Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Click chemistry wikipedia , lookup
Chemical reaction wikipedia , lookup
Marcus theory wikipedia , lookup
Electrolysis of water wikipedia , lookup
Chemical equilibrium wikipedia , lookup
Solar air conditioning wikipedia , lookup
Stoichiometry wikipedia , lookup
Countercurrent exchange wikipedia , lookup
George S. Hammond wikipedia , lookup
Thermodynamics wikipedia , lookup
Heat capacity wikipedia , lookup
Bioorthogonal chemistry wikipedia , lookup
Heat transfer wikipedia , lookup
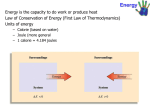
What do you already know about energy? Energy needs in the natural world? Comes from the sun Chemical bonds of photosynthesis Chemical bonds of cellular respiration Energy for life Heats the atmosphere – controls climate Why is Energy Important? Energy for human advancement and sustenance Fossil fuels Worth war? Extraction processes? Kinds of costs? Worth the waste? ○ Implications for earth as a whole? Alternative energy sources to Fossil Fuel Practical? Universal? Why hasn’t it already happened? What is Energy? The capacity to do work OR produce heat Law of Conservation of Energy Energy can be converted from one form to another but can be neither created nor destroyed Energy of the Universe is CONSTANT Potential Energy Energy due to position or COMPOSITION (chemical bonds) Position: water behind a dam converts eventually to electrical energy Composition: Gasoline burns to release energy (due to attractive and repulsive forces in the reactants and products of combustion) Kinetic Energy Energy of an object is due to the motion of the object Depends on the mass (m) Depends on its velocity (v) KE = 1/2mv2 Energy can be transferred orHow is PE converted to Converted KE? Are there other forces? Is any energy lost? If so, where did it go? Is there any frictional heating happening? What is the difference between heat and temperature? Temperature – measures the random motions of the particles in a substance Heat – TRANSFER of energy between TWO objects due to temperature differences. (heat is NOT contained in an object!!) Heat versus Temperature What is WORK?? Work is defined as force acting over a distance Is any work happening? There are TWO ways energy can be transferred! By WORK By HEAT The way energy is divided between work and heat depends on Pathway 1. If pathway is ROUGH, maybe NO work is done, but a lot of heat is generated 2. If pathway is SMOOTH, a lot of work can be performed, maybe with little What is a State Function/Property? SF/P – any property that only depends on its PRESENT state. Value doesn’t depend on how the system arrived. Change in SF/P is independent of the pathway Analogy: Imagine a trip from Chicago to Denver ○ Consider elevation. No matter how you travel the elevation change is the same (state function) ○ Consider distance. Distance totally depends on the pathway you take (not a state function) Are either of these pathways a state function? Compare System and Surroundings System – is place of focus (example: calorimeter) Surroundings – anyplace outside the system (outside the calorimeter) Two heat possibilities in reactions Exothermic – heat is REMOVED from the system Endothermic- heat is added to the system Where does heat come from in exothermic reactions? Difference in potential energies between the products and the reactions. If reactant has higher potential energy than reactant – heat will be given off from the system Energy gained by the surrounding must be equal to the energy lost by the system Example of exothermic event CH4 + 2 O2 CO2 + 2H2O + (energy)heat CH4 + 2O2 have MORE potential energy in the chemical bonds than CO2 + 2H2O have in their bonds Heat flow results from lowering the potential energy All exothermic reactions result in thermal energy release via heat ExoThermic Heat flow diagram: Endothermic Energy flows INTO the system Products have more energy (PE) than reactants Endothermic Heat Flow Thermodynamics Study of energy and its interconversions First Law of Thermodynamics The energy of the universe is constant Internal Energy (E) Sum of the KE + PE of all “particles” of the system E can be changed by a flow of work, heat or