Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Silencer (genetics) wikipedia , lookup
Gene expression wikipedia , lookup
Amino acid synthesis wikipedia , lookup
Non-coding DNA wikipedia , lookup
Ancestral sequence reconstruction wikipedia , lookup
Proteolysis wikipedia , lookup
Deoxyribozyme wikipedia , lookup
Two-hybrid screening wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
Nucleic acid analogue wikipedia , lookup
Biochemistry wikipedia , lookup
Protein structure prediction wikipedia , lookup
Genetic code wikipedia , lookup
Point mutation wikipedia , lookup
Forschungsvorschlag: BLAST
Leistungskennzeichnung, Leistungsanalyse, und
Leistungsverbesserung
Craig Stewart
License Terms
•
•
•
•
Please cite this presentation as: Stewart, C.A. 2006. Forschungsvorschlag: BLAST
Leistungskennzeichnung, Leistungsanalyse, und Leistungsverbesserung.
(Presentation). 11 April 20076. Technische Universität Dresden, Germany.
Available from: http://hdl.handle.net/###___
Portions of this document that originated from sources outside IU are shown here and
used by permission or under licenses indicated within this document.
Items indicated with a © are under copyright and used here with permission. Such items
may not be reused without permission from the holder of copyright except where license
terms noted on a slide permit reuse.
Except where otherwise noted, the contents of this presentation are copyright 2007 by
the Trustees of Indiana University. This content is released under the Creative
Commons Attribution 3.0 Unported license
(http://creativecommons.org/licenses/by/3.0/). This license includes the following terms:
You are free to share – to copy, distribute and transmit the work and to remix – to adapt
the work under the following conditions: attribution – you must attribute the work in the
manner specified by the author or licensor (but not in any way that suggests that they
endorse you or your use of the work). For any reuse or distribution, you must make clear
to others the license terms of this work.
Introduction to some important
bioinformatics codes
• BLAST – Basic Local Alignment Search Tool
• Smith Waterman alignment – alignment by dynamic
programming (several implementations)
• Clustal-W, T-Coffee – Multiple alignment
• HMMER - profile HMMs for protein sequence analysis
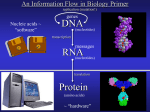
Central dogma of biology
•
The central dogma of biology
is that genes act to create
phenotypes through a flow of
information form DNA to RNA
to proteins, to interactions
among proteins (regulatory
circuits and metabolic
pathways), and ultimately to
phenotypes. Collections of
individual phenotypes
constitute a population (first
put forward by Crick in 1958)
http://www.ncbi.nlm.nih.gov/About/primer/genetics_cell.html
http://www.ornl.gov/sci/techresources/Human_Genome/graphics/slides/images/molecularmachine.jpg
Translating DNA to RNA and
Transcribing RNA to Proteins
DNA
AAAAAGGAGCAAATT
1
RNA
One possible amino
acid string
2
4
3
6
5
UUUUUCCUCGUUUAA
Phe
Asn
Asp
Ala
Genetic Code
Ala Alanine
Arg Arginine
Asn Asparagine
Asp Aspartic acid
Cys Cysteine
Glu Glutamic acid
Gln Glutamine
Gly Glycine
His Histidine
Ile Isoleucine
http://www.ncbi.nlm.nih.gov/Class/MLACourse/
Original8Hour/Genetics/geneticcode.html
Leu Leucine
Lys Lysine
Met Methionine
Phe Phenylalanine
Pro Proline
Ser Serine
Thr Threonine
Trp Tryptophan
Tyr Tyrosine
Val Valine
How do sequences differ?
• Differences in individual bases
CGTACCGTTAATAT
CGTACCGATAATAT
• Bases may be added to a
sequence
CGTACCCCGTAATAT
CGTACC . .GTAATAT
• Bases may be deleted from a
sequence
CGTACCGTTAATAT
CGTACCG . . .ATAT
Sickle Cell
Normal RBC
• GAG codes for Glutamine
• disc-Shaped, soft
• easily flow through small blood
vessels
• lives for 120 days
Sickle RBC
• GTG codes for Valine
• sickle-Shaped, hard
• often get stuck in small blood
vessels
• lives for 20 days or less
Malaria vs. Anemia!
http://www.nlm.nih.gov/medlineplus/
ency/imagepages/1223.htm
Goal: given a DNA or Amino acid
sequence, find something like it in a
large database
• For proteins, 95% similarity is ~ identical, 80% similarity is a
lot. Even less similarity than that needed for DNA
• Database techniques inadequate – they are too precise!
