Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Discovery and development of proton pump inhibitors wikipedia , lookup
Polysubstance dependence wikipedia , lookup
Neuropsychopharmacology wikipedia , lookup
Orphan drug wikipedia , lookup
Psychopharmacology wikipedia , lookup
Compounding wikipedia , lookup
Neuropharmacology wikipedia , lookup
Pharmacognosy wikipedia , lookup
Pharmaceutical industry wikipedia , lookup
Theralizumab wikipedia , lookup
Drug design wikipedia , lookup
Prescription costs wikipedia , lookup
Pharmacogenomics wikipedia , lookup
Drug discovery wikipedia , lookup
Plateau principle wikipedia , lookup
Pharmacokinetics
Dr. S A Ziai
Pharmacokinetics examines the
movement of a drug over time
through the body
Pharmacological as well as
toxicological actions of drugs are
primarily related to the plasma
concentrations of drugs
The goal of therapeutics is to achieve
a desired beneficial effect with
minimal adverse effects
concentration-effect
dose-concentration
ADME
The speed of onset of
drug action, the intensity
of the drug’s effect, and
the duration of drug
action are controlled by
four fundamental
pathways of drug
movement and
modification in the body
The "standard" dose of a drug is
based on trials in healthy
volunteers and patients with
average ability to absorb,
distribute, and eliminate the drug
Pharmacokinetic Principles
• In practical therapeutics, a drug should be
able to reach its intended site of action after
administration by some convenient route
• Drugs:
– The active drug molecule
– Prodrug
ROUTES OF DRUG ADMINISTRATION
• Enteral
– Oral
– Sublingual
• Parenteral
– IV
– IM
– SC
• Other
–
–
–
–
–
–
Inhalation
Intranasal
Intrathecal/Intraventricular
Topical
Transdermal
Rectal
• Oral
– Simple, Reversible
– First-pass metabolism
• Sublingual
– Enteric coated
• Parentral
–
–
–
–
–
–
–
–
Heparin, Insulin
Unconscious patients
Rapid onset of action
Highest bioavailability (IV)
Irreversible, Pain, Infection
Rate of administration (IV)
Depot and suspension (IM)
Pumps, Implants (SC)
Basic parameters in PK
• Clearance, the measure of the ability of the
body to eliminate the drug;
• Volume of distribution, the measure of the
apparent space in the body available to
contain the drug
VOLUME OF DISTRIBUTION
• The volume of distribution is a
hypothetical volume of fluid into
which a drug is dispersed.
• Water compartments in the body
1. Plasma compartment (6%)
• Size and PB (Heparin)
2. Extracellular fluid (20%)
•
LMW and hydrophil Aminoglycosides
3. Total body water (60%)
•
Ethanol
4. Other sites
•
Fetus
Physical Volumes (in L/kg Body Weight) of Some Body
Compartments into Which Drugs May Be Distributed
Compartment and Volume Examples of Drugs
Water
Total body water (0.6 L/kg1) Small water-soluble
molecules: eg, ethanol.
Total
body
water
in
a
young
lean
man
might
Extracellular water (0.2
Larger water-soluble
be
woman,eg,0.5
L/kg
L/kg)0.7 L/kg; in an obesemolecules:
gentamicin.
Blood (0.08 L/kg); plasma Strongly plasma protein(0.04 L/kg)
bound molecules and very
large molecules: eg, heparin.
Fat (0.2–0.35 L/kg)
Highly lipid-soluble
molecules: eg, DDT.
Bone (0.07 L/kg)
Certain ions: eg, lead,
fluoride.
Determination of Vd
• C = D/Vd or Vd = D/C
• C = the plasma concentration
of the drug
• D = the total amount of drug
in the body.