both ∆E=q+w ○ ∆ E= internal energy ○ Q = heat ○ W = work What does this Flow chart mean?? The lines represent exo and endo thermic reactions. Which color is which? Thermodynamics symbology TWO numbers Magnitude = quantity Sign= perspective ○ OURS – from the system point of view Exothermic = negative sign (energy LEAVES the system) Endothermic = + sign (energy is GAINED by the system) Same conventions for work ○ Engineers - reversed Now problem pg 233- Sample problem 6.1 Sample Exercise 6.1 (p. 233): Calculate ΔE for a system undergoing an endothermic process in which 15.6 kJ of heat flows and where 1.4 kJ of work is done on the system. We write the magnitudes and signs of the heat and the work as: q = +15.6 kJ and w = +1.4 kJ Both signs are positive because heat flows into the system and work is performed on the system. We then plug these quantities into the First Law (∆ E = q + w) to obtain the answer: ΔE = +15.6 kJ +1.4 kJ = 17.0 kJ The system has gained energy, so ΔE is a positive quantity. This is consistent with the fact that the process is endothermic. Work of GAS Expansion and Compression Example pistons move due to expansion of gas (work is done) Work of GAS Expansion and Compression The pressure (P) of the gases is related to the force (F) and the area (A) of the piston by the following equation: P = F/A We can solve this for the force; it is simply: F = P∙A Work is defined as force exerted over a distance. In this case, the distance is represented as Δh, so we can write the work as: work = F∙Δh Substituting for the force, we obtain: work = P∙A∙Δh = P∙ΔV Here ΔV is the difference between the final volume and the initial volume. What is the sign of this work? In this case, our system is expanding its volume against the surroundings, hence it is performing work on the surroundings. Thus the sign of the work should be negative, even though P and ΔV are both positive quantities. This forces us to write: w = — P∙ΔV More details p. 233 for the work done against the surroundings by an expanding gas. Note that this same expression gives us the correct sign in the case of the surroundings doing work on the system to compress a gas. In this case, ΔV is a negative quantity, making w a positive quantity. Sample Exercise 6.2 (p. 234): Calculate the work associated with the expansion of a gas from 46 L to 64 L at a constant external pressure of 15 atm. We start with our expression for work: w = — P∙ΔV Since ΔV is given by: ΔV = Vfinal — Vinitial We obtain for w: w = — P∙( Vfinal — Vinitial) = —15∙(64 — 46) L∙atm = —270 L∙atm The negative sign on this result is consistent with the fact that the system does work on the surroundings by its expansion. Sample Exercise 6.3 A balloon is being inflated by heating the air inside it. In the final states of the process, the volume of the balloon expands from 4.00x106 L to 4.50x106 L through the addition of 1.3x108 J of energy to the balloon in the form of heat. Assume that the external pressure remains at a constant 1.0 atm. Calculate ΔE for the process. To convert between J and L● atm, use 1 L ● atm = 101.3 J We will start with the First Law: ΔE = q + w Since 1.3x108 J of energy was added as heat, we can write that: q = +1.3x108 J And we can calculate the work: w = — P∙ΔV = — (1.0 atm)∙(0.50x106 L) = — 0.50x106 L∙atm The sign is negative, since the gas is expanding and doing work on the surroundings. Now we want to convert the work from L∙atm to J. We can use the unit factor: (101.3 J)=(1 L∙● atm) Thus the work is: w = — (0.50x106 L∙atm)∙(101.3 J)/(1 L∙atm) = — 0.51x108 J Now we are ready to plug the work and the heat into the First Law: ΔE = +1.3x108 J — 0.51x108 J = +0.8x108 J Enthalpy The Enthalpy (H) of a system is defined in terms of the internal energy (E), pressure (P) and volume (V) of the system: H = E + PV Since internal energy, pressure, and volume are all state functions, enthalpy is also a state function. But what is enthalpy? Consider a process carried out at constant pressure and such that the only work is pressure-volume work. (w = — P∙ΔV for pressure-volume work.) If we define qP as the heat of a process run at constant pressure, we can write the First Law: ΔE = qP + w = qP — P∙ΔV