• Datasets very large to search
Alignment
• An alignment is an arrangement of two sequences opposite
one another
• It shows where they are different and where they are similar
• We want to find the optimal alignment - the most similarity
and the least differences
• Alignments have two aspects:
– Quantity: To what degree are the sequences similar
(percentage, other scoring method)
– Quality: Regions of similarity in a given sequence
Alignment
• Methods:
– dynamic programming
– Hidden Markov Models
– Pattern matching
• Key problem: keeping the calculation time manageable
• Some alignment packages:
– BLAST (http://www.ncbi.nlm.nih.gov/BLAST/)
– FASTA (http://gcg.nhri.org.tw/fasta.html)
Scoring Alignments
GCTAAATTC
++ x x
GC AAGTT
• Matches are good: they get a positive value
• Mismatches are bad: they get a negative value
• Gaps are bad: they get a negative value
– Gap opening penalty
– Gap extension penalty
– Score = Matches –Mismatches
-∑{gap opening penalty +(length)*gap length penalty}
(Original) BLAST Algorithm
• Original algorithm does not permit gaps
• The original BLAST algorithm is a local (heuristic) alignment
tool
• Given a search sequence, e.g. ACGTAGGCATGAA
• BLAST first makes a list of all “words” of a given length that
would possibly have a score of at least T against the search
string.
• In the case of this example there would be (at least) the
following:
– ACGTAGGCATG
– CGTAGGCATGA
– GTAGGCATGAA
(Original) BLAST Algorithm, 2
•
•
•
•
BLAST takes the list of all words with a score of at least T against
the string one is trying to match…. and then searches a database
for any matches to these words. So if one were using the example
and the NR database, BLAST would search NR for all occurrences
of the words:
– ACGTAGGCATG
– CGTAGGCATGA
– GTAGGCATGAA
Suppose BLAST finds in the NR database an exact match to
– ACGTAGGCATG
BLAST then attempts to extend the match in both directions
– ACGTAGGCATGA
– ACGTAGGCATGA
So now we have an exact match of 12 letters
(Original) BLAST algorithm,3
• So BLAST keeps going, and in this case would stop at an
exact match of 13 letters (if one existed), since 13 letters was
the entire initial search string:
– ACGTAGGCATGAA
– ACGTAGGCATGAA
• BLAST has a stopping algorithm for dropping particular
search directions, or stopping altogether
Scoring of DNA
A
C
G
T
R
Y
M
W
S
K
D
H
V
B
N
4
-3
-3
-3
1
-1
1
1
-2
-2
1
1
1
-2
1
A
C
G
T
4
-3
-3
-1
1
1
-2
1
-2
-2
1
1
1
1
4
-3
1
-1
-2
-2
1
1
1
-2
1
1
1
4
-1 1
1 -3
-2 0
1 0
-2 0
1 0
1 1
1 0
-2 1
1 0
1 0
R
Y
1
0
0
0
0
0
1
0
1
0
M
1
0
0
0
0
1
1
0
0
1
0
0
1
1
0
0
0
W
S
1
0
0
0
1
1
0
K
1
1
0
0
1
0
D
1
0
0
0
0
H
1
0
0
0
V
1
0
0
B
1
0
N
1
BLAST algorithm in more detail
•
•
•
•
•
•
•
•
The BLAST algorithm searches for MSPs – Maximal Scoring Pairs –
such that the score of sequences cannot be improved either by
lengthening it or shortening it. “Pairs” here refers to a string – or a
substring – of the initial string used as the search string – and one or
more strings or substrings found in a database.
The search starts with the creation of all possible subwords of a given
length (default typically 11 for DNA sequences, 3 amino acids for protein
sequences) that would score at least T when matched against the
original search string. (T is short for Threshold)
BLAST then goes through the database being searched against looking
for any occurrence of each of these words that have a score of at least
T. This is a “hit” – or a “High Scoring Pair (HSP)”
The search then continues by trying to extend these HSPs.
Suppose “S” is the best score found for a word of length k. BLAST
stops trying to extend words when the score drops a certain amount
below the best value S in the previous round.
BLAST continues on and on until it is no longer possible to improve the
score of HSPs by making them longer.