• For example, if 25 mg of a
drug (D = 25 mg) are
administered and the plasma
concentration is 1 mg/L, then
Vd = 25 mg/1 mg/L = 25 L
Volume of Distribution
The volume of distribution may be defined
with respect to blood, plasma, or water
(unbound drug)
apparent volume
Clearance
Clearance, like volume of distribution,
may be defined with respect to blood
(CLb), plasma (CLp), or unbound in
water (CLu), depending on the
concentration measured
• Kidneys
• Liver
• Clearance of unchanged drug in the urine
represents renal clearance
• In liver, drug elimination occurs via
biotransformation of parent drug to one or
more metabolites, or excretion of unchanged
drug into the bile, or both.
First order kinetic
• For most drugs, clearance is constant over the
concentration range encountered in clinical
settings, ie, elimination is not saturable, and
the rate of drug elimination is directly
proportional to concentration
Total body clearance
• It is not possible to measure and sum these
individual clearances. However, total
clearance can be derived from the steadystate equation
Clearance is calculated from the dose divided by the AUC
Rate of elimination
• The elimination of a drug usually follows first-order kinetics, and
the concentration of drug in plasma drops exponentially with time.
• The elimination rate for most drugs is first-order and shows a linear
relationship with time if lnC (where lnC is the natural log of C,
rather than C) is plotted versus time
• This is because the elimination processes are not saturated.
• This can be used to determine the half-life, t1⁄2, of the drug (the
time during which the concentration of a drug at equilibrium
decreases from C to 1⁄2C):
• ke = the first-order rate constant for drug elimination from the total
body and CL = clearance
Half-Life
• Half-life (t1/2) is the time required to change
the amount of drug in the body by one-half
during elimination (or during a constant
infusion).
• Half-life is useful because it indicates the time
required to attain 50% of steady state—or to
decay 50% from steady-state conditions—
after a change in the rate of drug
administration.
Time required to reach the steadystate drug concentration
• Disease states can affect both of the volume
of distribution and clearance
• Patients with chronic renal failure have
decreased renal clearance of digoxin but also
a decreased volume of distribution
Capacity-Limited Elimination
• Phenytoin, ethanol, aspirin
• Other names: saturable, dose- or
concentration-dependent, nonlinear, and
Michaelis-Menten elimination.
has no
real meaning
for drugselimination
with capacity-limited
• IfClearance
dosing
rate
exceeds
capacity,
elimination, and AUC cannot be used to describe the elimination of
such drugs state cannot be achieved: The
steady
concentration will keep on rising as long as
dosing continues.
Flow-Dependent Elimination
• Most of the drug in the blood perfusing the organ
is eliminated on the first pass of the drug through
it
• They called "high-extraction" drugs since they are
almost completely extracted from the blood by
the organ.
• It depends on:
– Blood flow to the organ
– Plasma protein binding
– Blood cell partitioning
Drug Accumulation
• In practical terms, if the dosing interval is shorter than
four half-lives, accumulation will be detectable.
• For a drug given once every half-life, the accumulation
factor is 1/0.5, or 2.
• The accumulation factor predicts the ratio of the
steady-state concentration to that seen at the same
time following the first dose.
• Thus, the peak concentrations after intermittent doses
at steady state will be equal to the peak concentration
after the first dose multiplied by the accumulation
factor.
ABSORPTION OF DRUGS
Transport of a drug from the GI tract
• IV 100%
• Passive diffusion
–
–
–
–
Concentration gradient
Lipid soluble
Water soluble
Facilitated diffusion (by carrier)
• No energy, Saturated, Inhibited
• Active transport
– ATP
– Saturated
– Moving against concentration
gradient
• Endocytosis and exocytosis
– Vit B12
Permeation
Aqueous Diffusion
• Across epithelial membrane tight junctions
and the endothelial lining of blood vessels
through aqueous pores that—in some
tissues—permit the passage of molecules as
large as MW 20,000–30,000.
• Concentration gradient (Fick's law)
• Bound to large plasma proteins (eg, albumin)
• Electrical fields
Lipid Diffusion
• The lipid:aqueous partition coefficient of a
drug determines how readily the molecule
moves between aqueous and lipid media.
• In the case of weak acids and weak bases
(which gain or lose electrical charge-bearing
protons, depending on the pH), the ability to
move from aqueous to lipid or vice versa
varies with the pH of the medium
• A weak acid is a neutral molecule that can
reversibly dissociate into an anion (a
negatively charged molecule) and a proton (a
hydrogen ion).
