate of entropy number of arrangements Boltzmann constant Entropy Change of Mixing one distinguish arrangement 4! 4! S A k ln 0, S B k ln 0 4! 4! N N B ! number of arrangement increased 8 7 65 A B 70 N A!NB ! 4 3 2 1 S A B k ln 70 A In general mixing NA, NB S mix N A N B ! k ln N A! N B ! Entropy change of mixing Stirling’s approximation: ln N! N ln N + 0(N) for large N S mix k N A N B ln N A N B N A ln N A N B ln N B k N A N B ln N A N B X A ln N A X B ln N B k N A N B X A ln X A X B ln X B 0 From classical thermodynamics, isothermal reversible expansion of gases A & B n A RT V A VB qrev Wrev ln T T A T A VA V A VB n A R ln VA V A VB qrev nB R ln T B VB V A VB V A VB nB R ln S mix n A R ln VA VA nT R X A ln X A X B ln X B 0 Assignment (due on 06/12/1999) 2.4, 2.5, 2.37, 3.5, 3.23, 4.10, 4.29 Page 133 * Extensions of 2nd Law TdS dq Clausius Inequality For adiabatic process, TdS 0 or dS 0 Entropy will always attain maximum in adiabatic processes. A similar function for other processes? * Page 149 Define Helmholtz free energy A = U - TS Thermodynamic State Function dA = dU - TdS - SdT Substitute into Clausius Inequality 0 dq TdS 0 dq dA dU SdT * 0 PdV dA SdT for isothermal, isochoric (constant volume) process, 0 dA * A will tend to a minimum value isothermal, isochoric process, dA 0 equilibrium dA 0 spontaneous If only isothermal, 0 dA PdV PdV dA W dA isothermal reversible expansion V2 W RT ln A V1 change in Helmholtz free energy = maximum isothermal work Example of isothermal, isochoric process: combustion in a bomb calorimeter O2 + fuel Temp. bath heat given out O2, CO2, H2O Higher P * Gibbs Free Energy Define Gibbs free energy G = H - TS Page 149-155 Thermodynamic state function = U + PV -TS dG = dU +PdV + VdP -TdS -SdT substitute into Clausius inequality 0 dq TdS 0 dU PdV TdS 0 dG VdP SdT dG VdP SdT * constant pressure, constant temperature 0 dG * G will tend to a minimum value dG 0 equilibrium dG 0 spontaneous change G Page 154 time More applications since most processes are isothermal, isobaric chemical reactions at constant T, P Reactants Products endothermic H is positive exothermic H is negative H is not a criteria for spontaneity S only isolated system G G = H - TS S H G + ve - ve - ve spontaneous dissociation of unstable compounds - ve + ve + ve nonspontaneous + ve + ve ? ? dissociation of a strong compound - ve - ve ? ? recombination reaction e.g. H + H H2 R forming unstable compounds P rxn. Co-ord. Change of Gibbs free energy with temperature (constant pressure) dG SdT VdP 0 G2 G1 SdT G S T P Change of Gibbs free energy with pressure (constant temperature) dG VdP SdT 0 G2 G1 VdP P2 RT ln P1 Ideal gas f2 RT ln f1 Real gas fugacity P G G o RT ln o P Ideal gas f G G o RT ln o P Real gas Page 168-174 Gibbs free energy *G G o RT ln pi i o P fi o G RT ln o P Ideal Gases Real Gases Total Gibbs free energy of a mixture of gases G nAG A nBGB pA pB o nAG nA RT ln o nBGB nB RT ln o P P o A n X AG A,i X BGB ,i X A RT ln X A X B RT ln X B n X i G X i RT ln X i o i i Gmix TS mix X i RT ln X i i H mix 0 for ideal gases Chemical Equilibria aA + bB cC + dD consider G for a pass of the reaction at constant T & P G = cGC + dGD - aGA - bGB molar Gibbs free energy Gi G RT ln f i o i pure state fi in the unit of bar fugacity G cGCo dGDo aG Ao bGBo cRT ln f C dRT ln f D aRT ln f A bRT ln f B G RT ln o c fC f D d a b f A fB i G RT ln f i o i i At equilibrium, G = 0 * G o f cf d ln C a D b ln K RT f A fB 0 = SJ J J equilibrium constant for ideal gaseous mixture c G o RT d pC pD ln a b p A pB fi = pi ln K p const . K p const . pi ni RT ci RT V d ci = concentration c c D cC n n K p b a RT K c RT cB c A pi Xi pT n = c+d-a-b varies with total pressure d c d c X D X C PT PT n Kp K P x a b a b X A X B PT PT For liquids, use activity aC a D G ln a b ln Keq RT a A aB o c d Gibbs - Helmholtz Equation G VdP SdT G S T P G Also, G H TS H T T P G H G T T T P GT T P GT T P G 1 G 2 T T P T H 2 T G T H or 1 TP Gibbs - Helmholtz Equation For a reaction G T H 2 T T P G/T G T H 1 T P Similarly, Slope =H 1/T G2 G/T A T U 1 T P H G1 1/T Change of equilibrium constant with temperature GT RT ln K o GT R