Then it generates a list of the best HSPs. Default is a cutoff E-value of
10
BLAST (original) has an infinite gap penalty
BLAST over the web from NCBI
www.ncbi.nlm.nih.gov/
BLAST over the web from NCBI, 2
www.ncbi.nlm.nih.gov/
BLAST Statistics
•
•
•
•
•
•
BLAST reports E values rather than P values, but it turns out that
when E < 0.01, E~P
What do we do about the fact that we have done many tests?
If the sequence is length n, and the total length of the database
being searched is N, then a reasonable approach is to multiply E by
N/n
Edge effects – statistics tend to be conservative for short sequences
Problems:
– Highly repetitive segments
– Low complexity regions
– Bias in composition
Solution: low complexity regions can be excluded
Protein Sequence Alignment
• What most people do most of the time
• DNA sequences are useful for relationships that are close,
but DNA sequences are not nearly as well conserved as
Amino Acid sequences
• Now we need to talk about the characteristics of Amino Acids
and ways to compare what is similar and what is not!
• Amino acids can have similar chemical properties, and similar
functions as part of a protein, without being identical!
Point Accepted Mutations (PAM)
•
•
•
•
•
For scoring amino acid sequence
alignments
Dayhoff, M.O., Schwartz, R.M., Orcutt,
B.C. 1978. "A model of evolutionary
change in proteins." In Atlas of Protein
Sequence and Structure 5(3) M.O. Dayhoff
(ed.), 345 - 352, National Biomedical
Research Foundation, Washington.
PAM N corresponds to N mutations in
DNA sequence per 100 amino acids. N
can be greater than 100.
PAM 250 is most commonly used; PAM
100 is also used. PAM 250 => chains with
~20% identity
PAM matrix calculator at
www.cmbi.kun.nl/bioinf/tools/pam.shtml
http://www.psc.edu/biomed/training/
tutorials/sequence/db/index.html
BLOSUM Matrices
• Henikoff and Henikoff (1992) Proc Natl Acad Sci
89(22):10915-9
• Based on analysis of the BLOCKS database
(http://www.blocks.fhcrc.org/)
• BLOSUM = BLOcks SUM database
• Based on analysis of conserved and variable regions of
proteins Naming convention is different than for PAM
matrices.
• BLOSUMxy is based on likelihood ratios for two chains of
amino acids that are xy% identical
• BLOSUM62 is the ‘typical default’
• PAM250 is roughly equivalent to BLOSUM45
How many BLASTs?
Parallel?
Blastn
www.ncbi.nlm.nih.gov/B
LAST/
pthreads
nucleotide BLAST
BLASTx
„
„
Translated query vs. protein
database
tBLASTn
“
“
Protein query vs. translated
database
tBLASTx
“
“
Translated query vs. translated
database
BLASTp
“
“
Protein-protein BLAST
PSI- & PHI-BLAST
“
“
Position-Specific Iterated and
Pattern-Hit Initiated BLAST
WU-BLAST
blast.wustl.edu/
“
Washington University BLAST
Vector Versions
Vectorversion
Vectortyp
Seriellversionen
versorgt
url
Verweisung
AG-BLAST
Apple
Veloc
ity
Engi
ne
BLASTn
www.apple.com/acg/
Apple 2006.
BLAST with
VMX
Support
VMX
BLAST 2.2.10: BLASTn,
BLASTp,
BLASTx,tBLAST,
tBLASTx
www.ciri.upc.es/cela_pblade/
BLAST.htm
CIRI 2005
Altivec-enabled
BLAST
VMX/Altiv
ec
BLAST 2.2.10 & 2.2.13:
nur BLASTn
Chen & Tzen
2006
High Throughput Versions
High Throughput version
Seriellversionen
versorgt
url
High-Throughput BLAST
Verweisung
Camp, Cofer, &
Gomperts 2001
BLAST on split databases
ftp://saf.bio.caltech.edu
Mathog 2003
BLAST with SMBL
hydra.indiana.edu/
Hart et al. 2003.
Strategies for parallelization
• Cut the input string into pieces – each piece is assigned to a
process, and the piece is used as an input string against the
entire database
• Cut the database into pieces – each of the pieces of the
database is put in a different server, and each server
processes the input data against that piece of the database.