• A weak base is a neutral molecule that can
form a cation (a positively charged molecule)
by combining with a proton.
Effect of pH on drug absorption
• More of a weak acid will be in the lipid-soluble
form at acid pH
• More of a basic drug will be in the lipidsoluble form at alkaline pH
The pKa is a measure of the strength
of the interaction of a compound with
a proton
The lower the pKa of a drug, the more acidic it is.
Conversely, the higher the pKa, the more basic is the drug
Determination of how much drug will
be found on either side
of a membrane
Special Carriers
• Peptides, amino acids, sugars
• ABC (ATP-binding cassette) family
– less selective
– P-glycoprotein, multidrug-resistance type 1
(MDR1) transporter, multidrug resistanceassociated protein (MRP)
– the solute carrier [SLC] family, do not bind ATP but
use ion gradients for transport energy
Special Carriers
Transporter
NET
SERT
VMAT
MDR1
MRP1
Physiologic Function
Norepinephrine reuptake from
synapse
Serotonin reuptake from
synapse
Pharmacologic Significance
Target of cocaine and some
tricyclic antidepressants
Target of selective serotonin
reuptake inhibitors and some
tricyclic antidepressants
Target of reserpine
Transport of dopamine and
norepinephrine into adrenergic
vesicles in nerve endings
Transport of many xenobiotics Increased expression confers
out of cells
resistance to certain anticancer
drugs; inhibition increases blood
levels of digoxin
Leukotriene secretion
Confers resistance to certain
anticancer and antifungal drugs
Physical factors influencing
absorption
• Blood flow to the absorption site
– Blood flow to the intestine is much greater than the
flow to the stomach
– Shock severely reduces blood flow to cutaneous
tissues
• Total surface area available for absorption
– Intestine has a surface rich in microvilli, it has a
surface area about 1000-fold that of the stomach
• Contact time at the absorption surface
– Diarrhea, Parasympathetic, Food
Bioavailability
• Bioavailability is defined as the fraction of
unchanged drug reaching the systemic
circulation following administration by any
route
• The area under the blood concentration-time
curve (AUC) is a common measure of the
extent of bioavailability for a drug given by a
particular route
Factors that influence bioavailability
–
–
–
–
First-pass metabolism
Solubility of the drug
Chemical instability
Nature of the drug
formulation
– Particle size, salt form,
crystal polymorphism,
enteric coatings and the
presence of excipients (such
as binders and dispersing
agents) can influence the
ease of dissolution
Route
Bioavailability (%) Characteristics
Intravenous (IV)
100 (by definition) Most rapid onset
Intramuscular (IM) 75 to ≤ 100
Large volumes often
feasible; may be painful
Subcutaneous (SC) 75 to ≤ 100
Smaller volumes than IM;
may be painful
Oral (PO)
5 to < 100
Most convenient; firstpass effect may be
significant
Rectal (PR)
30 to < 100
Less first-pass effect than
oral
Inhalation
5 to < 100
Often very rapid onset
Transdermal
80 to ≤ 100
Usually very slow
absorption; used for lack
of first-pass effect;
prolonged duration of
action
Bioequivalence
• Two related drugs are bioequivalent if they
show:
1. comparable bioavailability
2. similar times to achieve peak blood
concentrations
Therapeutic equivalence
• Two similar drugs are therapeutically
equivalent if they have comparable efficacy
and safety
Extent of Absorption
• After oral administration, a drug may be
incompletely absorbed
– too hydrophilic (eg, atenolol)
– too lipophilic (eg, acyclovir)
– P-glycoprotein
– First-Pass Elimination
• In the gut wall (eg, by the CYP3A4 enzyme system)
• in the liver
• liver can excrete the drug into the bile
• The effect of first-pass hepatic elimination on
bioavailability is expressed as the extraction ratio
(ER):
• where Q is hepatic blood flow, normally about 90
L/h in a person weighing 70 kg.
