Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Oxidation state wikipedia , lookup
Hydroformylation wikipedia , lookup
Cluster chemistry wikipedia , lookup
Metal carbonyl wikipedia , lookup
Jahn–Teller effect wikipedia , lookup
Evolution of metal ions in biological systems wikipedia , lookup
Spin crossover wikipedia , lookup
Metalloprotein wikipedia , lookup
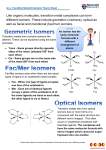
Chemistry 3211 – Coordination Chemistry Part 1 Introduction to Coordination Chemistry Covered in Chapter 8 of S & A 4Ed The elements arising from the filling of the 3d, 4d and 5d subshells, and located in the periodic table following the alkaline earth metals, are commonly referred to as the transition elements or the d-block elements. However, specifically speaking, a transition element is defined as one that has an incomplete d subshell in either the neutral atom or its ions. Thus, the Group 12 elements (Zn, Cd, Hg) are members of the d-block, but they are not transition elements. Sometimes, the term transition element is also extended to those elements possessing electrons in 4f and 5f orbitals, namely the lanthanide and actinide (or inner transition) elements. They exhibit a number of characteristic properties, which together distinguish them from other groups of elements: 1) They are all metals, and are therefore lustrous and malleable and have high electrical and thermal conductivities. Also, their melting and boiling points tend to be quite high and they are generally hard and strong. 2) Most of them display numerous oxidation states, which vary by steps of 1 rather than 2 as is usually the case with those main group elements that exhibit more than one oxidation state. 3) They generally have Lewis acid properties and, therefore, have a strong tendency for forming coordination compounds with Lewis bases. Coordination complexes have been known for over a century. They were originally called “complex compounds” because of the difficulties they caused for chemists studying them. These materials are stable (more or less) and stoichiometrically reliable, but they are often made up of combinations of other compounds that are independently stable. For example, Cu2+ ions combine with water to give Cu[H 2 O] 4 2+ ions. The Cu2+ ion coordinates four water molecules. The water molecules, which coordinate to the copper ion, are called ligands. It is important to realize that the interaction between the cation (Lewis acid) and the ligand (Lewis base) is not merely an intermolecular interaction based on electrostatics, like ion-dipole interactions for Na+ and Cl- ions in aqueous solution. Removal of solvent in an aqueous solution of sodium chloride just regenerates the solid sodium chloride. In coordination complexes, the cation-ligand interaction is actually a chemical bond. The bond possesses a highly variable, but nonetheless substantial bond enthalpy. Removal of water from such solutions of coordination complexes yields, for example, “hydrate” complexes, where the coordinated water remains with the ion in the solid state. Remember, that water is just another potential ligand. Much of traditional coordination chemistry is carried out in water, and the competition for metal binding between water and various ligands is a central theme. Review the concepts in Chapter 4 S & A 4Ed on complex ion equilibria and acid-base donor acceptor properties. If you wish to consult Housecroft & Sharpe 2nd Ed. (H & S) on introductory principles of coordination chemistry, see Section 6.11. Types of ligand Ligands are most commonly classified according to the number of donor atoms that they contain. They most often can be charge neutral or anionic. Simple monodentate or unidentate (meaning “onetoothed”) ligands include NH 3 , H 2 O, HS-, RO- and many others (see Table 8-1 in S & A 4Ed). Bidentate ligands are very common. Examples include ethylenediamine, 1.10-phenanthroline and oxalate. These are often “chelating” ligands (from Greek χηλ, crab’s claw). C. M. Kozak – Chemistry 3211 1 Tridentate and multidentate ligands are also an important class, especially