Solving for qP we obtain: qP = ΔE + PΔV Now we can relate qP to a change in enthalpy. Since we defined enthalpy as: H = E + PV We can write a change in enthalpy (ΔH) as: ΔH = ΔE + Δ(PV) Since our process runs at constant pressure, we can rewrite this as: ΔH = ΔE + PΔV But this is the same as our earlier expression for qP, the heat of a process run at constant pressure such that the only work is PV work. Thus: ΔH = qP Thus at constant pressure (and where only PV work is allowed), the change in enthalpy (ΔH) of the system is equal to the energy flow as heat. Because of this equality, we can say that in a reaction run at constant pressure, the change in enthalpy is the same as the heat of the reaction. Sample Exercise 6.4 (p. 236): Sample Exercise 6.4 (p. 236): When 1 mole of methane (CH4) is burned at constant pressure, 890 kJ of energy is released as heat. Calculate ΔH for a process in which 5.8 g of methane is burned at constant pressure: We can write the heat of reaction (at constant pressure) as: qp = ΔH = -890 kj/mol CH4 Since the number of moles of methane is:.36 mol The heat of the process where 5.8 g of methane burns at constant pressure thus is: .36 mol CH4 x -890kj/mol = -320kj Calorimetry Calorimetry is an experimental process for measuring heats of reaction. calorimeter. The device used for measuring heats of reactions Heat Measurements, Temperature, and Heat Capacity We do not have a way to make direct measurements of heat. What we actually measure is temperature, or, more specifically, the change in temperature resulting from a chemical reaction. Through knowledge of the heat capacity of the substance containing the heat generated by the process. We define the heat capacity (C) of a substance as: C = heat absorbed/ increase in temperature Influence of the heat capacity of water: Units of Heat Capacity If we heat a substance, the rise in temperature we obtain from adding a particular amount of heat depends on the amount of substance that is present. Specific Heat Capacity: When we express a heat capacity in terms of the mass of substance that is heated, we use units of joules per C° per gram, or equivalently, joules per Kelvin per gram per Kelvin (J/K∙g). Some specific heat capacities are given in Table 6.1: •Molar Heat Capacity: When we express a heat capacity in terms of the number of moles of substance that is heated, we use units of joules per C° per mole, or equivalently, joules per Kelvin per mole (J/K∙mol). Constant Pressure Calorimetry: Constant pressure calorimetry is nothing more than calorimetry carried out under conditions of constant pressure. A simple device for doing calorimetric measurements at ambient pressure is illustrated in Figure 6.5. It is made from Styrofoam coffee cups (hence the name, coffee cup calorimeter), but it is accurate enough for a student to use in chemistry lab Constant Pressure Calorimetry: Used for reactions in aqueous solutions. (Recall: heat generated in an exothermic reaction run at constant pressure is the change in enthalpy of the system.) We’ll follow the example in the text on pages 237-9, where we take 50.0 mL of 1.0 M HCl at 25.0 °C and 50.0 mL of 1.0 M NaOH also at 25.0 °C and mix them in the calorimeter. After we stir the mixed solutions, we measure a temperature of 31.9 °C. From these data we wish to determine the heat of the reaction that has just taken place inside the calorimeter. The reaction is the neutralization of a strong acid by a strong base: H+ (aq) + OH— (aq) ——> H2O (l) + qP Since we observed an increase in temperature, we know that the reaction generated heat, and we include this heat (qP) on the right hand side. At this point you might argue that the heat still remains in the system; that it has yet to flow to the surroundings. We can answer this argument two ways (They both yield the same result.): 1) We can define the system as consisting only of the ions that have reacted and the small amount of water that they generated in the reaction. Then the surroundings include the solvent water, and essentially all the heat produced by the reaction now resides in that portion of the surroundings that is inside the calorimeter. 