• The systemic bioavailability of the drug (F) can be
predicted from the extent of absorption (f) and
the extraction ratio (ER):
Rate of Absorption
• The rate of absorption is determined by the
site of administration and the drug
formulation
• zero-order when the rate is independent of
the amount of drug remaining in the gut
– when it is determined by the rate of gastric
emptying or by a controlled-release drug
formulation.
Alternative Routes of Administration &
the First-Pass Effect
• The hepatic first-pass effect can be avoided to
a great extent by use of sublingual tablets and
transdermal preparations and to a lesser
extent by use of rectal suppositories
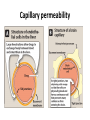
DRUG DISTRIBUTION
DRUG DISTRIBUTION
• Drug distribution is the process by which a drug
reversibly leaves the bloodstream and enters the
interstitium (extracellular fluid) and/or the cells
of the tissues.
• The delivery of a drug from the plasma to the
interstitium primarily depends on:
• Blood flow
• Capillary permeability
• Protein binding
• Hydrophobicity of the drug
Capillary permeability
BINDING OF DRUGS TO PLASMA
PROTEINS
Albumin has the strongest affinities
for anionic drugs (weak acids)
and hydrophobic drugs
Competition for binding between
drugs
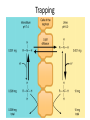
Trapping
Body Fluids with Potential for Drug "Trapping"
through the pH-Partitioning Phenomenon.
Body Fluid
Range of pH Total Fluid: Blood
Total Fluid: Blood
Concentration Ratios for
Concentration Ratios
Sulfadiazine (acid, pKa 6.5) for Pyrimethamine
(base, pKa 7.0)
Urine
Breast milk
5.0–8.0
6.4–7.6
0.12–4.65
0.2–1.77
72.24–0.79
3.56–0.89
Jejunum, ileum contents
7.5–8.0
1.23–3.54
0.94–0.79
Stomach contents
1.92–2.59
0.11
85,993–18,386
Prostatic secretions
6.45–7.4
0.21
3.25–1.0
Vaginal secretions
3.4–4.2
0.114
2848–452
DRUG METABOLISM
DRUG METABOLISM
• Drugs are most often eliminated by
biotransformation and/or excretion into the
urine or bile.
• The process of metabolism transforms
lipophilic drugs into more polar readily
excretable products.
• The liver is the major site for drug metabolism
Kinetics of metabolism
1. First-order kinetics
• [C], is much less than, Km
• That is, the rate of drug
metabolism is directly
proportional to the concentration
of free drug
• This means that a constant
fraction of drug is metabolized per
unit of time
Zero-order kinetics
• With a few drugs, such as aspirin, ethanol, and
phenytoin, the doses are very large. Therefore
[C] is much greater than Km
• A constant amount of drug is metabolized per
unit of time
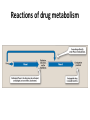
Reactions of drug metabolism
Phase I reactions utilizing the P450
system
• The Phase I reactions most frequently
involved in drug metabolism are catalyzed by
the cytochrome P450 system (also called
microsomal mixed function oxidase):
Drug + O2 + NADPH + H+ → Drugmodified + H2O + NADP+
Drug
NADP+
CYP
R-Ase
CYP
e-
Fe+3
Drug
Drug
OH
NADPH
Fe+3
CYP
Fe+2
CYP
Drug
Drug
eO2
CYP
O2
Drug
Fe+2
H2O
2H+
Electron flow in microsomal drug oxidizing system
OH
Summary of the P450 system
• Metabolism of many endogenous compounds
(steroids, lipids, etc.) and biotransformation of
exogenous substances (xenobiotics).
• Cytochrome P450, designated as CYP
• Composed of many families of hemecontaining isozymes
Summary of the P450 system
• There are many different genes, and many
different enzymes
• Six isozymes are responsible for the vast majority
of P450-catalyzed reactions: CYP3A4 (60%),
CYP2D6 (25%), CYP2C9/10 (15%), CYP2C19 (15%),
CYP2E1 (2%), and CYP1A2 (2%).