as bioinorganic ligands. Porphyrins, corrins, etc. are a fascinating class of biological tetradentate macrocyclic (ring-type) ligands. Proteins themselves have Lewis basic sites throughout which can act as ligands for metal ions in organisms. The research being conducted in the design of new metal-ligand combinations is vast and varied. The virtually infinite combination of “designer” ligands and metals means that the area is continually finding new complexes capable of myriad transformations, catalyses and other applications, in addition to expanding our knowledge of the fundamental behaviour and properties of inorganic compounds. Below is a small sampling of the more common multidentate ligands being used today. The possibility to make derivatives of these ligands means that their chemistry can be “fine tuned” for their desired purpose. NH2 NH2 O O PPh2 O O PPh2 NH NH2 NH2 Ethylenediamine, en R Diethylenetriamine, dien Oxalate R O O N R N O AsMe2 O AsMe2 R βDiketiminate, nacnac Acetylacetonate, acac 1,2-Bis(diphenylphosphino) ethane, dppe Tropolonate 1,2-Bis(dimethylarsino) benzene, diars N N N N N N N N Terpyridine, terpy 1,10-Phenanthroline, phen Porphorinate H O O N N O O B N N O N O O N N N O Ethylenediaminetetraacetate, EDTA C. M. Kozak – Chemistry 3211 N Hydrotris(pyrazolyl)borate 2 Historical Development Geometries of d-metal complexes were identified by Alfred Werner (Swiss Chemist, 1866-1919, 1st independent research paper published at age 25). He studied Co3+ complexes with ammonia ligands: CoCl 3 ·6NH 3 While structures weren’t known, these compounds were known according to their colour. This method was thought to be less random than the previous approach of naming complexes after their discoverer. In a sense, chemists still use their colour properties to characterize coordination complexes (e.g. UVvisible spectroscopy). Original Formulation Name Modern Form Cr(SCN) 3 ·NH 4 SCN·2NH 3 Reinecke’s Green Salt NH 4 [Cr(NCS) 4 (NH 3 ) 2 ] 2PtCl 2 ·2NH 3 Magnus’s Green Salt [Pt(NH 3 ) 4 ][PtCl 4 ] Co(NO 2 ) 3 ·KNO 2 ·2NH 3 Erdmann’s Salt K[Co(NO 2 ) 4 (NH 3 ) 2 ] PtCl 2 ·KCl·C 2 H 4 Zeise’s Salt K[PtCl 3 (C 2 H 4 )] Later, complexes where named using Latin to describe their colour. Originally Colour Name Modern Form 1) CoCl 3 ·6NH 3 Yellow Luteocobaltchloride [Co(NH 3 ) 6 ]Cl 3 2) CoCl 3 ·5NH 3 Purple Purpureocobaltchloride [CoCl(NH 3 ) 5 ]Cl 2 3) CoCl 3 ·4NH 3 Green Praseocobaltchloride trans-[CoCl 2 (NH 3 ) 4 ]Cl 4) CoCl 3 ·4NH 3 Violet Violeocobaltchloride cis-[CoCl 2 (NH 3 ) 4 ]Cl Roseocobaltchloride [Co(NH 3 ) 5 (H 2 O)]Cl 3 5) CoCl 3 ·5NH 3 ·H 2 O Red How did Werner characterize and separate these apparently very similar formulas? He used silver nitrate, AgNO 3 to precipitate out AgCl. The equivalence of AgCl precipitate indicates the strength of Cl- association. With compound 1), all of the chloride precipitated as solid AgCl. With compound 2), only two thirds of the chloride precipitated, and with compounds 3) and 4), only one third equivalent of AgCl formed. Werner accounted for this by proposing that these complexes possessed six ligands (either chloride or ammonia molecules) attached to each Co3+ ion. Using this relationship he used the terms “primary valence” and “secondary valence” to distinguish between strong metal ligand bonds and weaker ones. These terms have since evolved into oxidation state and coordination number, respectively. Modern Rules for Naming Complexes 1. The positive ion (cation) comes first, followed by the negative ion (anion). This is the common order for simple salts as well. Examples: diamminesilver(I) chloride, [Ag(NH 3 ) 2 ]Cl potassium hexacyanoferrate(III), K 3 [Fe(CN) 6 ] C. M. Kozak – Chemistry 3211 3 2. Within the coordination sphere, the ligands are named before the metal, but in formulas the metal ion is written first. The inner coordination sphere is enclosed in square brackets in the formula. Examples: tetraamminecopper(II) sulfate, [Cu(NH 3 ) 4 ]SO 4 hexaamminecobalt(III) chloride, [Co(NH 3 ) 6 ]Cl 3 3. Two systems exist for designating charge or oxidation number: a. The