ln K T Gibbs Helmholtz equation o o G T H T 2 T T o H T ln K R 2 T T O ln K HTo T RT 2 Van’t Hoff Equation o If H rxn constant (i.e. HP -HR is constant) H o ln K cons tan t RT endothermic, H + ve, K exothermic, H - ve, K with T with T * Relation between Thermodynamic functions dU = TdS-PdV dH = TdS+VdP, dA = -SdT-PdV, dG = -SdT+VdP, 1st law H = U+PV A = U-TS G = H-TS Page 149 From multivariable differential calculus dz = M dx + N dy total differential, i.e. z depends on x & y M N y x x y Maxwell Relations T P T V , V S S V P S S P S P , V T T V Page 163 S V P T T P Phase equilibrium Clapeyron equation G=0 molar Gibbs free energy G=G-G G G dG dG P liq V dP S dT V dP S dT dP S S S H dT V V V TV s Clausius- Clapeyron equation For vaporization and sublimation vap T H vap PH vap dP H vap dT TVvap TVvap RT 2 H vap dP d ln P 2 dT P RT H vap sat ln P sat const . RT P2 H vap T2 T1 ln P1 RT1T2 In P 1/T Example: What is the change in the boiling point of water at 100oC per torr change in atmospheric pressure? Hvap = 9725 cal mol-1 Vliq = 0.019 l mol-1 Vvap = 30.199 l mol-1 sat dP dT sat 9725 cal mol 0.04129 l atmcal T V V 37315 . K 30180 . l mol H vap 1 1 1 v l 0.03566 atm K 1 27.10 torr K 1 dT 0.0369 K torr 1 dP Example: Calculate the change in pressure required to change the freezing point of water 1oC. At 0oC, the heat of the fusion of ice is 79.7cal g-1, the density of water is 0.9998 g cm-3 and that of ice is 0.9168 g 1 1 Vl Vs 9.06 10 5 l g 1 0.9998 0.9168 H fus P sat T T Vl Vs sat 79.7 cal g 1 0.04129 l atmcal 1 2731 . K 9.06 10 5 l g 1 133 atm K 1 50kg P liq solid skate blade vap T ice liq. H2O Example 2.1 A certain electric motor produced 15 kJ of energy each second as mechanical work and lost 2 kJ as heat to the surroundings. What was the change in the internal energy of the motor and its power supply each second? Example 2.2 Calculate the work done when 50 g of iron reacts with hydrochloric acid in: (a) a closed vessel of fixed volume; (b) an open beaker at 25oC. Example 2.3 The internal energy change when 1.0 mole CaCO3 in the form of calcite converts to aragonite is 0.21 kJ. Calculate the difference between the enthalpy change and the change in internal energy. Example 2.4 The enthalpy change accompanying the formation of 1.00 mole NH3(g) from its elements at 298 K is -46.1 kJ. Estimate the change in internal energy. Example 2.5 Water is heated to boiling under pressure of 1.0 atm. When an electric current of 0.50 A from 12-V supply is passed for 300 s through a resistance in thermal contact with it, it is found that 0.798 g of water is vaporized. Calculate the molar internal energy and enthalpy changes at the boiling point (373.15 K). Exercise 2.6 At very low temperatures the heat capacity of a solid is proportional to T3, and we can write Cv=aT3. What is the change in enthalpy of such a substance when it is heated from 0 to T? Example 3.6 A sample of argon at 1.0 atm pressure and 25oC expands reversibly and adiabatically from 0.50 L to 1.00 L. Calculate its final temperature, the work done during the expansion, and the change in internal energy. The molar heat capacity of argon at constant volume is 12.48 JK-1 mol-1. Example 4.1 Calculate the entropy change in the surroundings when 1.00 mol H2O(l) is formed from its elements under standard conditions at 298.15 K. Example 4.4 Calculate the entropy change when argon at 25oC and 1.00 atm in a container of volume 500 cm3 is allowed to expand to 1000 cm3 and is simultaneously heated to 100oC. Example 5.4 The pressure deep inside the Earth is probably greater than 3×103 kbar, and the temperature is around 4×103 oC. Estimate the change in G on going from crust to core for a process in which V=1.0 cm3 mol-1 and S = 2.1 JK-1 mol-1. Example 5.5 Calculate the change in molar Gibbs energy when water vaporizes at 1 bar and 25 oC. Note that the molar Gibbs energies of formation are -237.13 and -228.57 kJ mol-1 for water and its vapor, respectively. Example 5.6 Suppose that the attractive interactions between gas particles can be neglected and find an expression for the fugacity of a van der Waals gas in terms of the pressure.