• An individual drug may be a substrate for more
than one isozyme
• Considerable amounts of CYP3A4 are found in
intestinal mucosa
Summary of the P450 system
• CYP2D6, exhibit genetic
polymorphism
• Some individuals, for example,
obtain no benefit from the
opioid analgesic codeine
because they lack the enzyme
that O-demethylates and
activates the drug
• The frequency of this
polymorphism is in part
racially determined, with a
prevalence of 5-10% in
European Caucasians as
compared to <2% of Southeast
Asians
Summary of the P450 system
Inducers
• Consequences of increased drug metabolism
include:
1. decreased plasma drug concentrations
2. decreased drug activity if metabolite is inactive
3. increased drug activity if metabolite is active
4. decreased therapeutic drug effect
• Polycyclic hydrocarbons (found as air pollutants)
induce CYP4501A2, which decreases the
therapeutic concentrations of amitriptyline and
warfarin
Inhibitors
• An important source of drug interactions that
leads to serious adverse events
• Some drugs, however, are capable of
inhibiting reactions for which they are not
substrates (ketoconazole)
• Omeprazole is a potent inhibitor of three of
the CYP isozymes responsible for warfarin
metabolism
Inhibitors
• The more important CYP inhibitors are
erythromycin, ketoconazole, and ritonavir,
because they each inhibit several CYP
isozymes
• Cimetidine blocks the metabolism of
theophylline, clozapine, and warfarin
• Grapefruit juice inhibits CYP3A4 and, thus,
drugs such as amlodipine, clarithromycin, and
indinavir
Phase I reactions not involving the
P450 system
• These include:
– amine oxidation (for example, oxidation of
catecholamines or histamine)
– alcohol dehydrogenation (for example, ethanol
oxidation)
– esterases (for example, metabolism of pravastatin
in liver)
– hydrolysis (for example, of procaine)
Phase II
• Conjugation reaction with an endogenous
substrate, such as glucuronic acid, sulfuric acid,
acetic acid, or an amino acid, results in polar,
usually more water-soluble compounds that are
most often therapeutically inactive
• A notable exception is morphine-6-glucuronide,
which is more potent than morphine
• Glucuronidation is the most common and the
most important conjugation reaction
Phase II
• Neonates are deficient in this conjugating
system, making them particularly vulnerable
to drugs such as chloramphenicol
• Drugs already possessing an –OH, –NH2, or –
COOH group may enter Phase II directly and
become conjugated without prior Phase I
metabolism
• The highly polar drug conjugates may then be
excreted by the kidney or bile
Entero-hepatic Recirculation – clinical significance
Oral contraceptive failure when an antibiotic is taken
An antibiotic such as rifampin also induces CYP enzymes that metabolize the contraceptive
hormones and thus reduces their effectiveness even more
DRUG ELIMINATION
DRUG ELIMINATION
• The most important route is through the
kidney into the urine.
• Other routes include the bile, intestine, lung,
or milk in nursing mothers
Renal elimination of a drug
• Glomerular filtration
– The glomerular filtration rate (125
mL/min) is normally about twenty
percent of the renal plasma flow (600
mL/min)
– Lipid solubility and pH do not influence
the passage of drugs into the glomerular
filtrate
• Proximal tubular secretion
– By two energy-requiring active transport
(carrier requiring) systems, one for
anions and one for cations
– Premature infants and neonates have an
incompletely developed tubular
secretory mechanism
Renal elimination of a drug
• Distal tubular reabsorption
– As a general rule, weak acids
can be eliminated by
alkalinization of the urine,
whereas elimination of weak
bases may be increased by
acidification of the urine. This
process is called “ion trapping.”
• For phenobarbital
(weak acid) overdose
we can give
bicarbonate
• For cocaine,
acidification of the
urine with NH4Cl is
useful
Quantitative aspects of renal drug
elimination
• Plasma clearance is expressed as the volume
of plasma from which all drug appears to be
removed in a given time—for example, as
mL/min
• Clearance equals the amount of renal plasma
flow multiplied by the extraction ratio, and
because these are normally invariant over
time, clearance is constant
The Time Course of Drug Effect
• Immediate Effects
• The effect will not usually be linearly
proportional to the concentration
• The half-life of enalapril is about 3 hours. After
an oral dose of 10 mg, the peak plasma
concentration at 3 hours is about 64 ng/mL.