Stock system puts the calculated oxidation number of the metal ion as a Roman numeral in parentheses after the name of the coordination sphere. This is the more common convention, although there are cases where it is difficult to assign oxidation numbers. b. The Ewing-Bassett system puts the charge on the coordination sphere in parentheses after the name of the coordination sphere. This convention is used by Chemical Abstracts and offers an unambiguous identification of the species. In either case, if the charge is negative, the suffix -ate is added to the name of the coordination sphere. Examples: tetraammineplatinum(II) or tetraammineplatinum(2+), [Pt(NH 3 ) 4 ]2+ tetrachloroplatinate(II) or tetrachloroplatinate(2-), [PtCl 4 ]2hexachloroplatinate(IV) or hexachloroplatinate(2-), [PtCl 6 ]2- 4. Ligands are named in alphabetical order (according to the name of the ligand, not the prefix), although exceptions to this rule are common. An earlier rule gave anionic ligands first, then neutral ligands, each listed alphabetically. Examples: tetraamminedichlorocobalt(III), [Co(NH 3 ) 4 Cl 2 ]+ amminebromochloromethylamineplatinum(II), [Pt(NH 3 )BrCl(CH 3 NH 2 )] 5. Anionic ligands are given an o suffix. Neutral ligands retain their usual name. Coordinated water is called aqua. Examples: chloro, Clbromo, Brsulfato, SO 4 2methylamine, CH 3 NH 2 ammine, NH 3 (the double m distinguishes NH 3 from alkyl amines) aqua, H 2 O 6. The number of ligands of one kind is given by the following prefixes. If the ligand name includes these prefixes or is complicated, it is set off in parentheses and the second set of prefixes is used. 2 3 4 5 6 di tri tetra penta hexa bis tris tetrakis pentakis hexakis C. M. Kozak – Chemistry 3211 4 7 8 9 10 Examples: hepta octa nona deca heptakis octakis nonakis decakis Simple ligands are given above. dichlorobis(ethylenediamine)cobalt(III), [Co(NH 2 CH 2 CH 2 NH 2 ) 2 Cl 2 ]+ tris(bipyridine)iron(II), [Fe(C 5 H 4 N–C 5 H 4 N) 3 ]2+ 7. The prefixes cis- and trans- designate adjacent and opposite geometric locations. Examples are given in the figure below. Other prefixes are used as well and will be introduced as needed in the text. cis- and trans-diamminedichloroplatinum(II), [PtCl 2 (NH 3 ) 2 ] cis- and trans-tetraamminedichlorocobalt(III), [CoCl 2 (NH 3 ) 4 ]+ Examples: NH3 H3 N H3N Pt H3 N Cl H 3N Cl Cl Cl Pt H3 N Cl H3N Cl H3N NH3 NH3 cis- and trans-dichlorodiammineplatinum(II) [PtCl 2 (NH 3 ) 2 ] Co Cl Co NH3 NH3 Cl cis- and trans-dichlorotetraamminecobalt(III) [CoCl 2 (NH 3 ) 4 ]+ 8. Bridging ligands between two metal ions as in the figure below have the prefix μ-. tris(tetraammine-μ-dihydroxocobalt)cobalt(6+), [Co(Co(NH 3 ) 4 (OH) 2 ) 3 ]6+ μ-amido-μ-hydroxobis(tetraamminecobalt)(4+), [(NH 3 ) 4 Co(OH)(NH 2 )Co(NH 3 ) 4 ]4+ Examples: NH3 H3N NH3 H3N H3N Co H O Co NH3 NH3 HO OH Co O NH3 H HO OH Co H3N NH3 NH3 H3N NH3 H3N NH3 tris(tetraammine-μ-dihydroxocobalt)cobalt(6+) [Co(Co(NH 3 ) 4 (OH) 2 ) 3 ]6+ C. M. Kozak – Chemistry 3211 Co H O O NH3 H NH3 Co NH3 NH3 NH3 μ-amido-μ-hydroxodi-(tetraamminecobalt)(4+), [(NH 3 ) 4 Co(OH)(NH 2 )Co(NH 3 ) 4 ]4+ 5 Structures of Coordination Complexes Coordination numbers (the number of metal-ligand interactions) and isomerism are important characteristics of coordination complexes. Inner sphere complexes are those with ligands directly attached to the central metal, the primary (1o) coordination sphere (shown by ligands within the square brackets). Outer sphere complexes possess ion pairs and a secondary (2o) coordination sphere (shown by ligands outside the square brackets). Three factors determine the coordination number (C.N.): i) the size of central atom/ion; ii) the steric interaction between ligands; iii) electronic interactions. Complexes may exhibit coordination numbers ranging from 1 to 12 or higher! The most commonly observed coordination numbers for transition metals are 4 to 6. Some examples of complexes with various coordination numbers are given below. Examples of the Variety of Coordination Numbers Found in Transition Metal Complexes a) C.N. of 1 or 2 is exceedingly