2) Alternatively we could argue that the system includes the solvent, and the surroundings stop at the inside wall of the calorimeter. The heat that produced the rise in temperature has been temporarily captured within the system, and we can measure it before it flows out of the system into the surroundings. Either way, the heat of the reaction was sufficient to raise the temperature of 100.0 mL of water by 6.9 Kelvins. But how much heat is this? If we consult Table 6.1, we see that the heat capacity of water (Cwater) is 4.18 J/K∙g. Thus: q = Cwater ∙ m water ∙ ΔT = (418 J/K ∙ g) (100.0 g) (6.9K) = 2.9 x 103 Here q is the heat actually produced inside the calorimeter by the reaction of the HCl (aq) with the NaOH (aq). (We have assumed that the volume of the solution is exactly the combined volumes of the HCl (aq) and the NaOH (aq) and that the density of the solution is exactly 1.000 g/mL.) What if we had reacted twice as much acid with twice as much base? We would have obtained twice as much heat. What if we had reacted 1.00 mole of HCl (aq) with 1.00 mole of NaOH (aq)? We can calculate this from the number of moles that actually reacted and the heat (q) that was produced: H HCl = VHCl MHCl = (0.0500 L) (1.0 mol/L) = .050 mol ΔH = -q/nHCl = -2.9 x 103 J/.050mol = 58kJ/mol Here ΔH represents the enthalpy of reaction for 1 mole of HCl (aq) reacting with 1.00 mole of NaOH (aq). Sample 6.5 pg. 239 1.00 L of 1.00 M Ba(NO3)2 solution at 25.0 °C is reacted with 1.00 L of 1.00 M Na2SO4 solution, also at 25.0 °C in a constant pressure calorimeter. A white, solid precipitate of BaSO4 forms and the temperature of the mixture rises to 28.1 °C. Calculate the enthalpy change per mole of BaSO4 that forms. You may assume that the calorimeter absorbs a negligible amount of heat, that the specific heat capacity of the solution is 4.18 J/K∙g, and that the density of the solution is 1.0 g/mL. First we write the net ionic reaction: Ba2+ (aq) + SO42— (aq) ——> BaSO4 (s) We observed a rise in temperature, indicating that the reaction generated heat. Thus the reaction is exothermic and the change in enthalpy (ΔH) will be negative. The volume of the combined solution is 2.00x103 mL, so the mass of the solution is 2.00x103 g. The change in temperature is 3.1 K. Thus the heat evolved by the reaction is: q = Cwater ∙ m water ∙ ΔT = (4.18 J/K∙ g) (2.00 x 103 g) (3.1 K) = 2.5 x 104 J Since the reaction produced 1 mole of BaSO4 (s), the molar enthalpy change is ΔH = -q = -2.6 x 104 J/mol = -26kj/mol Bomb Calorimeter You are not required to do problems associated with this type of calorimeter, but it is cool to contemplate! Hess’s Law law used to predict the enthalpy change and conservation of energy (denoted as state function ΔH) regardless of the path through which, it is to be determined. The law states that because enthalpy is a state function, the enthalpy change of a reaction is the same regardless of what pathway is taken to achieve the products. In other words, only the start and end states matter to the reaction, not the individual steps between. Hess’s Law Example:(see schematic next slide) Example 1. The oxidation of nitrogen to produce nitrogen dioxide. The enthalpy change ΔH of the reaction is 68 kJ N2(g) + 2O2(g) = 2NO2(g) ΔH = 68 kJ OR This reaction also can be carried out in two distinct steps: N2(g) + O2(g) = 2NO(g) ΔH1 = 180 kJ 2NO(g) + O2(g) = 2NO2(g) ΔH2 = -112 kJ ____________________________________________ _ Net reaction: N2(g) + 2O2(g) = 2NO2(g) ΔH = ΔH1 + ΔH2 = 68 kJ Hints for Using Hess’s Law Calculations involving Hess’s law typically require that several reactions be manipulated and combined to finally give the reaction of interest. In doing this procedure you Should: • Work backward from the required reaction, using the reactants and products to decide how to manipulate the other given reactions at you disposal • Reverse any reactions as needed to give the required reactants and products • Multiply reactions to give the correct numbers of reactants and products Hess Rules and More If the reaction is multiplied (or divided) by some factor, H must also be multiplied (or divided) by that same factor. If the reaction is reversed (flipped), the sign of H must also be reversed. Sample