Complicating issue: getting the statistics right
• Use of threads or vector operations to calculate the actual
similarity scores
Parallel Versions
Parallelversion
Seriellversionen
versorgt
url
Verweisung
MPI-BLAST
BLASTn, BLASTp,
BLASTx,tBLAST,
tBLASTx
mpiblast.lanl.gov/
Darling, Carey, & Feng 2003;
Feng 2003
www.paracel.com/
Boyson & Rieffel 2004;
Rieffel, Gill & White 2004;
Striking Developments
2006
Paracel BLAST
PARBLAST
NCBI 2.2.11
From IU, not yet
published
mpiBLAST http://mpiblast.lanl.gov/
mpiBLAST performance
• Scaling can be superlinear when pieces are small enough
that they fit into memory
• Scalability limitations due to communication, implicit barrier
before assembly of results
• If pieces of data distributed out to workers are larger than
available RAM, then scaling is still good but not superlinear
• Blast is the most heavily used bioinformatics tool in existence.
Parallelization of BLAST has huge payoff for practicing
biologists
Motivation: BLAST with Low Memory
• Standard BLAST running on a system with 128 MB of
memory.
Slide courtesy of and © Wu-chun Feng
[email protected] Los Alamos National Laboratory
mpiBLAST: Low-Memory Performance
• Environment
– 1, 2, or 4 nodes.
– Each node w/ dual
550-MHz CPUs and
128-MB memory.
– Same query and
database used.
• Conclusions
– blastn is I/O bound.
Superlinear speed-up
possible.
– tblastx is CPU bound.
Slide courtesy of and © Wu-chun Feng
[email protected] Los Alamos National Laboratory
mpiBLAST
on Green Destiny
BLAST Run Time for 300-kB Query against nt
Nodes
Runtime (s)
Speedup over 1 node
1 80774.93
1.00
4
8751.97
9.23
8
4547.83
17.76
16
2436.60
33.15
32
1349.92
59.84
64
850.75
94.95
128
473.79
170.49
The Bottom Line: mpiBLAST reduces search time
from 1346 minutes (or 22.4 hours) to under 8
minutes!
Slide courtesy of and © Wu-chun Feng
[email protected] Los Alamos National Laboratory
PARBLAST
•
•
•
•
From IU
We think it I s better engineered than mpiBLAST
Parallelization by subdividing input string works just fine
Subdivision of atabase presently capable of generating core
dumps
Paracel BLAST
• Have made queries with the vendor – not clear if they care to
have their code benchmarked.
Vector Processing – from IBM
Vector processing – from Apple
Ideas for research for now and for the
long term
•
•
•
Leistungskennzighnung und leistingsanalyse. Es ist einfach zu
finden Websieten oder papieren uber Hochleistungsversionen uber
BLAST. Alle sind da selber: Unser version ist schneller, und mit meh
prozoessoren ist schneller als mit veniger processoren. Aber wenn
man finden wollen, was Prozent Leistungsfähigkeit von theoretische
Hochgrenze – das kann man nicht. Es ist klar, das die „super linear
speedups“ sind nur Speicherwirkungen. Was Prozent von
theoreticshe Hochgrenze benutz verschiedenes versionen von
BLAST?
Leistungsanalyse und Leistungsverbesserung mit MPI-BLAST
und/oder PARBLAST.
Vektorfähigkeiten:
– Mit VMS bei PowerPC Prozessoren
– Mit SSE bie Intel oder AMD Prozessoren
Und fuer April:
•
•
Für ein Einmonat Forschungsbesuch, es waere gut das folgende zu
komplett machen:
– Zwei oder drei Versionen zu vergleichen, mit verschiedene
Suchsequenzen, Datenbanken, und Prozessorennummer.
(BLAST Versionen: mpiBLAST, PARBLAST, und Paracel
BLAST velleicht).
– Leistenganalyse und Leistungsverbesserung mit ein oder zwei
Versionen von BLAST (mpiBLAST und PARBLAST)
Es gibt dann ministens ein, aber besser zwei, Ergebnisse:
– Kentnisse uber was verschiedine Versionen von BLAST macht
für Flieβkommarechnungen, Integerrechnung, und
Narchrichtenverbindungzeit mit verschiedene Eingabe und
Prozessorennummer.
– Hoffentlich versionen von PARBLAST und/oder MPIBLAST die
schneller laufen.
– Velleicht ein Sammlung von Eingabedaten fur Leistungsanalyse
von BLAST