rare and requires very bulky ligands to protect the coordination sphere of the metal, mostly later transition metals (Groups 11 or 12). The complex bearing the bulky ligand, [2,4,5-Ph 3 C 6 H 2 Cu] shown below is an example of a 1-coordinate copper(+1) compound. The complexes [CuX 2 ]- (where X = Cl, Br), AuCl(PEt 3 ), and Hg(CH 3 ) 2 are examples of 2-coordinate species. Cu b) C.N. of 3 is also preferred by the later transition metals. The coordination number may not always be apparent from the written formula, e.g. K[Cu(CN) 2 ] appears to be 2 coordinate, but CN- is a ligand which may bridge two metals, therefore in the solid state we see sheets of trigonal planar Cu centres. Cu C N Cu C N Cu Me3Si C N Cu Me3Si N n SiMe3 Fe N SiMe3 N Me3Si SiMe3 Ph3P Au PPh3 PPh3 c) Four coordinate complexes are very common and favoured for early transition metals, especially 3d, and for large ligands. Two geometries are possible: tetrahedral and square planar, as well as intermediate/distorted geometries. Ideal tetrahedral geometry occurs for homoleptic complexes (those possessing all the same ligand). Classic examples are early-mid transition metals in high oxidation states with oxide, O2-, ligands and late transition metal halide complexes in the +2 oxidation state. C. M. Kozak – Chemistry 3211 6 O 2- Cl Mn O Cu O Cl O Cl Cl Werner’s early observations of four coordinate Pt2+ complexes led to the surprising conclusion that two isomers exist for complexes such as Pt(NH 3 ) 2 Cl 2 . This shows that their structure cannot be tetrahedral. However, a planar structure can have two isomers, named cis and trans. Square planar complexes of 3d metals are typically formed by metal ions with d8 electron configurations (such as Ni2+) with ligands that can form π bonds by accepting electrons from the metal atom (more on this later). For 4d and 5d metals with d8 configurations, they are almost always square planar. Cl H3N Cl H3N Pt H3N Pt Cl Cl NH3 trans cis d) Five coordinate complexes are less common, but important for their involvement reactivity and reaction mechanisms (discussed later). Two geometries exist, square pyramidal and trigonal bipyramidal, and the ligands strongly influence which one is observed. Energies of these geometries are often similar, therefore, fluxional processes may interconvert them. The most common mechanism of interconversion is Berry pseudorotation, which can be studied spectroscopically using NMR (discussed later). 3- Cl Cl Cu 3- CN Cl NC Cl NC Cl Ni CN CN e) Six coordinate complexes are the most common and almost all are octahedral in geometry. Trigonal prismatic geometries are much less common. Recall that Werner proposed that 6 groups were arranged in a symmetrical fashion about the Co3+ ion. Based on the number of observed isomers, Werner concluded that the most likely arrangement is as an octahedron. Distortions from ideal octahedral geometry are also possible. In particular, d9 complexes may undergo tetragonal, rhombic and trigonal distortions (more later). Octahedral complexes can possess two types of isomerism: cis/trans and facial/meridional, fac/mer. H3N Co H3N Cl H3N Cl Cl NH3 cis purple C. M. Kozak – Chemistry 3211 Co NH3 trans green NH3 Cl NH3 NH3 Cl H3N NH3 H3N Co NH3 fac facial Cl H3N Cl Cl Co Cl Cl NH3 mer meridional 7 f) Higher coordination numbers are also possible, such as 7 and 8 coordinate species in heavier d block metals (4d and 5d) and the lanthanides. Seven coordinate geometries are described as pentagonal bipyramidal, capped octahedral, and monocapped triangular prismatic. Formal Oxidation States of Metals in Coordination Complexes To figure out the oxidation state or oxidation number of the central metal atom in a complex is very important. Proceed as follows: • identify the charges on the ligands • look at the total charge on the molecule ⇒ charge of ligands + formal oxidation state = total charge e.g., [FeCl 4 ]2- has 4 Cl- ligands and overall 2- charge, so it must contain Fe+2 or Fe(II). Electron Configurations Consult Section 1.6 to 1.9 in S & A 4Ed to review the principles of electron