Exercise 6.7 Two forms of carbon are graphite, the soft, black, slippery material used in “lead” pencils and as a lubricant for locks, and diamond, the brilliant, hard gemstone. Using the enthalpies of combustion for graphite (-394 kJ/mol) and diamond (-396 kJ/mol), calculate Δ H for the conversion of graphite to diamond: C graphite(s) Cdiamond See page 244 for solution Sample Exercise 6.8 Calculate the enthalpy of formation of diborane from its elements according to the equation: 2B(s) + 3H2(g) B2H6(g) from the following data: a.2B(s) + 3/2O2(g) B2O3(s) DH = -1273 kJ b.B2H6(g) + 3O2(g) B2O3(s) + 3H2O(g) DH = -2035 kJ c.H2(g) + 1/2O2(g) H2O(l) DH = -286 kJ d.H2O(g) H2O(l) DH = -44 kJ Solution: This kind of problem is best approached by working backwards from the required products given the information with which you are provided. There is no set "formula". Remember to be very careful to keep your equations balanced and make sure they sum to the required final equation. Focus on the boron and diborane: equations (a) and -(b) do what we want but involve a whole collection of junk not in the final equation: FIRST: (a) 2B(s) + 3/2O2(g) B2O3(s) DH = -1273 kJ -(b) B2O3(s) +3H2O(g) B2H6(g) + 3O2(g) DH = 2035 kJ ¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯ Sum: B2O3(s) + 2B(s) + 3/2O2(s) + 3H2O(g) B2O3(s) + B2H6(g) + 3O2(g) DH = 762 kJ Or: 2B(s) + 3H2O(g) B2H6(g) + 3/2O2(g) DH = 762 kJ Next : we have to get rid of the H2O(g) and O2(g) and introduce H2. 2B(s) + 3H2O(g) B2H6(g) + 3/2O2(g) DH = 762 kJ 3x(c) 3H2(g) + 3/2O2(g) 3H2O(l) DH = 3x(-286) kJ ¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯ Sum: 2B(s) + 3H2(g) + 3/2O2(g) + 3H2O(g) B2H6(g) + 3/2O2(g) + 3H2O(l) DH = -96 kJ The 3/2O2(g) cancels but not the water which is gas on one side and liquid on the other: 2B(s) + 3H2(g) + 3H2O(g) B2H6(g) + 3H2O(l) DH = -96 kJ 3x(-d) 3H2O(l) 3H2O(g) DH = 3x44 kJ ¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯ Sum: 2B(s) + 3H2(g) + 3H2O(g) + 3H2O(l) B2H6(g) + 3H2O(l) + 3H2O(g) DH = 36 kJ Or: 2B(s) + 3H2(g) B2H6(g) DH = 36 kJ Standard Enthalpies of Formation Many reactions cannot really be studied by calorimetry. The conversion of graphite to carbon requires enormous pressure and high temperature. Other processes would be dangerous: the decomposition of an explosive like nitroglycerine or diazomethane. The diborane example showed how it is possible to calculate enthalpies from other reactions which can be studied. A standard method of doing this is by starting with heats of formation. The Standard Enthalpy of Formation Δ Hfo of a compound is defined as the change in enthalpy that occurs when a mole of the compound is formed from its elements with all substances in their standard states. The Standard State for a substance is a precisely defined reference state: ○ The standard state for a gas is 1 atm pressure (101 305 Pa) or 1 bar (100 000 Pa). ○ For normally condensed states, liquid or solid is the standard state. ○ For solutions the standard concentration is 1 M. ○ For an element, the standard state is the normal form of the element at 1 atm and 25oC. Look again at the reaction for the formation of NO2 from its elements: 1/2N2(g) + O2(g) + energy(heat) D Hfo = 34 kJ/mol One mole of product is formed and all reactants are in their standard states. See Table 6.2 for typical heats of formation NO2(g) What about methane Here is how the enthalpy change for the combustion of CH4 can be calculated from the standard enthalpies of formation ( for this to make sense: see Fig 6.8): CH4(g) + 2O2(g) CO2(g) + 2H2O(l) Now: C(s) + H2(g) CH4(g) ΔHfo = -75 kJ Reaction (a) in the figure is the reverse of this. Reaction (b) is actually no change so ΔH = 0. Reaction (c) is another standard formation reaction: C(s) + O2(g) CO2(g) ΔHfo = -394 kJ/mol and reaction (d) is double the formation of water from its elements: 2H2(g) + O2(g) 2H2O(l)2. ΔHfo = -572 kJ So ΔHoreaction = ΔHo(a) +Δ Ho(b) + ΔHo(c) +Δ Ho(d) = [-Hfo Δfor CH4] + 0 + [ΔHfo for CO2] + [2. ΔHfo for H2O] = -891 kJ/mol In General: where np and nr are numbers of moles: ΔHoreaction = ∑npHfo(products) - ∑nr ΔHfo(reactants) Check out A-20 in the Appendix for LOTS of enthapies of formation