• Enalapril is usually given once a day
• Enalapril’s EC50 is about 1 ng/mL
Note that plasma concentrations of
enalapril change by a factor of 16 over
the first 12 hours (four half-lives) after
the peak, but ACE inhibition has only
decreased by 20%.
• The key factor is a high initial concentration in
relation to the EC50
• When concentrations are in the range between
one fourth and four times the EC50, the time
course of effect is essentially a linear function of
time—13% of the effect is lost every half-life over
this concentration range.
• At concentrations below one fourth the EC50, the
effect becomes almost directly proportional to
concentration
Delayed Effects
• Changes in drug effects are often delayed in
relation to changes in plasma concentration.
• This delay may reflect the time required for the
drug to distribute from plasma to the site of
action
• The delay due to distribution is a
pharmacokinetic phenomenon that can account
for delays of a few minutes.
• Slow turnover of a physiologic substance that is
involved in the expression of the drug effect take
many hours or even days to occur (eg. warfarin).
Maintenance Dose
• Clearance is the most important
pharmacokinetic term to be considered in
defining a rational steady state drug dosage
regimen.
• At steady state, the dosing rate ("rate in")
must equal the rate of elimination ("rate
out").
Example: Maintenance Dose
Calculation
• A target plasma theophylline concentration of
10 mg/L is desired to relieve acute bronchial
asthma in a patient
• clearance is 2.8 L/h/70 kg
• intravenous infusion, F = 1
• Oral theophylline every 12 hours using an
extended-release formulation
Loading Dose
• Volume of distribution is the proportionality
factor that relates the total amount of drug in the
body to the concentration in the plasma (Cp)
• For the theophylline example given in Example:
Maintenance Dose Calculation, the loading dose
would be 350 mg (35 L x 10 mg/L) for a 70-kg
person.
• For intravenous doses of theophylline, initial
injections should be given over a 20-minute
period to reduce the possibility of high plasma
concentrations during the distribution phase.
Examples of loading dose
• L = TC.Vd/ F
e.g. lidocaine t1/2 = 1-2 h
Post MI arrythmias –> life threatening –> can’t wait 4-8 h –>
Loading dose is used
• Another example: Digoxin for heart failure
If only maintenance dose is given it takes 10 days t1/2 = 61 h
L = 1.5 ng/ml x 580 / 0.7 = 1243 µg ~ 1 mg
It can be given iv divided – 0.5 mg, after 6-8 h 0.25 mg, after 6h
0.125 mg and then 0.125 mg (to avoid overdigitalization and
toxicity)
Multiple
IV
injections
Orally administered
drugs
D = the dose,
F = the fraction absorbed
(bioavailability)
T = dosage interval
Influence of the rate of drug infusion
on the steady state
• Css = the steady-state
concentration of the drug,
Ro = the infusion rate (for
example, mg/min)
• Because ke, CLt, and Vd are
constant for most drugs
showing first-order kinetics,
Css is directly proportional
to Ro
Influence of the clearance on the
steady state
• The steady-state concentration is inversely
proportional to the clearance of the drug, CLt
• liver or kidney disease, increases the steadystate concentration of an infused drug
• increased metabolism, decrease the steadystate concentrations of an infused drug
Therapeutic Drug Monitoring
Relating Pharmacokinetics &
Pharmacodynamics
• Diseases may modify all of the following
parameters:
– pharmacokinetic variables: absorption, clearance,
and volume of distribution (and the derived
variable, half-life)
– pharmacodynamic variables: maximum effect
attainable in the target tissue and the sensitivity
of the tissue to the drug
The Target Concentration Strategy
1. Choose the target concentration, TC.
2. Predict volume of distribution (Vd) and clearance (CL)
based on standard population values with adjustments for
factors such as weight and renal function.