configuration, shielding and effective nuclear charge. It also covers periodic trends in Atomic Parameters, such as size, ionization energy, electron affinity, electronegativity, polarizability. You MUST fully understand these basic concepts to grasp some of the more complex ideas that will be presented later on. A. Atoms For elements up to Argon (Z=18), we fill the 1s, 2s, 2p, 3s, and 3p levels in that order. Filling these orbitals causes various changes to the energies of the higher and as yet unfilled orbitals. Not all orbitals are affected equally by the screening of the nuclear charge provided by these s and p electrons. In particular, the 3d orbitals, which penetrate the Ar core rather little, are relatively unaffected. In contrast, the 4p level and especially the 4s level penetrate the core quite a bit and drop substantially in energy. The next two electrons are added to the 4s level. Potassium (Z=19) and Calcium (Z=20) result. What level do we fill next? Recall that we have increased the nuclear charge by 2 and that the 4s orbitals are quite diffuse (i.e., ineffective at screening the nuclear charge), the 3d orbitals drop in energy and are filled next. Thus, Scandium (Sc) has the configuration [Ar]4s23d1. Titanium is [Ar]4s23d2. Chromium (Cr) is peculiar, being [Ar]4s13d5, not [Ar]4s23d4. Copper (Cu) is also anomalous, being [Ar]4s13d10, not [Ar]4s23d9. This is due to electron-electron repulsion and results in higher stability for “half-filled” and “filled” shells. The diagram below illustrates the approximate energy levels for the orbitals with C. M. Kozak – Chemistry 3211 8 increasing principle quantum number, n, and atomic number, Z. Note the relatively similar energies of the 3d and 4s orbitals for the first row transition metals, Z = 21 to 30. B. Complexes The above-described process for filling orbitals (Aufbau process) applies only to atoms, and therefore only represents their ground state electron configurations. In transition metal complexes, the nd orbitals end up lower in energy than the (n + 1) s orbitals (ie. 3d is filled before 4s). e.g., in Vanadium complexes: V(0) 3d5 not 4s23d3, as in free atoms! V(I) 3d4 V(II) 3d3 V(III) 3d2 V(IV) 3d1 C. M. Kozak – Chemistry 3211 9 General electron counting rules for number of d electrons: 1. Count total number of valence electrons from the periodic table. 2. Deduce oxidation state. 3. Subtract. Examples: Fe2+ is d6, Fe3+ is d5, Co2+ is d7, Co3+ is d6. Isomerism In order to assist your understanding of isomerism and chirality as it relates to coordination complexes, it is strongly recommended that you review the material on Symmetry in M&T Chapter 4 (specifically, Sections 4-1 and 4-2 only). The material in Ch 4 on matrices and character tables is not important for this discussion. There are many types of isomers in coordination chemistry. Some isomers are structurally similar and possess the same basic formula, but posssess different ligands attached to the metal centre. These may be solvento isomers, which vary the solvent such as water (aquo), or other coordinating solvents. Common examples are tetrahydrofuran (THF), pyridine (py), acetone, methanol, etc. Ionization isomers are ion pairs in which the counter ion to the metal containing ion may vary, such as substituting the outer sphere Cl in [Co(NH 3 ) 4 Cl 2 ]Cl with a Br to give [Co(NH 3 ) 4 Cl 2 ]Br. Coordination isomers are related but substitute the complex ion in the structure. Linkage isomerism or ambidentate isomerism refers to the same ligand bonding to the metal centre via different atoms. Stereoisomers possess the same ligands and metals, but differ in the geometric or spatial arrangement of the ligands. We have previously discussed some forms of stereoisomerism, namely cis/trans and fac/mer orientations of ligands. An additional type of isomerism is noted for a complex closely related to cis[Co(NH 3 ) 4 Cl 2 ]+. If the four NH 3 ligands are replaced with two ethylenediamine (en) ligands, we now have the possibility of forming a chiral complex that can exhibit two enantiomers. Recall that chiral complexes cannot be superimposed on their own image and have the ability to polarize light (they are optically active). The resulting [Co(en) 2 Cl 2 ]+ complex may now have three possible confirmations. Werner studied en complexes of Co3+ and observed the presence of three related systems. Mirror Plane N Cl Co N N N Cl N trans, green colour Internal mirror plane Achiral, optically inactive C. M. Kozak – Chemistry 3211 N Co N N N Cl Cl Cl Cl Co N N N cis, violet colour External mirror plane Chiral, optically active 10 Complexes such as [Co(en) 3 ]3+ and [Fe(ox) 3 ]3- (ox = oxalate) can exist as two optical isomers. All complexes of this general type are called tris chelate complexes, and their structures can be represented schematically: Λ Δ These two molecules are enantiomers. The chiral complex [Co(en) 3 ]3+ is yellow in colour and optically active. Official nomenclature differentiates between the two conformations be labelling them lambda (Λ) and delta (Δ), based on their “screw axis”. Lambda represents a left-handed (counter clockwise) screw, and delta represents a right-handed (clockwise) screw axis. These labels have no correlation to the actual optical rotation, only regards to the absolute configuration. That is, a Δ configuration may cause a d (+) or l (-) rotation in polarized light. Enantiomers have the same physical properties of solubility, melting point, etc., therefore they cannot be separated by these means. How does one isolate one enantiomer selectively? Current technology allows the use of chiral separation media (columns). These columns behave like silica or alumina columns used in chromatography. They are able to separate two enantiomers of a complex based on their geometry, rather than on their polarity. This is a powerful technique when used with modern instruments such as HPLC. Classically, however, the first person to separate optical isomers was Louis Pasteur, who crystallized tartaric acid. Tartaric acid occurs in the meso and rac forms. The racemic mixture contains two enantiomers which Pasteur separated using a polarized microscope to “crystal pick” one enantiomer over another. The availability of the enantiomerically pure tartaric acid allowed for its use as a chiral counterion in coordination complexes. The incorporation of pure tartaric acid with the mixed isomers of a metal complex results in the formation of two diastereomers. The resulting diastereomers have different physical properties (ie. solubility) because the “fit” or packing of crystals will be different. Basically, one becomes more soluble than the other. Consult Box 8.2 (p. 234) in S & A 4Ed for a more detailed discussion. Similar isomers are possible with multidentate ligands, such as the tri- and tetradentate amines diethylenetriamine (dien) and triaminotriethylamine (tren), respectively. N [Co(dien)2]3+ N Co N N fac N N N N N Co N dien = N NH2 H N H2N N mer Complexes possessing tetradentate ligands, such as tren for example, have three forms based on the relative positions of the planes of the chelate rings. Other isomers are possible when the two remaining coordination sites of the octahedron possess two different ligands leading to very complicated arrays of conformations. Consult Sections 8.10 in S & A 4Ed for a detailed discussion. Multidentate ligands that generate 5 or 6-membered rings are the most preferred. However, even in these preferred systems there may exist a degree of strain due to steric effects. The resulting minimum C. M. Kozak – Chemistry 3211 11 energy angle that the ligand possesses is called the bite angle. A small bite angle may cause further distortion in the complex, for example, a true octahedral complex may distort to adopt a trigonal prismatic geometry if the ligand present has a small bite angle. Ligand Ring Conformation The ligand chelate rings themselves may not be planar and this results in another source of different conformations in complexes. The notation used to describe this chirality is similar to clockwise and counter clockwise notation used for the “propeller” shaped isomers described above. Chelate ring