3. Give a loading dose or maintenance dose calculated from
TC, Vd, and CL.
4. Measure the patient's response and drug concentration.
5. Revise Vd and/or CL based on the measured
concentration.
6. Repeat steps 3–5, adjusting the predicted dose to achieve
TC.
Pharmacokinetic Variables
• Absorption
– Patient's compliance
– Abnormalities in the small bowel
• Clearance
– function of the kidney, liver, & heart
– Creatinine clearance is a useful quantitative indicator
of renal function
– hepatic disease does not always affect the hepatic
intrinsic clearance
– there is no reliable marker of hepatic drugmetabolizing function
Pharmacokinetic Variables
• Volume of Distribution
– Older people have a relative decrease in skeletal muscle
mass and tend to have a smaller apparent volume of
distribution of digoxin (which binds to muscle proteins)
– The volume of distribution may be overestimated in obese
patients if based on body weight and the drug does not
enter fatty tissues well, as is the case with digoxin
– In contrast, theophylline has a volume of distribution
similar to that of total body water even in obese patients
– Abnormal accumulation of fluid—edema, ascites, pleural
effusion—can markedly increase the volume of
distribution of drugs such as gentamicin that are
hydrophilic and have small volumes of distribution
Pharmacokinetic Variables
• Half-Life
– For example, the half-life of diazepam increases
with age. The increasing half-life for diazepam
actually results from changes in the volume of
distribution with age; the metabolic processes
responsible for eliminating the drug are fairly
constant
Pharmacodynamic Variables
• Maximum Effect
– If increasing the dose in a particular patient does not
lead to a further clinical response, it is possible that
the maximum effect has been reached.
• Sensitivity
– The sensitivity of the target organ to drug
concentration is reflected by the concentration
required to produce 50% of maximum effect, the EC50.
– This may be a result of abnormal physiology—eg,
hyperkalemia diminishes responsiveness to digoxin—
or drug antagonism—eg, calcium channel blockers
impair the inotropic response to digoxin.
Timing of Samples for Concentration
Measurement
• It is important to avoid drawing blood until
absorption is complete (about 2 hours after an
oral dose)
– Some drugs such as digoxin and lithium take several
hours to distribute to tissues. Digoxin samples should
be taken at least 6 hours after the last dose and
lithium just before the next dose (usually 24 hours
after the last dose). Aminoglycosides distribute quite
rapidly, but it is still prudent to wait 1 hour after giving
the dose before taking a sample.
Initial Predictions of Volume of
Distribution
• In patients with edema, ascites, or pleural
effusions, the weight should be corrected as
follows: Subtract an estimate of the weight of
the excess fluid accumulation from the
measured weight. Use the resultant "normal"
body weight to calculate the normal volume
of distribution. Finally, this normal volume
should be increased by 1 L for each estimated
kilogram of excess fluid.
Initial Predictions of Clearance
• Cockcroft-Gault equation
• Because of the difficulty of obtaining complete
urine collections, creatinine clearance calculated
in this way is at least as reliable as estimates
based on urine collections.
• Fat-free mass should be used for obese patients,
and correction should be made for muscle
wasting in severely ill patients.
Revising Individual Estimates of
Volume of Distribution & Clearance
• If a patient is taking 0.25 mg of digoxin a day, a
clinician may expect the digoxin concentration
to be about 1 ng/mL.
– bioavailability of 70% and total clearance of about
7 L/h (CLrenal 4 L/h, CLnonrenal 3 L/h).
• In heart failure, the nonrenal (hepatic)
clearance might be halved (5.5 L/h). The
concentration is then expected to be about
1.3 ng/mL.
Revising Individual Estimates of
Volume of Distribution & Clearance
• Suppose that the concentration actually
measured is 2 ng/mL. Common sense would
suggest halving the daily dose to achieve a
target concentration of 1 ng/mL.
• This approach implies a revised clearance of
3.5 L/h, may reflect additional renal functional
impairment due to heart failure.
• This technique will often be misleading if
steady state has not been reached.
• Blog.sbmu.ac.ir/ziai