chirality is described by the lower case characters for delta (δ) and lambda (λ). The diagram below shows a chelate ring formed by an ethylenediamine (en) ligand on a metal. The chirality can be assigned based on the rotation of the plane formed by the chelate. This is done by imagining a line connecting the two donor atoms, N, which we label Line 1 in the diagram. Line 2 connects the two atoms adjacent to the donor atoms; in this case, it is the C-C bond. The chirality is assigned based on the direction of rotation required to translate Line 1 onto Line 2. The side view illustrations show that a clockwise rotation of Line 1 results in its alignment with Line 2 to give a δ conformation, while a counter clockwise rotation yields a λ conformation. Top View M N N Line 1 Line 2 C C Side View N M N C δ C C M N C N λ The presence of one ligand ring conformation over another may be thermodynamically favoured for a particular complex chirality. For example, in a [Co(en) 3 ]3+ complex bearing a Δ chirality, the three individual ligand ring conformations will favour λ conformations in order to achieve minimum steric hindrance. The Δλλλ form was found to be more stable than the Δδδδ orientation by 7.5 kJ/mol. The exact nature of this preference, of course, is strongly dependant upon the ligands and the size of the metal ion. Also, in solution, this small difference in energy allows rapid interconversion between δ and λ and mixtures of ligand ring conformations may result. The interconversion between Δ and Λ, however, often requires the breaking and forming of metal-ligand bonds, and is therefore much higher in energy, but interconversion may still occur via a trigonal prismatic intermediate. C. M. Kozak – Chemistry 3211 12 Constitutional Isomers Hydrate (or solvent) isomerism is not common, but it played an important role in the foundation of Werner’s theory. It describes variations between isomers possessing differing quantities of water (or other solvent molecules) present in either the inner or outer coordination sphere. This occurs not only in transition metal coordination complexes, but is main group salts as well, such as Na 2 SO 4 , which may be anhydrous, or possess 7 or 12 equivalents of water molecules in its “hydrate” form. The classic coordination complex example is CrCl 3 ·6 H 2 O (chromium(III)chloride hexahydrate). It can occur as three distinct crystalline forms depending upon the number of H 2 O molecules in the inner coordination sphere. [Cr(H 2 O) 6 ]Cl 3 is violet, [CrCl(H 2 O) 5 ]Cl 2 ·H 2 O is blue-green and [CrCl 2 (H 2 O) 4 ]Cl·2 H 2 O is dark green. Ionization Isomerism Compounds that have the same formula but which differ in their ion arrangement in solution are ionization isomers. The difference occurs in whether the different ions are in the inner or outer coordination sphere of the complex, for example, [Co(NH 3 ) 4 (H 2 O)Cl]Br 2 and [Co(NH 3 ) 4 Br 2 ]Cl·H 2 O. Note that these two complexes are also hydrate isomers. Coordination Isomerism Best described for complexes bearing two or more metal ions. The overall ratio of ligands to metals remains constant, but the arrangement of the coordination sphere around each metal varies. The metals may be the same or different, and the oxidation state of each metal may also vary. For example, [Pt(NH 3 ) 2 Cl 2 ] may also crystallize as [Pt(NH 3 ) 4 ][PtCl 4 ] depending on the conditions. Ion pairs, such as [Co(en) 3 ][Cr(CN) 6 ] and [Cr(en) 3 ][Co(CN) 6 ], are another example of coordination isomers. Linkage Isomerism (Ambidentate Ligands) Some ligands possess different potential donor atoms. Ligands such as SCN- (thiocyanate), NO 2 (nitrite), or (CH 3 ) 2 SO (dimethylsulfoxide, DMSO) are common examples of simple ambidentate ligands. The atom to which the metal binds depends on whether the metal is a hard or soft Lewis acid. In thiocyanate, hard acids will bind to the nitrogen atom, a hard base. Soft acids will prefer to bind the the sulphur atom, which is a soft base. Other factors, such as the solvent used, will also influence the bonding. C. M. Kozak – Chemistry 3211 13